Introduction
Oral cancer is the eighth most common cancer
worldwide as reported by the International Agency for Research on
Cancer of the World Health Organization (1). The 5-year survival rate of oral cancer
patients has remained at 50% for the past decades (2). Information regarding the development
of oral cancer remains largely unknown.
The oral cavity is a viable environment for millions
of bacteria. Some of them, such as Genera fusobacterium,
Treponema denticola, Porphyromonas endodontics and
Porphyromonas gingivalis, are oral resident endogenetic
bacteria, which have a capacity to produce hydrogen sulfide
(H2S) (3).
H2S was previously known as a toxic gas. To date,
H2S is considered as the third gaseotransmitter along
with nitric oxide and carbon monoxide, and exerts important
physiological and pathological functions in the entire body. For
example, H2S is involved in the process of oxidative
stress-induced cytotoxicity (4–6),
cardioprotection (7–9), inflammation (10–12)
and cancer development (13,14).
However, the underlying mechanisms regulating the multiple
functions of H2S in many tissues and organs remain
unknown.
Considering that H2S is one of the main
causes of halitosis (3) and
represents an index of oral hygiene (15) and gastrointestinal health (16), H2S is thought to be
associated with oral diseases including periodontitis (17) and oral cancer (18,19).
Previous studies have shown that H2S can enhance the
severity of periodontitis (17,20).
In addition, an H2S controlled-releasing drug has been
developed for the treatment of cardiovascular disease (21). If oral cancer patients use a
controlled release H2S drug to manage their
cardiovascular disease, it is important to elucidate the effects of
H2S on oral cancer. The effects of H2S on
oral cancer remain largely unknown.
In this study, we investigated the effect of
H2S on oral cancer cell proliferation and the mechanisms
involved using three oral squamous cell carcinoma cell lines,
WSU-HN6 CAL27 and Tca83, through CCK-8, EdU incorporation, flow
cytometry, pathway inhibition, real-time PCR and western blot
assays.
Materials and methods
Reagents
Sodium hydrosulfide (NaHS) was obtained from
Sigma-Aldrich (St. Louis, MO, USA). Freshly made NaHS solution was
used as a hydrogen sulfide donor. Antibodies against cyclooxygenase
2 (COX2), phosphorylated ERK1/2 (p-ERK), total ERK1/2 (t-ERK),
phosphorylated AKT (p-AKT), and total AKT (t-AKT), were purchased
from Cell Signaling Technology (Danvers, MA, USA). An antibody
against GAPDH was purchased from Santa Cruz Biotechnology (Santa
Cruz, CA, USA). Dulbecco's modified Eagle's medium (DMEM),
RPMI-1640 and fetal bovine serum (FBS), trypsin-EDTA solution and
1% penicillin-streptomycin solution were purchased from Invitrogen
(Grand Island, NY, USA). COX2 inhibitor niflumic acid (NA), ERK
pathway inhibitor U0126 and AKT pathway inhibitor GSK690693 were
purchased from Sigma-Aldrich. Cell Counting Kit-8 (CCK-8) was
purchased from Dojindo Laboratories (Kumamoto, Japan).
Click-iT® EdU HCS Assay kit and TRIzol were purchased
from Invitrogen (Carlsbad, CA, USA). GoScript™ reverse
transcription system was purchased from Promega (Madison, WI, USA).
SYBR Green PCR Master Mix was obtained from Roche Diagnostics
(Indianapolis, IN, USA). RIPA buffer was a product of Applygen
Technologies, Inc. (Beijing, China). Bicinchoninic acid (BCA)
protein assay kit was purchased from Thermo Fisher Scientific Inc.
(Rockford, IL, USA). Polyvinylidene difluoride membranes were
purchased from Millipore Corporation (Billerica, MA, USA).
Cell culture
Human head and neck squamous cell carcinoma cell
lines WSU-HN6 and CAL27 were maintained in Dulbecco's minimum
essential medium (DMEM) supplemented with 10% FBS and 1%
penicillin-streptomycin solution. Human head and neck squamous cell
carcinoma cell line Tca83 was maintained in RPMI-1640 supplemented
with 10% FBS and 1% penicillin-streptomycin solution in a
humidified incubator at 37°C in an atmosphere of 5%
CO2.
Optimal NaHS dose finding assay
To determine the optimal dose of hydrogen sulfide
for the following experiments, we used NaHS, a commonly used donor
of H2S, to treat the oral cancer cells for 5 h in a T25
flask with 4 ml medium, and then detected the H2S
concentration in the atmosphere of a T25 flask by a halimeter. The
optimal concentration of NaHS was based on the range of the
H2S concentration found in patients with halitosis.
CCK-8 assay
Cell proliferation ability was evaluated using the
CCK-8 assay which was performed as previously described (18). Briefly, cells were cultured in
96-well tissue culture plates (1×104 cells/well) with
10% FBS overnight. Then, the cells were treated with 0–1,000
µM NaHS (different doses of NaHS were used in different
plates) and sealed with parafilm for an additional 5 h. The cell
proliferation was measured by the CCK-8 assay according to the
manufacturer's instructions.
Crystal violet assay staining assay
Cell proliferation ability was confirmed by crystal
violet assay. Briefly, 1×105 cells were plated in a T25
flask with 5 ml complete medium overnight. On the following day,
the cells were treated with various doses of NaHS for 3 days. The
cells were fixed with 95% alcohol, stained with crystal violet, and
photographed using a digital camera. Then crystal violet was
discolored from the stained cells using 33% glacial acetic acid.
The optical density (OD)570 of the crystal violet
correlates with the number of stained cells.
EdU incorporation assay
Cell proliferation ability was also detected using
the 5-ethynyl-2-deoxyuridine (EdU) incorporation assay. Briefly,
5×105 cells were plated in a T25 flask with 5 ml
complete medium overnight, and then the cells were serum-starved.
At 24 h post-starvation, the cells were treated with various doses
of NaHS in the presence of EdU for an additional 5 h. The labeled
cells were trypsinized for EdU incorporation assay using the
Click-iT EdU HCS assay kit (Invitrogen). EdU-positive cells were
detected by flow cytometry.
Real-time PCR
Cells in mid-logarithmic growth were used for
real-time PCR. Total RNA was extracted from the tumor cells using
TRIzol reagent, and cDNA was obtained by reverse transcription with
the GoScript™ reverse transcription system. Relative quantitative
real-time PCR reactions were performed with SYBR Green PCR Master
Mix in an ABI Prism 7500 sequence detection system (Applied
Biosystems Life Technologies, Foster City, CA, USA).
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the
endogenous control. The sequences of the COX2 primers were
5′-CTGGCGCTCAGCCATACAG-3′ and 5′-ACACTCATACATACACCTCGGT-3′. The
thermal cycling consisted of 10 min at 95°C, followed by 40 cycles
at 95°C for 15 sec, and at 60°C for 1 min. All amplifications were
performed in triplicate for each sample and repeated three times
independently. Relative expression of the target genes was analyzed
using the 2−ΔΔCt method.
Western blot analysis
Cells were lysed on ice in RIPA buffer supplemented
with protease inhibitors (Roche Diagnostics). The concentration of
total protein was determined using the BCA protein assay kit. Equal
quantities of protein (30 µg) were separated by sodium
dodecyl sulfate-polyacrylamide gel electrophoresis and transferred
to polyvinylidene difluoride membranes. The membranes were blocked
in 5% nonfat dry milk for 1 h and probed with primary antibodies
against GAPDH, COX2, p-ERK, t-ERK, p-AKT, t-AKT, respectively, at
4°C overnight. After incubation with horseradish peroxidase
(HRP)-linked secondary antibodies, the protein bands were
visualized using an enhanced chemiluminescent substrate (Applygen
Technologies).
Pathway inhibition assay
To investigate the role of COX2, ERK and AKT
pathways in hydrogen sulfide-induced cancer cell proliferation,
pathway inhibition assays were carried out using an inhibitor for
each molecule/pathway. Based on our preliminary data and previous
studies (22–24), we found that COX2 could be markedly
downregulated by 100 µM niflumic acid (NA) at 24 h
post-treatment; p-ERK and p-AKT could be significantly blocked by
50 µM U0126 and 1 µM GSK690693 at 1 h post-treatment,
respectively. Therefore, in the present study, WSU-HN6 cells were
pretreated with either COX2 inhibitor NA for 24 h, ERK pathway
inhibitor U0126 for 1 h, or AKT pathway inhibitor GSK690693 for 1
h, under serum-starvation condition, and then exposed to NaHS for 5
h for the real-time PCR and western blot assays or with EdU reagent
for the EdU incorporation assay.
Statistical analysis
The results are expressed as mean ± SD. Differences
between groups were analyzed by one-way ANOVA. Significance was
established at the P<0.05 level.
Results
Determination of the optimal NaHS
concentration
The NaHS dose finding assay showed that the
concentration of H2S in the atmosphere of 1,000
µM NaHS-treated cells was 698.33±18.93 ppb, which is equal
to 1073.34±29.09 µg/l. Based on previous studies, the
concentration of H2S in patients with halitosis is
810.30±204.09 µg/l (25).
Therefore, the H2S concentration released from 250, 500
and 1,000 µM NaHS was comparable with the H2S
concentration found in the oral cavity of patients with halitosis,
and 1,000 µM NaHS was in the range of the highest
concentration of H2S in the oral cavity of halitosis
patients (Fig. 1A).
H2S promotes the growth of
oral cancer cells
To evaluate the effect of H2S on oral
cancer cell proliferation, the CCK-8, crystal violet staining and
EdU incorporation assays were performed. The CCK-8 assay showed
that H2S significantly promoted the proliferation of the
WSU-HN6 and CAL27 cells in a dose-dependent manner. The WSU-HN6,
CAL27 and Tca83 cells treated with 1,000 µM NaHS exhibited a
22.94, 33.79 and 32.20% increase in proliferation, respectively
(Fig. 1B, P<0.05). Crystal
violet staining assay showed that the cell growth of WSU-HN6 and
CAL27 cells was accelerated by NaHS in a dose-dependent manner
(Fig. 1C–F). The CCK-8 results were
further confirmed in the WSU-HN6 cells using the EdU incorporation
assay, which showed a 16.19±1.89% increase in the proliferation of
the WSU-HN6 cells following treatment with 1,000 µM NaHS
(Fig. 1G, P<0.05).
H2S promotes oral cancer cell
proliferation through the activation of COX2, ERK and AKT signaling
pathways
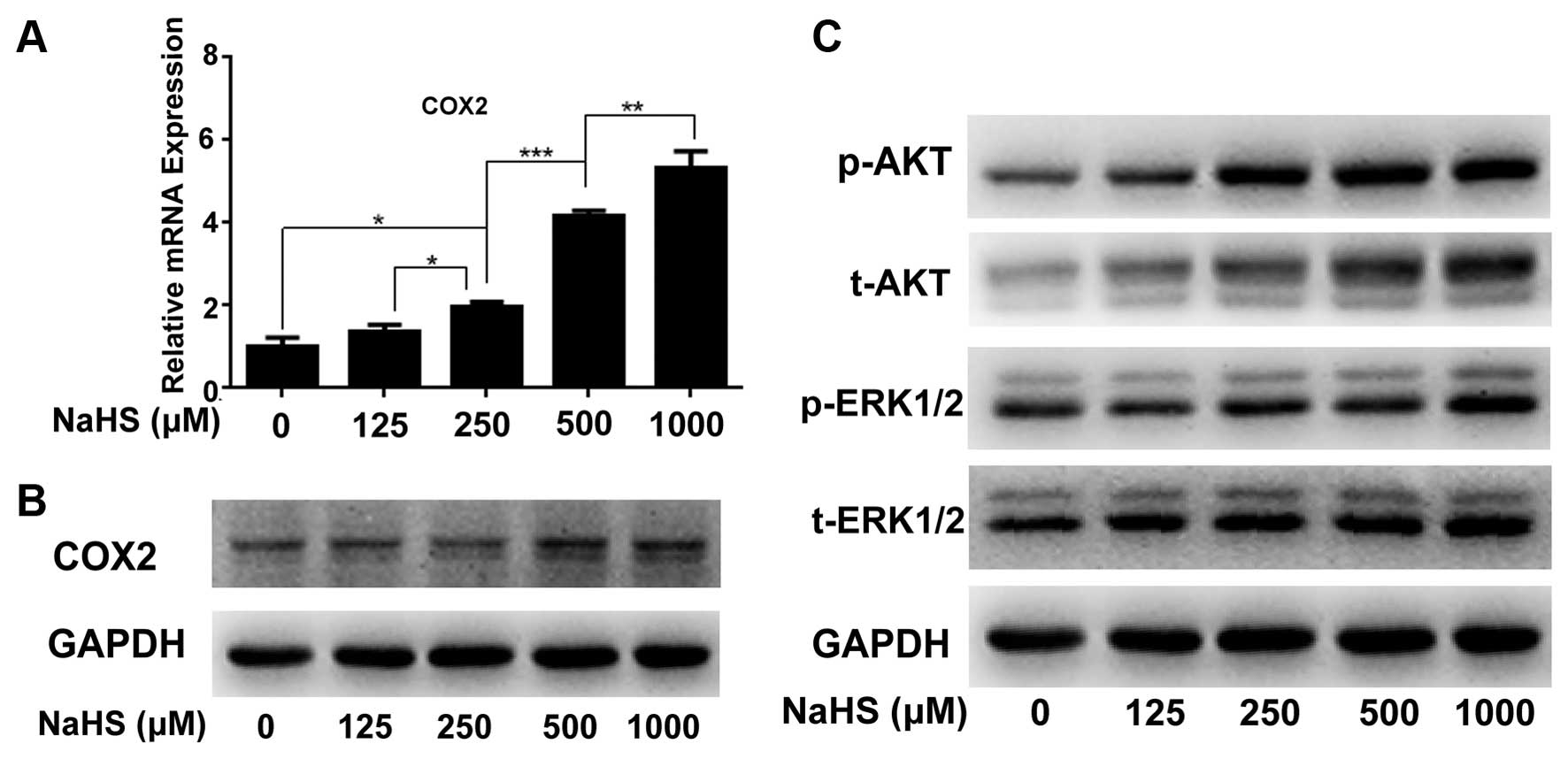
To analyze the molecular changes during NaHS-induced
cancer cell proliferation, we detected the changes in cancer
proliferation-related molecule COX2 by real-time PCR (Fig. 2A) and western blotting (Fig. 2B), and pathways including ERK and
AKT by western blotting (Fig. 2C).
The results showed that compared with the corresponding controls,
hydrogen sulfide markedly upregulated COX2 at the mRNA and protein
levels in a dose-dependent manner (Fig.
2A and B). Meanwhile, H2S significantly activated
p-ERK and p-AKT expression in a dose-dependent manner (Fig. 2C).
Inhibition of COX2, ERK and AKT by their
corresponding inhibitors significantly blocks
H2S-induced oral cancer cell proliferation
To investigate the role of COX2, the ERK and AKT
pathways in H2S-induced oral cancer cell proliferation,
we uses the CCK-8 and EdU cellular incorporation assays to detect
the change in proliferation. The proliferation of WSU-HN6 cells
caused by H2S was decreased by inhibition of COX2, ERK
and AKT pathways. CCK-8 assay showed that in the WSU-HN6 cells,
treatment of NA (COX2 inhibitor) resulted in a nearly 50% decrease
in growth caused by H2S. Meanwhile, U0126 (ERK pathway
inhibitor) and GSK690693 (AKT pathway inhibitor) achieved a less
significant decrease in proliferation as determined by the CCK-8
assay (Fig. 3A).
The EdU incorporation assay further confirmed the
CCK-8 results. Compared with the untreated control, all these
inhibitors alone decreased the percentage of EdU-positive cells
while NaHS alone increased the percentage of EdU-positive cells. NA
alone caused a 11.07±3.71% increase, while U0126 and GSK690693
caused a 3.9±0.98 and 3.22±1.00% decrease in the WSU-HN6 cells.
Compared with NaHS treatment alone, NaHS in combination with NA
yielded a 31.24±6.39% decrease; NaHS combined with U0126 yielded a
51.50±1.95% decrease; and NaHS plus GSK690693 yielded a 50.35±1.89%
decrease in Edu-positive cells. The results above indicated that
H2S-induced upregulation of COX2, p-ERK and p-AKT plays
a positive role in the process of H2S-induced
proliferation of oral cancer cells (Fig. 3B).
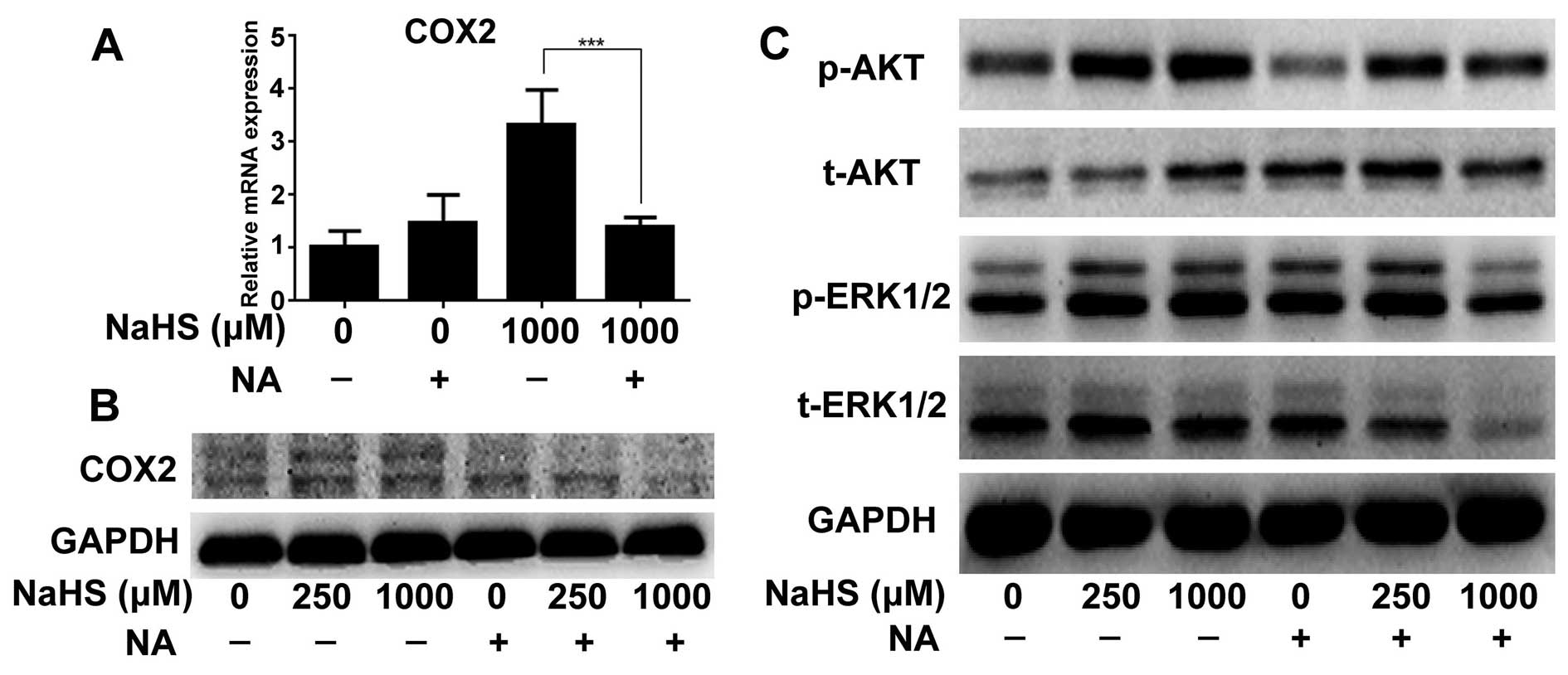
COX2 inhibitor downregulates NaHS-induced
p-AKT and p-ERK expression
To further verify the role of COX2 in the promotion
of proliferation, we detected the changes in the ERK and AKT
pathway folowing the treatment of NA. NA not only decreased COX2
expression caused by H2S at the mRNA and protein levels
(Fig. 4A and B), but also decreased
p-ERK and p-AKT expression when the cells were treated with 1,000
µM of NaHS (Fig. 4C).
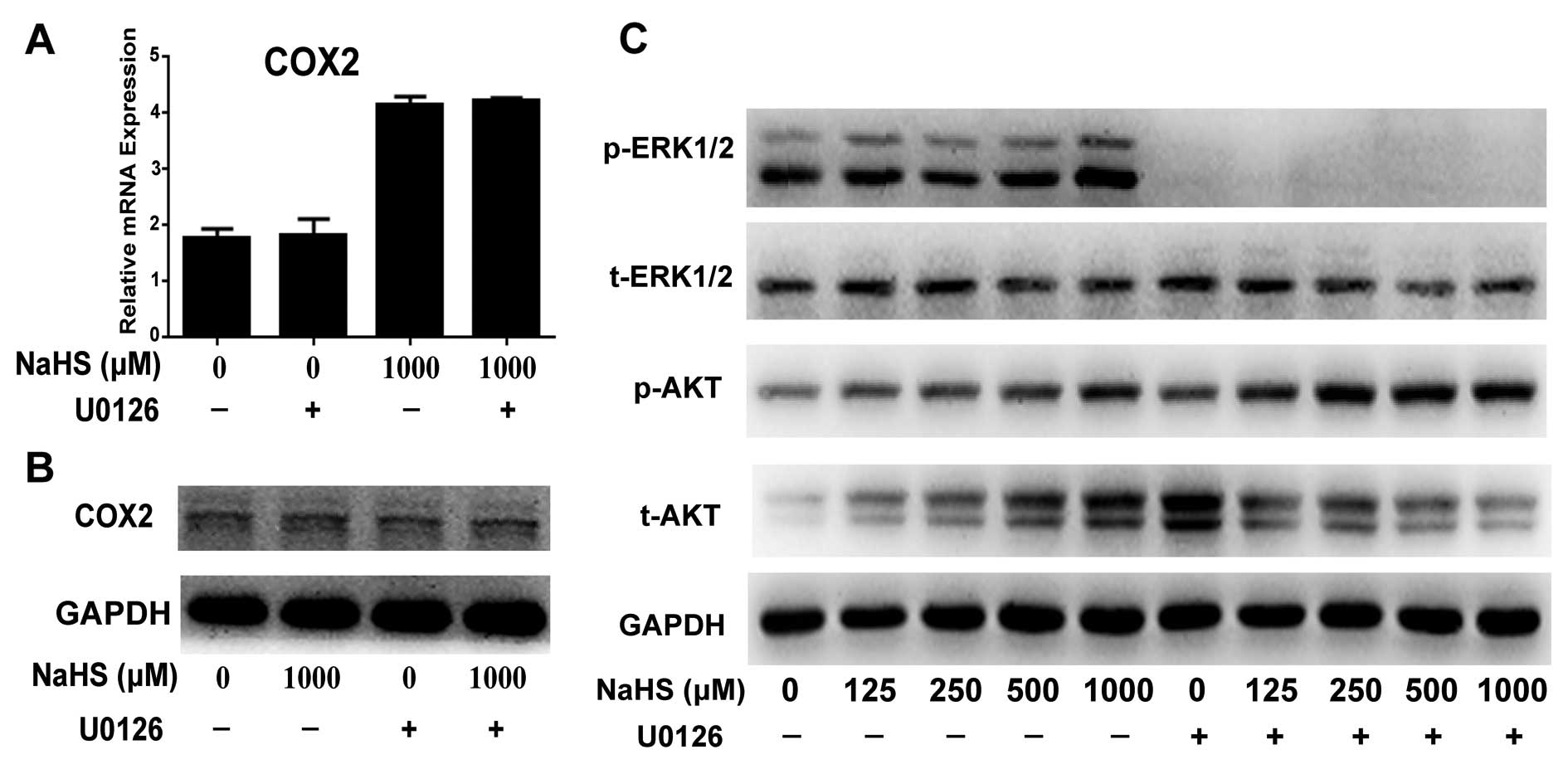
Blockade of the ERK pathway by U0126
upregulates H2S-induced p-AKT expression, but does not
affect H2S-induced COX2 expression
To investigate the role of the ERK pathway in
proliferation, U0126 was used to inhibit p-ERK. Western blotting
showed that U0126 completely blocked the activation of the ERK
pathway by eliminating p-ERK expression (Fig. 5C). Real-time PCR and western
blotting showed that inhibition of the ERK pathway by U0126 did not
change COX2 expression at the mRNA and protein levels (Fig. 5A and B). Furthermore, U0126
increased the activation of the H2S-induced p-AKT
pathway (Fig. 5C).
Blockade of the AKT pathway downregulates
H2S-induced p-ERK expression, but does not affect
H2S-induced COX2 expression
To verify the role of the AKT pathway in
H2S-induced cell proliferation, GSK690693 was used for
inhibition of p-AKT. Western blotting showed that GSK690693
decreased the expression of p-AKT (Fig.
6C). Real-time PCR results showed that GSK690693 did not change
H2S-induced COX2 expression at the mRNA and protein
levels (Fig. 6A and B), although
GSK690693 alone induced an increase in COX2 at the mRNA level. In
addition, inhibition of the AKT pathway decreased the expression
level of p-ERK (Fig. 6C).
Discussion
H2S is characterized by its rotten egg
odor yet exists widespread in the body. It is produced naturally by
either protein metabolism through cystathionine γ-lyase, (CSE),
cystathionine-β-synthase (CBS), and 3-Mercaptopyruvate
sulfurtransferase (3-MT), or bacteria existing in the digestive,
respiratory or genital tracts reducing compound-containing sulfur
element (26). Since it has been
identified as the third gaseotransmitter playing an important
physiological and pathological role in the body, more attention has
been given to its beneficial properties in many organs and systems.
However, in the oral cavity, H2S is commonly considered
to play a deleterious role. For example, H2S is taken as
one of the main contributors of bad mouth breath (halitosis), which
is often associated with dental plaque and periodontal disease
(17,20). Moreover, H2S can promote
cell cycle progression in oral cancer cells (18). However, the mechanisms involved in
the regulation of oral cancer growth by H2S are not
fully elucidated.
In the present study, we investigated the underlying
mechanism and found that the COX2-AKT-ERK1/2 axis is closely
associated the regulation of oral cancer growth by H2S.
We firstly identified that the concentrations of H2S
released from 250, 500 and 1,000 µM NaHS used in the T25
flask were comparable with the H2S concentrations found
in patients with halitosis. In addition, 1,000 µM NaHS is in
the range of the highest concentration of H2S in the
oral cavity of halitosis patients (25). Next, we confirmed that
H2S promoted oral cancer growth through CCK-8 crystal
violet staining and EdU incorporation assays. Meanwhile, we found
that COX2, ERK and AKT may be involved in this process through
real-time PCR and western blotting.
Along with the increase in NaHS concentration, an
increase in the dose-dependent proliferation of oral cancer cells
and upregulation of COX2 expression were concurrent. In addition,
when COX2 expression was blocked by the COX2 inhibitor, the effect
of H2S-induced oral cancer cell proliferation was
completely inhibited. These results indicated that the regulation
of oral cancer cell growth by H2S was closely related
with COX2 expression. It was previously found that COX2 expression
has a close relationship with the proliferation of other cancers
(23,24,26–28).
Activation of COX2 contributes to the progression from Barrett's
metaplasia to esophageal cancer (27). Upregulation of COX2 expression
promotes colorectal tumorigenesis and metastasis (28). Inhibition of COX2 decreases the
ability of breast cancer cell motility in breast cancer tissues
(23,24). Of importance, a meta-analysis showed
that COX2 expression in oral cancer tissues was significantly
higher than that in normal benign tissues, and positive COX2
expression may be a marker of worse prognosis in oral cancer
patients (29).
In addition to the COX2 molecule, we also found two
important pathways (AKT and ERK pathways) associated with the
H2S-induced oral cancer cell proliferation, AKT and ERK
were activated during H2S-induced cell proliferation,
and blockade of either ERK or AKT pathway reduced this effect. The
results indicate that H2S-mediated oral cancer growth is
also related with the AKT and ERK pathways. A previous study
reported that ERK activation is necessary for cancer cell
proliferation (30). Activation of
the ERK pathway enhanced the resistance of prostate cancer and
osteosarcoma cells to apoptosis (31). Scutellarein inhibits proliferation
of the human lung cancer via reducing the expression levels of
p-ERK (32). Baicalin
dose-dependently inhibited vascular smooth muscle cell
proliferation by blocking the ERK pathway (33). Activation of the AKT pathway
contributes to the proliferation of gastric cancer (34) and cervical cancer cells (35). Curcumin was found to play an
anticancer role by suppressing AKT phosphorylation (36). Furthermore, inactivation of the AKT
and ERK pathways contributes to the inhibition of prostate cancer
growth (37).
The results of this experiment are in accordance
with the proliferation-promoting role of the ERK and AKT pathways.
Through pathway inhibition and EdU incorporation assays, we can
conclude that the COX2, ERK and AKT pathways are involved in the
regulation of oral cancer growth by H2S. The next
question is whether COX2, ERK and AKT pathways have a regulatory
role in the hydrogen sulfide-regulated oral cancer growth. From the
pathway inhibition assays, we found that blockade of COX2 clearly
downregulated H2S-induced p-ERK and p-AKT expression.
Inactivation of the AKT pathway markedly decreased
H2S-induced p-ERK expression but did not alter the COX2
expression level. Inhibition of the ERK pathway significantly
increased H2S-induced p-AKT expression but did not
significantly alter the level of COX2 expression. Based on these
data (Fig. 7), we can deduce that
H2S promotes oral cancer cell proliferation through the
COX2/AKT/ERK1/2 axis. Upregulation of p-AKT expression by
H2S is due to the reactive response after blockade of
the ERK pathway. The cells express more p-AKT in order to enhance
the expression level of p-ERK, that is, to activate the ERK
pathway.
In conclusion, in the present study, we, for the
first time, identified a new mechanism elucidating that
H2S promotes oral cancer cell proliferation through the
COX2/AKT/ERK1/2 axis, which may provide new potential targets by
which to eliminate the effect of H2S on oral cancer
development.
Acknowledgments
The study was supported by the Research Fund for
Capital Medical Development (2011-0425-02), Research Grants from
the Nature Foundation of Heilongjiang Province (no. QC2014C107),
National Nature Science Foundation of China (grant no. 81470707),
National Natural Science Foundation of China (81300901), and
Tianjin Natural Science Foundation (14JCQNJC12500).
References
|
1
|
Edwards BK, Brown ML, Wingo PA, Howe HL,
Ward E, Ries LA, Schrag D, Jamison PM, Jemal A, Wu XC, et al:
Annual report to the nation on the status of cancer, 1975–2002,
featuring population-based trends in cancer treatment. J Natl
Cancer Inst. 97:1407–1427. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Persson S, Edlund MB, Claesson R and
Carlsson J: The formation of hydrogen sulfide and methyl mercaptan
by oral bacteria. Oral Microbiol Immunol. 5:195–201. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xie L, Feng H, Li S, Meng G, Liu S, Tang
X, Ma Y, Han Y, Xiao Y, Gu Y, et al: SIRT3 mediates the antioxidant
effect of hydrogen sulfide in endothelial cells. Antioxid Redox
Signal. 24:329–343. 2016. View Article : Google Scholar
|
|
5
|
Xie ZZ, Shi MM, Xie L, Wu ZY, Li G, Hua F
and Bian JS: Sulfhydration of p66Shc at cysteine59 mediates the
antioxidant effect of hydrogen sulfide. Antioxid Redox Signal.
21:2531–2542. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Heneberg P: Reactive nitrogen species and
hydrogen sulfide as regulators of protein tyrosine phosphatase
activity. Antioxid Redox Signal. 20:2191–2209. 2014. View Article : Google Scholar :
|
|
7
|
Calvert JW, Jha S, Gundewar S, Elrod JW,
Ramachandran A, Pattillo CB, Kevil CG and Lefer DJ: Hydrogen
sulfide mediates cardioprotection through Nrf2 signaling. Circ Res.
105:365–374. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Salloum FN, Chau VQ, Hoke NN, Abbate A,
Varma A, Ockaili RA, Toldo S and Kukreja RC: Phosphodiesterase-5
inhibitor, tadalafil, protects against myocardial
ischemia/reperfusion through protein-kinase g-dependent generation
of hydrogen sulfide. Circulation. 120(Suppl 11): S31–S36. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kondo K, Bhushan S, King AL, Prabhu SD,
Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G Sr, Gojon G Jr,
et al: H2S protects against pressure overload-induced
heart failure via upregulation of endothelial nitric oxide
synthase. Circulation. 127:1116–1127. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Badiei A, Rivers-Auty J, Ang AD and Bhatia
M: Inhibition of hydrogen sulfide production by gene silencing
attenuates inflammatory activity of LPS-activated RAW264.7 cells.
Appl Microbiol Biotechnol. 97:7845–7852. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miller TW, Wang EA, Gould S, Stein EV,
Kaur S, Lim L, Amarnath S, Fowler DH and Roberts DD: Hydrogen
sulfide is an endogenous potentiator of T cell activation. J Biol
Chem. 287:4211–4221. 2012. View Article : Google Scholar :
|
|
12
|
Mok YY and Moore PK: Hydrogen sulphide is
pro-inflammatory in haemorrhagic shock. Inflamm Res. 57:512–518.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Szabo C, Coletta C, Chao C, Módis K,
Szczesny B, Papapetropoulos A and Hellmich MR: Tumor-derived
hydrogen sulfide, produced by cystathionine-β-synthase, stimulates
bioenergetics, cell proliferation, and angiogenesis in colon
cancer. Proc Natl Acad Sci USA. 110:12474–12479. 2013. View Article : Google Scholar
|
|
14
|
Elsheikh W, Blackler RW, Flannigan KL and
Wallace JL: Enhanced chemopreventive effects of a hydrogen
sulfide-releasing anti-inflammatory drug (ATB-346) in experimental
colorectal cancer. Nitric Oxide. 41:131–137. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gokdogan O, Catli T and Ileri F: Halitosis
in otorhinolaryngology practice. Iran J Otorhinolaryngol.
27:145–153. 2015.PubMed/NCBI
|
|
16
|
Motta JP, Flannigan KL, Agbor TA, Beatty
JK, Blackler RW, Workentine ML, Da Silva GJ, Wang R, Buret AG and
Wallace JL: Hydrogen sulfide protects from colitis and restores
intestinal microbiota biofilm and mucus production. Inflamm Bowel
Dis. 21:1006–1017. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang JH, Dong Z and Chu L: Hydrogen
sulfide induces apoptosis in human periodontium cells. J
Periodontal Res. 45:71–78. 2010. View Article : Google Scholar
|
|
18
|
Ma Z, Bi Q and Wang Y: Hydrogen sulfide
accelerates cell cycle progression in oral squamous cell carcinoma
cell lines. Oral Dis. 21:156–162. 2015. View Article : Google Scholar
|
|
19
|
Balasenthil S, Rao KS and Nagini S:
Apoptosis induction by S-allylcysteine, a garlic constituent,
during 7,12-dimethylbenz[a] anthracene-induced hamster buccal pouch
carcinogenesis. Cell Biochem Funct. 20:263–268. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Morita M and Wang HL: Association between
oral malodor and adult periodontitis: A review. J Clin Periodontol.
28:813–819. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wen YD and Zhu YZ: The pharmacological
effects of S-propargyl-cysteine, a novel endogenous
H2S-producing compound. Handbook Exp Pharmacol.
230:325–336. 2015. View Article : Google Scholar
|
|
22
|
Kim BM, Maeng K, Lee KH and Hong SH:
Combined treatment with the Cox-2 inhibitor niflumic acid and PPARγ
ligand ciglitazone induces ER stress/caspase-8-mediated apoptosis
in human lung cancer cells. Cancer Lett. 300:134–144. 2011.
View Article : Google Scholar
|
|
23
|
Larkins TL, Nowell M, Singh S and Sanford
GL: Inhibition of cyclooxygenase-2 decreases breast cancer cell
motility, invasion and matrix metalloproteinase expression. BMC
Cancer. 6:1812006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brueggemeier RW, Díaz-Cruz ES, Li PK,
Sugimoto Y, Lin YC and Shapiro CL: Translational studies on
aromatase, cyclooxygenases, and enzyme inhibitors in breast cancer.
J Steroid Biochem Mol Biol. 95:129–136. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang W, Guo J, Huang D and Cheng W:
Correlation of volatile sulfide compounds in the oral cavity with
periodontitis and tongue coating. Zhongguo Shengwu Yixue Gongcheng
Xuebao. 18:149–152. 2012.In Chinese.
|
|
26
|
Moore PK, Bhatia M and Moochhala S:
Hydrogen sulfide: From the smell of the past to the mediator of the
future? Trends Pharmacol Sci. 24:609–611. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Looby E, Abdel-Latif MM, Athié-Morales V,
Duggan S, Long A and Kelleher D: Deoxycholate induces COX-2
expression via Erk1/2-, p38-MAPK and AP-1-dependent mechanisms in
esophageal cancer cells. BMC Cancer. 9:1902009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Greenhough A, Smartt HJ, Moore AE, Roberts
HR, Williams AC, Paraskeva C and Kaidi A: The COX-2/PGE2 pathway:
Key roles in the hallmarks of cancer and adaptation to the tumour
micro-environment. Carcinogenesis. 30:377–386. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang ZM, Liu J, Liu HB, Ye M, Zhang YF and
Yang DS: Abnormal COX2 protein expression may be correlated with
poor prognosis in oral cancer: A meta-analysis. Biomed Res Int.
2014:3642072014.PubMed/NCBI
|
|
30
|
Chambard JC, Lefloch R, Pouysségur J and
Lenormand P: ERK implication in cell cycle regulation. Biochim
Biophys Acta. 1773:1299–1310. 2007. View Article : Google Scholar
|
|
31
|
Rasola A, Sciacovelli M, Chiara F, Pantic
B, Brusilow WS and Bernardi P: Activation of mitochondrial ERK
protects cancer cells from death through inhibition of the
permeability transition. Proc Natl Acad Sci USA. 107:726–731. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cheng CY, Hu CC, Yang HJ, Lee MC and Kao
ES: Inhibitory effects of scutellarein on proliferation of human
lung cancer A549 cells through ERK and NFκB mediated by the EGFR
pathway. Chin J Physiol. 57:182–187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dong LH, Wen JK, Miao SB, Jia Z, Hu HJ,
Sun RH, Wu Y and Han M: Baicalin inhibits PDGF-BB-stimulated
vascular smooth muscle cell proliferation through suppressing
PDGFRβ-ERK signaling and increase in p27 accumulation and prevents
injury-induced neointimal hyperplasia. Cell Res. 20:1252–1262.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ye Y, Ge YM, Xiao MM, Guo LM, Li Q, Hao
JQ, Da J, Hu WL, Zhang XD, Xu J, et al: Suppression of SHIP2
contributes to tumorigenesis and proliferation of gastric cancer
cells via activation of Akt. J Gastroenterol. Jul 23–2015.(Epub
ahead of print). http://dx.doi.org/10.1007/s00535-015-1101-0.
PubMed/NCBI
|
|
35
|
Huang H, Song Y, Wu Y, Guo N, Ma Y and
Qian L: Erbin loss promotes cancer cell proliferation through
feedback activation of Akt-Skp2-p27 signaling. Biochem Biophys Res
Commun. 463:370–376. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yallapu MM, Khan S, Maher DM, Ebeling MC,
Sundram V, Chauhan N, Ganju A, Balakrishna S, Gupta BK, Zafar N, et
al: Anti-cancer activity of curcumin loaded nanoparticles in
prostate cancer. Biomaterials. 35:8635–8648. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rick FG, Schally AV, Szalontay L, Block
NL, Szepeshazi K, Nadji M, Zarandi M, Hohla F, Buchholz S and Seitz
S: Antagonists of growth hormone-releasing hormone inhibit growth
of androgen-independent prostate cancer through inactivation of ERK
and Akt kinases. Proc Natl Acad Sci USA. 109:1655–1660. 2012.
View Article : Google Scholar : PubMed/NCBI
|