Introduction
Esophageal carcinoma is the eighth most common type
of cancer worldwide, with high incidence rates and poor prognosis
among patients in developing countries (1,2). The
two most common types of esophageal carcinoma are esophageal
squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC),
with ESCC being the most dominant subtype of esophageal carcinoma
in East Asia and China, possibly due to smoking and eating habits
(3). During past decades, though
tremendous progress has been made toward targeted chemotherapy,
radiotherapy or advanced surgery to improve clinical outcomes, the
overall survival rate for patients with ESCC is still very poor,
normally below 30% (4).
MicroRNAs (miRNAs) are small (~18 to 22 nt long),
non-coding RNAs that post-transcriptionally inhibit gene production
or induce protein degradation by binding on the complimentary DNA
sequence on the 3′ untranslated region (3′ UTR) of the targeted
genes (5,6). Studies have shown that miRNAs are
differentially expressed in both serum and tumor tissues, thus,
playing important regulatory roles, acting as either oncogenes or
tumor suppressor genes in human esophageal carcinoma (7–9).
MicroRNA-181d (miR-181d), belongs to the family of miR-181, which
includes miR-181a, miR-181b, miR-181c and miR-181d. miR-181d has
been shown to be aberrantly expressed in various types of
carcinomas, and to play critical roles in regulating
carcinogenesis, cancer metastasis or cancer apoptosis in human
cancers. For example, miR-181d had tumor suppressive effect on
glioma migration, apoptosis and cell cycle transition through the
regulation of K-ras (10). In
addition, miR-181d was shown to be a reliable biomarker for human
glioblastoma, and acted as a tumor suppressor to inhibit cancer
migration by downregulating methylguanine-methyltransferase
(11). In human ESCC, miR-181d,
along with miR-335, miR-25, miR-7 and miR-495, were closely
associated with the patients gross pathologic classification
(12). However, the exact gene
expression pattern or possible cancer regulatory role of miR-181d,
have not been elucidated.
Gene of Derlin-1 (DERL1) is the partner of ATPase
complex, and initially found to regulate misfolding proteins at
endoplasmic reticulum (13,14). In human cancers, DERL1 has been
mostly reported to be an oncogene of regulating carcinoma
progression. In colon cancer, DERL1 was upregulated in carcinoma
tissues and promoted cancer proliferation (15). In pancreatic cancer, DERL1 was
overexpressed in tumor cells and direct targeted therapy on tumor
growth in animal models (16). In
addition, DERL1 was upregulated in non-small cell lung cancer, and
induced cancer cell progression and migration through the
regulation on EGFR signaling pathways (17,18).
Yet, the role of DERL1 in human ESCC has not been determined.
In the present study, we first assessed the gene
expression level of miR-181d in both ESCC cell lines and clinical
samples from ESCC patients. Secondly, we applied lentiviral
transduction in ESCC cell lines, ECA109 and Kyse30 to ectopically
upregulate miR-181d. Thirdly, we examined the effects of miR-181d
overexpression on ESCC proliferation, migration, cell cycle
transition in vitro and ESCC tumorigenicity in vivo.
Fourthly, we investigated whether DERL1 was the downstream target
gene of miR-181d in ESCC. Finally, we overexpressed DERL1 gene in
ESCC cells to assess its functions in regulating ESCC growth in
association with miR-181d overexpression.
Materials and methods
Ethics statements
In the present study, all protocols were approved by
the Ethics Committee of the Shandong Provincial Hospital Affiliated
to Shandong University (Jinan, Shandong, China). All participating
subjects provided a written consent. All procedures were conducted
in accordance with the Declaration of Helsinki, local and central
regulations for good clinical practice in China.
ESCC cell lines and clinical samples
In the present study, human ESCC cell lines, ECA109,
Kyse30, Kyse50, Kyse70, Kyse150 and Kyse510 were purchased from the
Cell Bank of Type Culture Collection of Chinese Academy of Sciences
(Shanghai, China). A control cell line, esophageal epithelial cell
line (HET-1A) was purchased from the American Type Culture
Collection (ATCC; Manassas, VA, USA). All cells were maintained in
RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS;
Thermo Fisher Scientific, Waltham, MA, USA) at 37°C with 95/5% of
O2/CO2.
Between March 2012 and March 2016, paired clinical
samples, including ESCC tissues (T) and their adjacent normal
esophageal epithelial tissues (ANT) were surgically obtained from
16 patients diagnosed with ESCC in the Department of Thoracic
Surgery at Shandong Provincial Hospital Affiliated to Shandong
University (Jinan, Shandong, China). Once extracted, all clinical
samples were immediately snap-frozen in a bath of liquid nitrogen,
labelled and stored at −70°C until RNA extraction for quantitative
RT-PCR.
RNA extraction and qRT-PCR
RNA extraction was carried out using a TRIzol kit
(Thermo Fisher Scientific) according to the manufacturer's
protocol. After confirming the quality of RNA extract by a
NanoDrop-3000 spectrophotometer (Thermo Fisher Scientific),
reverse-transcription was performed using a QuantiTect Reverse
Transcription kit (Qiagen Inc., Valencia, CA, USA) according to the
manufacturer's protocol. Quantitative real-time polymerase chain
reaction (qRT-PCR) was carried out on an ABI Prism 7700 Sequence
Detection system (Thermo Fisher Scientific). For quantification on
miR-181d expression, a TaqMan miRNA PCR real-time RT-PCR kit
(Applied Biosystems, Foster City, CA, USA) was used with U6 snRNA
as sampling control. For quantification of DERL1 expression, a
Brilliant SYBR-Green II qRT-PCR kit (Strategene, La Jolla, CA, USA)
was used with 18s as sampling control. Gene levels of miR-181d and
DERL1 were calculated as fold changes relative to U6 or 18s levels
and then normalized to the relative gene expression in control
samples using the 2−ΔΔCT method, where CT represents
cycle thresholds.
miR-181d overexpressing assay
The lentivirus including synthetic miR-181d mimic
oligonucleotides were purchased from SunBio Biomedical Technology
(Nanjing, China) (Lenti-miR181d-M). The control non-specific
lentiviral inhibitor vector (Lenti-C) was also purchased from
SunBio Biomedical Technology. ESCC cell lines, ECA109 and Kyse30
cells, were transduced with Lenti-C or Lenti-miR181d-M, along with
10 μg/ml Polybrene, at multiplicity of infection of 20–35.
48 h later, culture medium was changed to fresh one without
lentiviruses and cells were maintained for another 3–5 days to
stabilize lentiviral transduction. Transduction efficiency was then
confirmed by qRT-PCR.
ESCC in vitro proliferation assay
The in vitro cancer cell growth was assessed
by a proliferation assay. Briefly, ECA109 and Kyse30 cells were
plated at 1,000 cells/well in single-cell layer into a 96-well
plate. A Vybrant® MTT Cell Proliferation Assay kit
(Thermo Fisher Scientific) was then carried out for five
consecutive days according to the manufacturer's protocol. Each 24
h, MTT (5 μg/ml) was added to each well for 4 h.
Proliferation rates were characterized as recorded optical
absorbance at 490 nm using a V-5000 spectrophotometer reader
(Yuanxi Instrument Co., Ltd., Shanghai, China).
ESCC in vitro migration assay
The in vitro migrating capability of ESCC
cells was assessed by a Transwell migration assay (Thermo Fisher
Scientific) according to the manufacturer's protocol. Briefly, the
inserts of Transwell were pre-coated with 0.1% gelatin (StemCell,
USA), then, filled with 5,000 cells/well of ECA109 or Kyse30 cells
in culture medium without serum. The lower chambers of Transwell
were filled with culture medium with 15% FBS as migration inducer.
After 48 h, the migrated cells in lower chambers were fixed by 70%
ethanol, immune-stained by crystal violet, and photographed using
an Olympus IX51 inverted microscopy system (Olympus, Tokyo,
Japan).
ESCC in vitro cell cycle assay
The in vitro cell cycle transition of ESCC
was assessed by multicolor flow cytometry. Briefly, ECA109 and
Kyse30 cells were fixed in ice-cold 70% ethanol for 10 min, then
treated with DNA binding dyes propidium iodide (20 μg/ml;
Thermo Fisher Scientific) for 30 min at 37°C. Cell DNAs were
analyzed using an epics XL-MCL flow cytometer (Beckman Coulter,
Krefeld, Germany) according to the manufacturer's protocol. Cell
percentiles at G0/G1, S or M/G2 cycling stages were determined by
cytometry histograms using a MultiCycle AV DNA analysis software
(Phoenix Flow Systems, San Diego, CA, USA) according to the
manufacturer's protocol.
ESCC in vivo tumorigenicity assay
The growth of ESCC transplantation was assessed by
an in vivo tumorigenicity assay. Briefly, ECA109 cells were
subcutaneously injected into abdominal flanks of 6-week-old female
athymic nude mice (1 million cells/injection, n=7). The left flanks
were injected with cells transduced with Lenti-C, and the right
flanks were injected with cells transduced with Lenti-miR181d-M.
For five consecutive weeks, in vivo ESCC transplantations
were weekly monitored by measuring their sizes (mm3,
l × w × w/2, where l represents length
and w represents width).
Dual-luciferase reporter assay
Human DERL1 3′ UTR with putative hsa-miR-181d
binding sequences was cloned into a PmirGLO dual-luciferase
expression plasmid (Promega, Madison, WI, USA) to create DERL1
luciferase vector. A mutant DERL1 3′ UTR without hsa-miR-181d
binding sequences was synthesized by SunBio Biomedical Technology,
and cloned into PmirGLO to created Mu-DERL1 luciferase vector.
HEK293 cells were co-transfected with DERL1 or Mu-DERL1, along with
Lenti-C or Lenti-miR181d-M using DharmaFECT4 (Dharmacon) for 48 h.
A Dual-Luciferase reporter assay (Promega) was then carried out
according to the manufacturer's protocol.
DERL1 overexpressing assay
Full sequence of human DERL1 gene was amplified from
a human cDNA library then inserted into a recombinant eukaryotic
expression plasmid pcDNA3.1 to create DERL1 overexpressing vector
(pcDNA/DERL1). An empty expression plasmid, pcDNA/+, was used as
control vector. ECA109 and Kyse30 cells were transfected with
pcDNA/+ or pcDNA/DERL1 using Lipofectamine 2000 (Thermo Fisher
Scientific) for 48 h. Transfection efficiency was then confirmed by
qRT-PCR.
Statistical analysis
All experiments were carried out in triplicates and
the data are shown as mean ± standard errors. Statistical analyses
were performed using unpaired Student's t-test on SPSS software
(SPSS, Inc., Chicago, IL, USA). Statistical significance was
assigned at P-value of <0.05.
Results
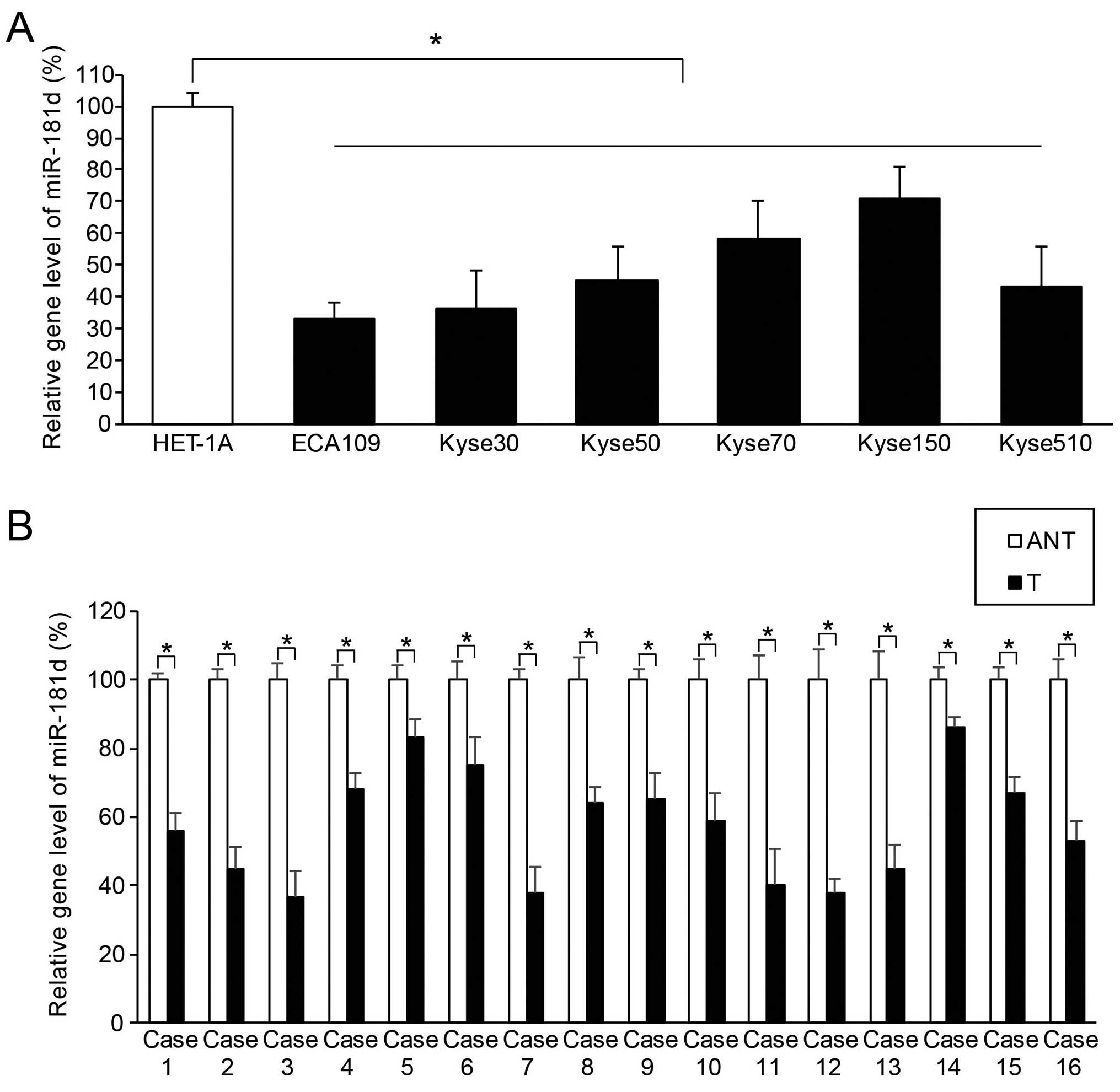
miR-181d is aberrantly downregulated in
both ESCC cell lines and clinical samples
The gene level of hsa-miR-181d in ESCC was
quantitatively assessed using method of qRT-PCR. We found that, in
the six human ESCC cell lines, ECA109, Kyse30, Kyse50, Kyse70,
Kyse150 and Kyse510, endogenous miR-181d expression was
significantly lower than the expression level in the control human
esophageal epithelial cell line, HET-1A (Fig. 1A; P<0.05). We also found that, in
clinical samples extracted from 16 cases of ESCC patients, miR-181d
expression in ESCC tissues (T) was significantly lower than
miR-181d expression in their adjacent normal esophageal epithelial
tissues (ANT) (Fig. 1B; P<0.05).
Thus, our data suggest that human miR-181d was aberrantly
downregulated in both ESCC cell lines and ESCC tumors in
patients.
miR-181d overexpression inhibits ESCC
proliferation in vitro
We transduced the ESCC cell lines, ECA109 and Kyse30
cells with Lenti-C or Lenti-miR181d-M to assess the mechanistic
regulation of miR-181d overexpression. Five or seven days after
lentiviral transduction, qRT-PCR confirmed transduction efficiency
by showing that endogenous gene level of miR-181d was significantly
upregulated in ESCC cells transduced with Lenti-miR181d-M compared
to in cells transduced with Lenti-C (Fig. 2A; P<0.05). The ECA109 and Kyse30
cells were re-suspended and re-seeded in 96-well plates. Cancer
growth was then assessed by an in vitro ESCC proliferation
assay for five consecutive days. We found in vitro cancer
proliferation was greatly inhibited in ESCC cells transduced with
Lenti-miR181d-M than in cells transduced with Lenti-C (Fig. 2B; P<0.05). Thus, our data suggest
that miR-181d is acting as a tumor suppressor in ESCC by inhibiting
cancer growth in vitro.
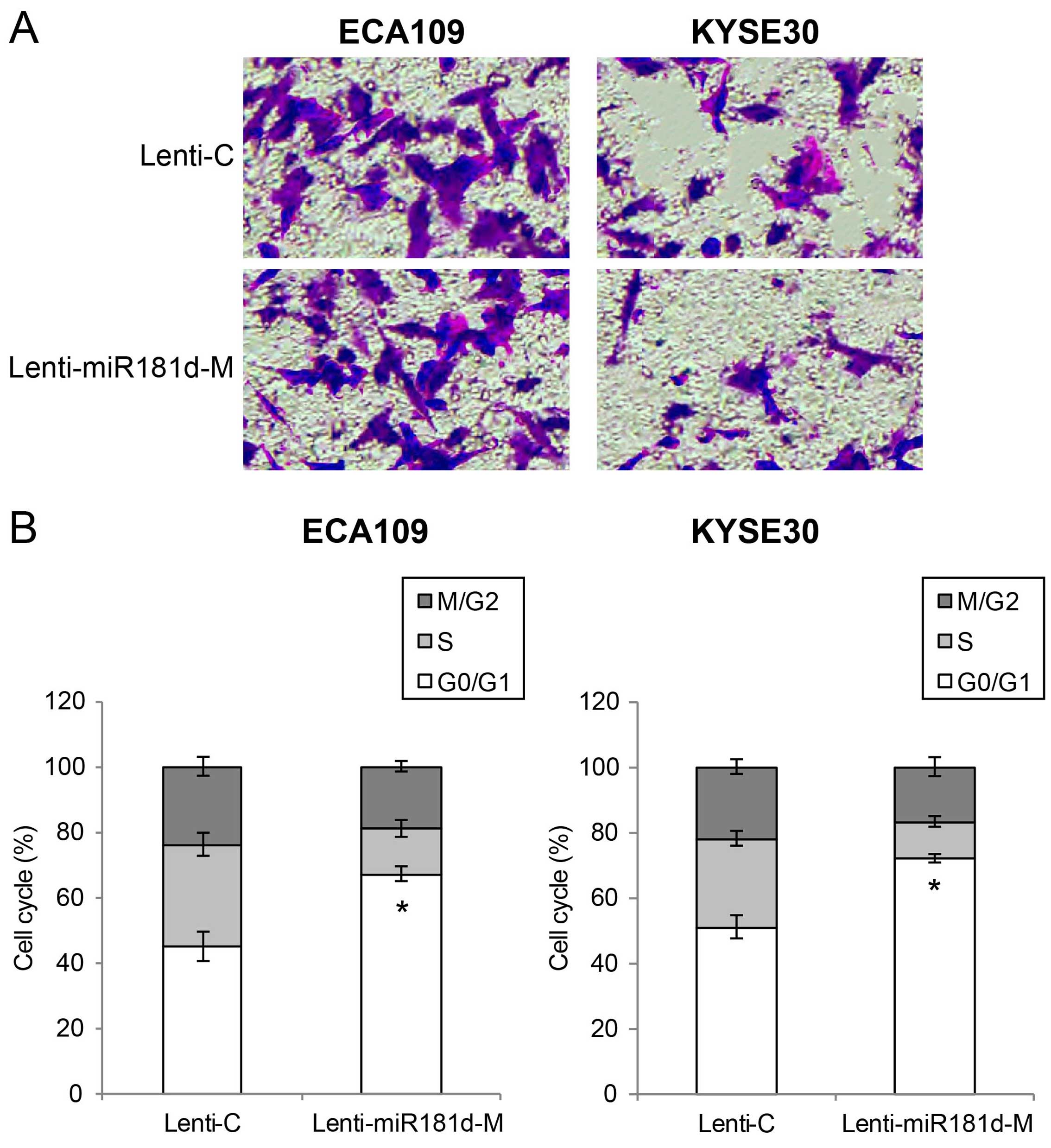
miR-181d overexpression inhibits ESCC
migration and cell cycle transition in vitro
We then assessed the effect of miR-181d
overexpression on ESCC migration and cell cycle transition in
vitro. ECA109 and Kyse30 cells with lentiviral transduction
were re-suspended and re-seeded in 96-well plates. An in
vitro migration assay was performed. We found that
significantly less ECA109 or Kyse30 cells migrated into Transwell
lower chambers while they were transduced with Lenti-miR181d-M,
than when they were transduced with Lenti-C (Fig. 3A). We also performed a cell cycle
assay by multicolor flow cytometry. It showed that significant
amount of ESCC cells were arrested at G0/G1 stage while they were
transduced with Lenti-miR181d-M, compared when they were transduced
with Lenti-C (Fig. 3B; P<0.05).
Thus, our data suggest that miR-181d overexpression also has tumor
suppressive effect on ESCC migration and cell cycle transition
in vitro.
miR-181d overexpression inhibits ESCC
growth in vivo
We then assessed the effect of miR-181d
overexpression on the in vivo growth of ESCC tumors. ECA109
cells with lentiviral transduction were subcutaneously injected
into the abdominal regions of female null mice. ECA109 cells with
Lenti-C transduction were injected on the left flanks and ECA109
cells with Lenti-miR181d-M transduction on the right flanks. For
five consecutive weeks, an in vivo tumorigenicity assay was
carried out by comparing the tumor sizes. The results showed that
in vivo tumor growth was significantly inhibited in explants
transduced with Lenti-miR181d-M than explants transduced with
Lenti-C (Fig. 4; P<0.05).
Therefore, our data suggest that miR-181d overexpression also had
tumor suppressive effect on inhibiting the in vivo
proliferation of ESCC tumors.
miR-181d directly regulates downstream
target gene of DERL1 in ESCC
We further investigated the downstream molecular
targets of miR-181d in ESCC. We found that human DERL1 gene was a
candidate gene with a putative miR-181d binding sequence on its 3′
UTR (Fig. 5A). To verify this
hypothesis, we carried out a dual-luciferase assay. PmirGLO
luciferase plasmids containing a wild-type DERL1 3′ UTR (DERL1,
consisting of miR-181d binding sequence), or a mutated DERL1 3′ UTR
(mu-DERL1, without miR-181d binding sequence) were co-transfected
with Lenti-C or Lenti-miR181d-M in HEK293 cells. Forty-eight hours
later, we used a dual-luciferase reporter assay to compare the
relative luciferase activities between HEK293 cells transfected
with Lenti-C and HEK293 cells transfected with Lenti-miR181d-M. The
result showed that relative luciferase activities were
significantly different with DERL1 co-transfection, than Mu-DERL1
co-transfection (Fig. 5B;
P<0.05), confirming that human DERL1 gene is the downstream
target of miR-181d.
We also assessed the effect of miR-181d
overexpression on gene regulation of DERL1 in ESCC. Analysis of
qRT-PCR demonstrated that DERL1 expression was markedly
downregulated in ESCC cells transduced with miR-181d-M than in ESCC
cells transduced with Lenti-C (Fig.
5C; P<0.05). Therefore, our data indicate that DERL1 gene
was directly modulated by miR-181d in ESCC.
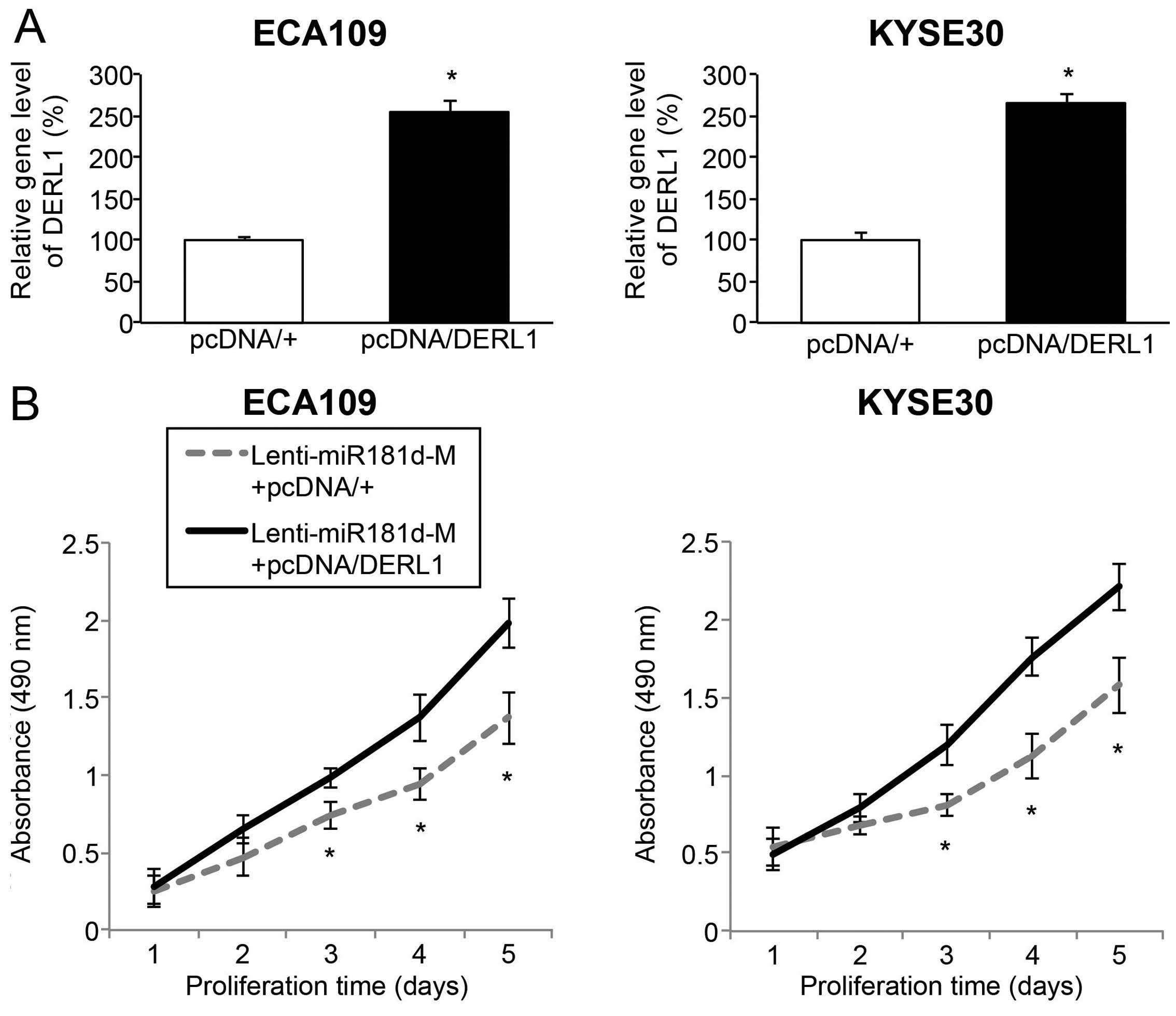
DERL1 upregulation reverses
tumor-suppressing effect of miR-181d overexpression in ESCC
Finally, we investigated whether DERL1 was involved
in tumor suppression of miR-181d overexpression in ESCC. To achieve
this goal, we re-seeded ECA109 and Kyse30 cells with Lenti-miR181d
transduction, and then transfected them with an DERL1
overexpressing vector, pcDNA/DERL1 or an empty overexpressing
vector, pcDNA/+. Analysis of qRT-PCR verified that, endogenous
DERL1 gene level was significantly upregulated by pcDNA/DERL1,
rather than pcDNA/+ in miR-181d overexpressed ESCC cells (Fig. 6A; P<0.05).
The double-transfected ECA109 and Kyse30 cells
(those transduced with Lenti-miR181d-M, then transfected with
pcDNA/+ or pcDNA/DERL1), were re-suspended and re-plated in 96-well
plates and assessed with an in vitro proliferation assay. It
showed that, in ESCC cells with miR-181d overexpression, DERL1
upregulation greatly promoted in vitro cancer proliferation
(Fig. 6B; P<0.05).
The double-transfected ECA109 and Kyse30 cells were
also examined through the in vitro migration assay. It
showed that, in ESCC cells with miR-181d overexpression, DERL1
upregulation markedly induced more cells to migrate into the lower
chambers (Fig. 7A). Finally, the
in vitro cell cycle assay revealed that cells arrested at
G0/G1 stages were markedly relived by DERL1 upregulation (Fig. 7B; P<0.05).
Thus, the data of our double-transfection assays
suggest that DERL1 upregulation could reverse the tumor suppressive
effects of miR-181d overexpression on ESCC proliferation, migration
and cell cycle transition.
Discussion
The expression pattern of miR-181d varies among
different types of cancer. In human glioma and glioblastoma,
miR-181d was reported to be downregulated in cancer tissues
(10,11). However, in human colorectal and
gastric cancer, upregulation of miR-181d was seen in tumor tissues
(19,20). In human esophageal squamous cell
carcinoma (ESCC), though miR-181d was reported to be linked to
pathological features of cancerous tissue (12), its exact expression pattern was
largely unknown. In the present study, we first applied qRT-PCR to
investigate the gene expression pattern of miR-181d between ESCC
cell lines and control esophageal epithelial cell line, as well as
between paired ESCC tumor tissues and adjacent non-cancerous
esophageal tissues in human patients. We found that miR-181d was
significantly downregulated in both ESCC cell lines and ESCC tumor
tissues. Thus, the results of the present study provided strong
evidence showing an aberrant expression pattern of miR-181d in
ESCC, suggesting that miR-181d could be associated with tumor
suppressive regulations in ESCC.
We then assessed the functional role of miR-181d in
regulating cancer development in ESCC. Lentiviral transduction was
shown to successfully upregulate miR-181d expression in ESCC cells
ECA109 and Kyse30. The subsequent functional experiments revealed
that miR-181d was actually acting as a tumor suppressor in ESCC as
it inhibited cancer proliferation, migration, cell cycle transition
in vitro and tumor growth in vivo. These data are in
line with similar tumor suppressive role of miR-181d previously
reported in glioma and glioblastoma, showing miR-181d inhibited
cancer proliferation, induced cell cycle arrest and cancer
apoptosis in glioma (10), as well
as induced temozolomide sensitivity in glioblastoma cells (11). It is worth noting that, though
several other studies reported contrary expression pattern,
i.e. upregulation of miR-181d in colorectal caner or gastric
cancer, no oncogenic functions of miR-181d were revealed in those
cancer types (19,20). Therefore, it is very likely that,
although the expression of miR-181d may exhibit different patterns,
either upregulated or downregulated in different cancers, the
regulatory function of miR-181d may be predominantly tumor
suppressive, rather than oncogenic.
In addition, in the present study, we explored the
molecular signaling pathway associated with tumor suppressive
effect of miR-181d in ESCC. Dual-luciferase reporter showed that
DERL1 was the downstream target gene of miR-181d. Subsequent
qRT-PCR analysis confirmed that DERL1 was inversely downregulated
by miR-181d overexpression in ESCC cells. Moreover, experiments of
double-transfection by upregulating DERL1 in miR-181d-overexpressed
ESCC cells revealed that DERL1 reversed the tumor suppression of
miR-181d overexpression on ESCC proliferation, migration and cell
cycle transition. DERL1 was often found to be over-expressed in
human cancers, including colon (15), lung (17) or pancreatic cancer (16). Thus, our data are in line with
previous studies, showing that DERL1 could be an active oncogenic
gene in various types of human cancers, including ESCC.
In conclusion, in the present study, we provided
strong evidence showing tumor suppressive role of miR-181d in human
ESCC. The molecular mechanism of miR-181d in ESCC progression may
very likely act through the inverse regulation of the oncogene
DERL1.
References
|
1
|
Herszényi L and Tulassay Z: Epidemiology
of gastrointestinal and liver tumors. Eur Rev Med Pharmacol Sci.
14:249–258. 2010.PubMed/NCBI
|
|
2
|
Napier KJ, Scheerer M and Misra S:
Esophageal cancer: A Review of epidemiology, pathogenesis, staging
workup and treatment modalities. World J Gastrointest Oncol.
6:112–120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang HZ, Jin GF and Shen HB:
Epidemiologic differences in esophageal cancer between Asian and
Western populations. Chin J Cancer. 31:281–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu J, Xie X, Zhou C, Peng S, Rao D and Fu
J: Which factors are associated with actual 5-year survival of
oesophageal squamous cell carcinoma? Eur J Cardiothorac Surg.
41:e7–e11. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
O'Malley MA, Elliott KC and Burian RM:
From genetic to genomic regulation: Iterativity in microRNA
research. Stud Hist Philos Biol Biomed Sci. 41:407–417. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guarnieri DJ and DiLeone RJ: MicroRNAs: A
new class of gene regulators. Ann Med. 40:197–208. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu SG, Qin XG, Zhao BS, Qi B, Yao WJ,
Wang TY, Li HC and Wu XN: Differential expression of miRNAs in
esophageal cancer tissue. Oncol Lett. 5:1639–1642. 2013.PubMed/NCBI
|
|
8
|
Gu J, Wang Y and Wu X: MicroRNA in the
pathogenesis and prognosis of esophageal cancer. Curr Pharm Des.
19:1292–1300. 2013.
|
|
9
|
Wu C, Wang C, Guan X, Liu Y, Li D, Zhou X,
Zhang Y, Chen X, Wang J, Zen K, et al: Diagnostic and prognostic
implications of a serum miRNA panel in oesophageal squamous cell
carcinoma. PLoS One. 9:e922922014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang XF, Shi ZM, Wang XR, Cao L, Wang YY,
Zhang JX, Yin Y, Luo H, Kang CS, Liu N, et al: MiR-181d acts as a
tumor suppressor in glioma by targeting K-ras and Bcl-2. J Cancer
Res Clin Oncol. 138:573–584. 2012. View Article : Google Scholar
|
|
11
|
Zhang W, Zhang J, Hoadley K, Kushwaha D,
Ramakrishnan V, Li S, Kang C, You Y, Jiang C, Song SW, et al:
miR-181d: A predictive glioblastoma biomarker that downregulates
MGMT expression. Neurooncol. 14:712–719. 2012.
|
|
12
|
Guo Y, Chen Z, Zhang L, Zhou F, Shi S,
Feng X, Li B, Meng X, Ma X, Luo M, et al: Distinctive microRNA
profiles relating to patient survival in esophageal squamous cell
carcinoma. Cancer Res. 68:26–33. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang F, Olson EM and Shyng SL: Role of
Derlin-1 protein in proteostasis regulation of ATP-sensitive
potassium channels. J Biol Chem. 287:10482–10493. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen SF, Wu CH, Lee YM, Tam K, Tsai YC,
Liou JY and Shyue SK: Caveolin-1 interacts with Derlin-1 and
promotes ubiquitination and degradation of cyclooxygenase-2 via
collaboration with p97 complex. J Biol Chem. 288:33462–33469. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tan X, He X, Jiang Z, Wang X, Ma L, Liu L,
Wang X, Fan Z and Su D: Derlin-1 is overexpressed in human colon
cancer and promotes cancer cell proliferation. Mol Cell Biochem.
408:205–213. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ran Y, Hu H, Hu D, Zhou Z, Sun Y, Yu L,
Sun L, Pan J, Liu J, Liu T, et al: Derlin-1 is overexpressed on the
tumor cell surface and enables antibody-mediated tumor targeting
therapy. Clin Cancer Res. 14:6538–6545. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dong QZ, Wang Y, Tang ZP, Fu L, Li QC,
Wang ED and Wang EH: Derlin-1 is overexpressed in non-small cell
lung cancer and promotes cancer cell invasion via EGFR-ERK-mediated
up-regulation of MMP-2 and MMP-9. Am J Pathol. 182:954–964. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu L, Wang ZH, Xu D, Lin G, Li DR, Wan T
and Guo SL: Expression of Derlin-1 and its effect on expression of
autophagy marker genes under endoplasmic reticulum stress in lung
cancer cells. Cancer Cell Int. 14:502014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Motoyama K, Inoue H, Takatsuno Y, Tanaka
F, Mimori K, Uuetake H, Sugihara K and Mori M: Over- and
under-expressed microRNAs in human colorectal cancer. Int J Oncol.
34:1069–1075. 2009.PubMed/NCBI
|
|
20
|
Ueda T, Volinia S, Okumura H, Shimizu M,
Taccioli C, Rossi S, Alder H, Liu CG, Oue N, Yasui W, et al:
Relation between microRNA expression and progression and prognosis
of gastric cancer: A microRNA expression analysis. Lancet Oncol.
11:136–146. 2010. View Article : Google Scholar
|