Introduction
It is well known that anti-neoplastic drugs
interfere with the structure and functions of DNA directly or
indirectly. However, they sometimes not only affect target cells
but also normal cells. In light of this, comprehension of the
anticancer functions still require investigation in order to reduce
the side effects before their use in the direct treatment of
patients. Thus, there is not only the need to evaluate the
impairment caused by anticancer drugs on the whole organism but
also to investigate the effects of genotoxic alterations at a
cellular level (1). Currently,
numerous compounds from natural plants have been shown to induce
cell death via the induction of cell apoptosis. However, the
interruption of cell DNA damage is also needed because these
effects can lead to cell death. Some anticancer drugs such as
cisplatin or etoposide have been shown to induce DNA damage and
eventually cell death (2). Thus,
focusing on the ability of these compounds to interfere with DNA
and produce DNA damage will be helpful and critical to understand
how these compounds induce cell death.
Casticin, one of the ingredients derived from
Fructus viticis (3), has
been shown to exhibit anticancer activity in prostate (4), breast (5), colon (6,7), lung
(8,9), cervical (10), gastric (11) and ovarian cancer (12), glioma (13) and leukemia (14) Recently it was reported that forkhead
box O3 (FOXO3a) is a critical mediator of the inhibitory effects of
casticin on apoptosis in breast cancer cells (3). Furthermore, casticin significantly
induced cell apoptosis through the activation of the apoptosis
signal-regulating kinase 1-c-Jun N-terminal kinase (ASK1-JNK)-Bim
signaling cascade and the accumulation of reactive oxygen species
(ROS) in colon cancer cells (15).
However, there is no available information to show that casticin
induces cell apoptosis in melanoma cancer cells. Furthermore, there
is no report showing that casticin induces DNA damage and affects
DNA repair-associated protein expression levels in melanoma
cells.
After melanoma becomes metastatic melanoma, it is
characterized by a high mortality rate (16) due to a universal resistance to
standard chemotherapy (17). Hence,
the motality rate from unresectable melanoma continues to rise
(18). Presently, the
ineffectiveness of the treatments available, encourage additional
studies to identify novel therapeutic molecules, delivery systems,
and/or combination therapies for the treatment of melanoma
(19). Casticin may be a potential
antitumor agent with both antitumor and anti-proliferative
activities. However, the effects of casticin on DNA damage and
repair with associated protein expression are not widely known.
Thus, the objective of this study was to investigate DNA damage and
repair of melanoma B16F10 cells and our results confirmed that
casticin induced DNA damage and affected DNA repair systems in
vitro.
Materials and methods
Chemicals and reagents
Casticin, dimethyl sulfoxide (DMSO), propidium
iodide (PI), Trypsin-EDTA, penicillin-streptomycin, anti-MGMT (cat
no. M3068), anti-PARP (cat no. P248), anti-p-ATMSer1981 (cat no.
SBA4300100) and anti-β-actin (cat no. A5316) were all purchased
from Sigma Chemical Co. (St. Louis, MO, USA). Anti-DNA-PK (cat no.
PC127) was purchased from Calbiochem (San Diego, CA, USA).
Anti-p-H2A.X (cat no. GTX80694) and anti-BRCA1 (cat no. GTX70111),
were purchased from GeneTex Inc. (Irvine, CA, USA). Anti-p-p53 (cat
no. sc-7997) was obtained from Santa Cruz Biotechnology (Santa
Cruz, CA, USA). Anti-MDC1 (cat no. 05-1572) was purchased from
Millipore (Billerica, MA, USA). Dulbecco's modified Eagle's medium
(DMEM) and fetal bovine serum (FBS) were purchased from
Gibco®/Invitrogen Life Technologies (Carlsbad, CA,
USA).
Cell culture
The murine melanoma cell line (B16F10) was purchased
from the Food Industry Research and Development Institute (Hsinchu,
Taiwan). Cells were grown in 75-cm2 flasks with DMEM
supplemented with 10% FBS, 100 U/ml penicillin and 100 µg/ml
streptomycin at 37°C in 5% CO2 humidified incubators
(20).
Cellular morphology and viability
examination
B16F10 cells were plated at a density of
1×105 cells/well into 12-well plates in DMEM. After the
required confluency was reached, cells were exposed to 0, 20, 30
and 40 µM of casticin for 24 and 48 h in a 5% CO2
incubator at 37°C. Cells were examined and their images were
captured by contrast phase microscopy at ×200 magnification.
Subsequently, the cells were collected, washed and stained with PI
(5 µg/ml) in phosphate-buffered saline (PBS) and were
analyzed by flow cytometry (Becton-Dickinson, San Jose, CA, USA)
for the total percentage of viable cells as previously described
(21).
4′,6-Diamidino-2-phenylindole
dihydrochloride (DAPI) staining for DNA condensation
examination
B16F10 cells (1.5×105 cells/well) were
plated onto a 6-well plate for 24 h and then were exposed to
casticin (30 µM) for 0, 6, 24 and 48 h. After treatment, 4%
formaldehyde in PBS was used to fix cells for 10 min and then DAPI
staining followed. After staining, the cells were examined and
their images were captured using a fluorescence microscope at ×200
magnification as previously described (21).
Western blotting for examination of
protein expression
B16F10 cells (1×106 cells/dish) were
plated onto a 10-cm dish and were incubated with 30 µM of
casticin for 0, 6, 24 and 48 h. Cells were collected, suspended in
sodium dodecyl sulfate (SDS) sample buffer, sonicated, boiled for
10 min as previously described (21) and were centrifuged at 12,000 rpm for
15 min. The supernatant was collected and the concentrations of the
total protein were determined by Bio-Rad protein assay kit (Bio-Rad
Laboratories, Hercules, CA, USA). The cells were electro-phoresed
by 10% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE) and
were transferred to polyvinylidene fluoride (PVDF) membranes
(Millipore, Bedford, MA, USA). Immune complexes were formed by
incubation of proteins with primary antibodies (anti-MGMT,
anti-BRCA1, anti-PARP, anti-p-p53, anti-MDC1, anti-DNA-PK,
anti-p-ATM and anti-β-actin) at 4°C (overnight) followed by
incubation with a secondary antibody. Immunoreactive protein bands
were visualized with a chemiluminescent detection system and the
protein expression levels were measured as described by the
manufacturer (20,21).
Confocal laser microscopy for examination
of protein translocation
B16F10 cells were plated at a density of
1.5×105 cells/well on a 6-well plate and were treated
with 30 µM of casticin for 48 h. After treatment, cells were
fixed in 4% formaldehyde in PBS for 15 min and 0.1% Triton X-100 in
PBS was added to permeable cells. Subsequently, cells were washed
with PBS and blocked with 1% BSA in PBS for 60 min and then they
were stained with primary anti-p-p53 and anti-p-H2A.X (green
fluorescence) overnight followed by staining with a secondary
antibody (FITC-conjugated goat anti-mouse IgG). After being washed,
cells were stained using PI (red fluorescence) for nuclei. All
samples were mounted and photomicrographed by using a Leica TCS SP2
confocal spectral microscope (Leica Microsystems, Heidelberg,
Mannheim, Germany) as previously described (21).
Statistical analysis
The comparisons between the casticintreated and the
untreated groups were performed using the Student's t-test, to
determine the statistical significance of the differences between
these groups. P<0.05 was considered to be significant.
Results
Casticin induces cell morphology and
decreases the total viability of the B16F10 cells
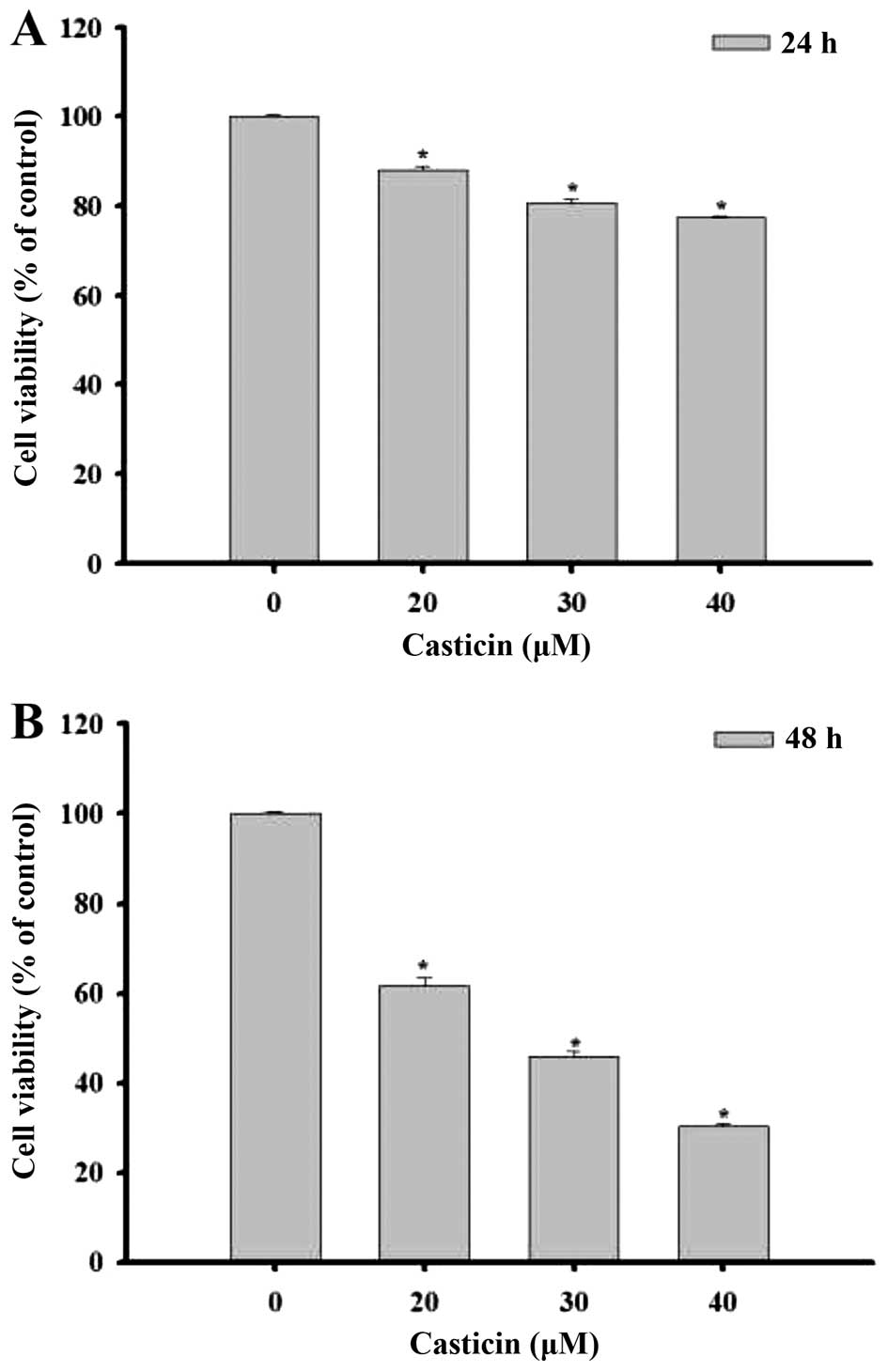
B16F10 cells were treated with various
concentrations of casticin (0, 20, 30 and 40 µM) at 24 and
48 h. Cells were examined for morphological changes and images were
captured using a phase contrast microscope at ×200 magnification
(Fig. 1). The results indicated
that casticin induced cell morphological changes in a
dose-dependent manner. Cells were collected in order to measure the
percentage of viable cells by flow cytometric assay (Fig. 2). The results indicated that the
percentage of total cell viability was decreased significantly
after treatment with casticin and that this effect was
dose-dependent. The treatment of casticin at 48 h had a higher
effect than that at 24 h.
Casticin induces nuclear DNA condensation
of B16F10 cells
In order to further confirm whether casticin induced
cell death via nuclear DNA condensation in the B16F10 cells, we
selected 30 µM of casticin for treatment with cells at 0, 6,
24 and 48 h, Subsequently the cells were stained with DAPI to
examine the formation of DNA condensation (Fig. 3A and B). The results indicated that
casticin induced nuclear DNA condensation in the B16F10 cells and
this effect was time-dependent.
Casticin affects DNA damage of the
associated proteins in the B16F10 cells
Cells were treated with 30 µM of casticin for
0, 6, 24 and 48 h and then DNA damage of the associated proteins
such as O6-methylguanine-DNA methyltransferase
(MGMT), p-H2A.X, breast cancer 1 and early onset (BRCA1),
poly(ADP-ribose) polymerase (PARP), phospho-p53 tumor suppressor
protein (p-p53), mediator of DNA damage checkpoint 1 (MDC1),
DNA-dependent protein kinase (DNA-PK) and phospho-ataxia
telangiectasia mutated kinase (p-ATM) were examined by western blot
analysis (Fig. 4). The results
indicated that casticin decreased the protein levels of MGMT and
BRCA1 (Fig. 4A) and MDC1 (Fig. 4B), but increased the levels of
p-H2A.X and PARP (Fig. 4A), p-p53
and p-ATM (Fig. 4B) in the B16F10
cells. These effects are associated with DNA damage and repair that
may lead to cell death.
Casticin affects the translocation of
p-p53 and p-H2A.X in the B16F10 cells
To further confirm whether casticin affects DNA
damage in associated protein translocation in the B16F10 cells,
cells were treated with 30 µM of casticin and then they were
examined by confocal microscopy (Fig.
5). The results revealed that casticin increased the p-p53
(Fig. 5A) and p-H2A.X (Fig. 5B) expression levels in the cytoplasm
when compared to the control groups and these observations indicate
that casticin induces DNA damage and repair and may also regulate
p-p53 and p-H2A.X in the cytoplasm in the B16F10 cells.
Discussion
Based on the review of the literature, casticin was
found to induce cell death (cytotoxic effects) via both induction
of cell cycle arrest and apoptosis in many types of human cancer
cells, but there is no available information showing that casticin
induces DNA damage and repair and affects associated protein
expression in human cancer cells. Therefore, in the present study,
we investigated the cytotoxic effects of casticin and whether,
through the induction of DNA damage, it affected DNA repair and
associated protein expression levels in mouse melanoma B16F10 cells
in vitro. After B16F10 cells were exposed to various
concentrations of casticin we found that i) casticin induced cell
morphological changes (Fig. 1) and
decreased the total cell viability (percentage of viable cells) in
a concentration- and time-dependent manner (Fig. 2); ii) a time-dependent increase in
nuclear DNA condensation was observed in the B16F10 cells after
exposure to casticin, which was assayed by DAPI staining (Fig. 3); iii) casticin decreased the
proteins levels of MGMT and BRCA1 (Fig.
4A), and MDC1 (Fig. 4B), and
increased the levels of p-H2A.X and PARP (Fig. 4A), p-p53 and p-ATM (Fig. 4B) in the B16F10 cells and these
effects were time-dependent; iv) casticin induced DNA damage and
repair and may also regulate p-p53 (Fig. 5A) and p-H2A.X (Fig. 5B) which are increased in the
cytoplasm when compared to the control groups in the B16F10
cells.
We observed that casticin induced cell morphological
changes and decreased the total percentage of viable B16F010 cells
in a dose-dependent manner at 20–40 µM. Thus, we further
examined whether casticin induced cell death and was associated
with induction of DNA damage in the B16F10 cells. Based on the
results from Fig. 3, it was
revealed that casticin induced DNA damage and condensation in the
B16F10 cells which were assayed by DAPI staining, respectively. It
has been reported that specific and bulky DNA lesions which trigger
cell apoptosis have been identified (22). It has been well documented that DAPI
staining can reveal DNA fragmentation and nuclear DNA condensation
and our results indicated that casticin induced nuclear DNA
condensation in a time-dependent manner. Recently, a new type of
anti-neoplastic therapy has emerged, whose aim is to manipulate DNA
damage response (DDR) (23–25) as DDR inhibition has been proven as
an effective treatment for cancer. It has been reported that
oxidative DNA damage has been recognized to be an etiological
factor in aging and in the development of systemic diseases
including cancer in the human population (26,27).
In cells, DNA repair enzymes monitor chromosomes and correct
damaged nucleotides to prevent these adverse effects (28).
In the present study, our findings are the first to
provide information regarding casticin-induced DNA damage and the
affect on the DNA repair system in the B16F10 cells (Fig. 3). Western blotting (Fig. 4) indicated that casticin induced DNA
damage and affected repair in associated protein expression levels
such as MGMT and BRCA1 (Fig. 4A),
MDC1 and DNA-PK (Fig. 4B) but
increased the levels of p-H2A.X and PARP (Fig. 4A) and p-p53 and p-ATM (Fig. 4B) in the B16F10 cells. It has been
reported that MGMT is a DNA repair enzyme which eliminates
O6 methylguanines (29) and that the inhibition of MGMT may be
strategic in increasing tumor susceptibility to chemotherapy
(30). In breast and ovarian
cancers, BRCA1 plays an important role in DNA repair in the
maintainance of genomic stability (31) and in breast cancer, BRCA1 promoter
methylation was found to be positively associated with increased
mortality (32). MDC1 and BRCA1
represent important assets in the repair of double-strand breaks
after DNA damage occurs (33,34).
Furthermore, MDC1 may affect the radiosensitivity of tumor cells
(35). It has been reported that
DNA-PK is a serine/threonine protein kinase and is expressed in
most mammalian cells (36). DNA-PK
plays an important role in the main repair pathway of DNA
double-strand breaks and cells deficient in DNA-PK exhibit
hypersensitivity to radio/chemotherapy (37–39).
It has also been reported that anticancer drugs such as 5-FU induce
DNA double-strand breaks, and the presence of p-H2A.X which is a
phosphorylated form of the histone H2A.X which has been shown to be
a specific marker for the detection of these DNA breaks was
observed (40). Based on this
observation, we suggest that casticin induced double-strand breaks
in the B16F10 cells.
Moreover, it has been reported that the ATM/p53
pathway is involved in the apoptosis of various cancer cells
induced by chemotherapy drugs (41). Herein, results from the western blot
analysis indicated that casticin increased the protein levels of
proteins p-p53 and p-ATM (Fig. 4B)
in the B16F10 cells which was also confirmed by confocal laser
system microscopy examination (Fig.
5). Notably, it has also been reported that antioxidant
N-acetylcysteine (NAC) pretreatment enhances ATM and p53
phosphorylation, p53 acetylation and H2A.X phosphorylation in
ovarian cancer cells (42). In view
of this, the possible signaling pathways involved in the
casticin-induced DNA damage and nuclear condensation in the B16F10
cells are summarized in Fig. 6.
Thus, further studies should be conducted to elucidate the exact
molecular mechanism of casticin-induced DNA damage and how to
affect DNA damage and repair-associated signaling pathways.
Acknowledgments
This study was supported by grant CMU103-ASIA-01
from China Medical University (Taichung, Taiwan) and by grant
103-08 and 103-41 from Cheng Hsin General Hospital (Taipei,
Taiwan). Experiments and data analysis were performed in part
through the use of the Medical Research Core Facilities Center,
Office of Research and Development at China Medical University
(Taichung, Taiwan).
References
|
1
|
Parrella A, Lavorgna M, Criscuolo E, Russo
C and Isidori M: Ecogenotoxicity of six anticancer drugs using
comet assay in daphnids. J Hazard Mater. 286:573–580. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li X, Tian J, Bo Q, Li K, Wang H, Liu T
and Li J: Targeting DNA-PKcs increased anticancer drug sensitivity
by suppressing DNA damage repair in osteosarcoma cell line MG63.
Tumour Biol. 36:9365–9372. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu LP, Cao XC, Liu F, Quan MF, Sheng XF
and Ren KQ: Casticin induces breast cancer cell apoptosis by
inhibiting the expression of forkhead box protein M1. Oncol Lett.
7:1711–1717. 2014.PubMed/NCBI
|
|
4
|
Weisskopf M, Schaffner W, Jundt G, Sulser
T, Wyler S and Tullberg-Reinert H: A Vitex agnus-castus extract
inhibits cell growth and induces apoptosis in prostate epithelial
cell lines. Planta Med. 71:910–916. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Haïdara K, Zamir L, Shi QW and Batist G:
The flavonoid casticin has multiple mechanisms of tumor
cytotoxicity action. Cancer Lett. 242:180–190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Imai M, Kikuchi H, Denda T, Ohyama K,
Hirobe C and Toyoda H: Cytotoxic effects of flavonoids against a
human colon cancer derived cell line, COLO 201: A potential natural
anti-cancer substance. Cancer Lett. 276:74–80. 2009. View Article : Google Scholar
|
|
7
|
Tang SY, Zhong MZ, Yuan GJ, Hou SP, Yin
LL, Jiang H and Yu ZY: Casticin, a flavonoid, potentiates
TRAIL-induced apoptosis through modulation of anti-apoptotic
proteins and death receptor 5 in colon cancer cells. Oncol Rep.
29:474–480. 2013.
|
|
8
|
Koh DJ, Ahn HS, Chung HS, Lee H, Kim Y,
Lee JY, Kim DG, Hong M, Shin M and Bae H: Inhibitory effects of
casticin on migration of eosinophil and expression of chemokines
and adhesion molecules in A549 lung epithelial cells via NF-κB
inactivation. J Ethnopharmacol. 136:399–405. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou Y, Peng Y, Mao QQ, Li X, Chen MW, Su
J, Tian L, Mao NQ, Long LZ, Quan MF, et al: Casticin induces
caspase-mediated apoptosis via activation of mitochondrial pathway
and upregu-lation of DR5 in human lung cancer cells. Asian Pac J
Trop Med. 6:372–378. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zeng F, Tian L, Liu F, Cao J, Quan M and
Sheng X: Induction of apoptosis by casticin in cervical cancer
cells: Reactive oxygen species-dependent sustained activation of
Jun N-terminal kinase. Acta Biochim Biophys Sin (Shanghai).
44:442–449. 2012. View Article : Google Scholar
|
|
11
|
Zhou Y, Tian L, Long L, Quan M, Liu F and
Cao J: Casticin potentiates TRAIL-induced apoptosis of gastric
cancer cells through endoplasmic reticulum stress. PLoS One.
8:e588552013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiang L, Cao XC, Cao JG, Liu F, Quan MF,
Sheng XF and Ren KQ: Casticin induces ovarian cancer cell apoptosis
by repressing FoxM1 through the activation of FOXO3a. Oncol Lett.
5:1605–1610. 2013.PubMed/NCBI
|
|
13
|
Liu E, Kuang Y, He W, Xing X and Gu J:
Casticin induces human glioma cell death through apoptosis and
mitotic arrest. Cell Physiol Biochem. 31:805–814. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shen JK, Du HP, Yang M, Wang YG and Jin J:
Casticin induces leukemic cell death through apoptosis and mitotic
catastrophe. Ann Hematol. 88:743–752. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qu L, Liu FX, Cao XC, Xiao Q, Yang X and
Ren KQ: Activation of the apoptosis signal-regulating kinase
1/c-Jun N-terminal kinase pathway is involved in the
casticin-induced apoptosis of colon cancer cells. Exp Ther Med.
8:1494–1500. 2014.PubMed/NCBI
|
|
16
|
Garbe C and Leiter U: Melanoma
epidemiology and trends. Clin Dermatol. 27:3–9. 2009. View Article : Google Scholar
|
|
17
|
Jilaveanu LB, Aziz SA and Kluger HM:
Chemotherapy and biologic therapies for melanoma: Do they work?
Clin Dermatol. 27:614–625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lillehammer T, Engesaeter BO, Prasmickaite
L, Maelandsmo GM, Fodstad O and Engebraaten O: Combined treatment
with Ad-hTRAIL and DTIC or SAHA is associated with increased
mitochondrial-mediated apoptosis in human melanoma cell lines. J
Gene Med. 9:440–451. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chang YM, Velmurugan BK, Kuo WW, Chen YS,
Ho TJ, Tsai CT, Ye CX, Tsai CH, Tsai FJ and Huang CY: Inhibitory
effect of alpinate Oxyphyllae fructus extracts on Ang II-induced
cardiac pathological remodeling-related pathways in H9c2
cardiomyoblast cells. BioMedicine. 3:148–152. 2013. View Article : Google Scholar
|
|
21
|
Chueh FS, Chen YL, Hsu SC, Yang JS, Hsueh
SC, Ji BC, Lu HF and Chung JG: Triptolide induced DNA damage in
A375.S2 human malignant melanoma cells is mediated via reduction of
DNA repair genes. Oncol Rep. 29:613–618. 2013.
|
|
22
|
Roos WP and Kaina B: DNA damage-induced
cell death by apoptosis. Trends Mol Med. 12:440–450. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hsu YC, Weng HC, Lin S and Chien YW:
Curcuminoids cellular uptake by human primary colon cancer cells as
quantitated by a sensitive HPLC assay and its relation with the
inhibition of proliferation and apoptosis. J Agric Food Chem.
55:8213–8222. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ireson C, Orr S, Jones DJ, Verschoyle R,
Lim CK, Luo JL, Howells L, Plummer S, Jukes R, Williams M, et al:
Characterization of metabolites of the chemopreventive agent
curcumin in human and rat hepatocytes and in the rat in vivo, and
evaluation of their ability to inhibit phorbol ester-induced
prostaglandin E2 production. Cancer Res. 61:1058–1064.
2001.PubMed/NCBI
|
|
25
|
Ireson CR, Jones DJ, Orr S, Coughtrie MW,
Boocock DJ, Williams ML, Farmer PB, Steward WP and Gescher AJ:
Metabolism of the cancer chemopreventive agent curcumin in human
and rat intestine. Cancer Epidemiol Biomarkers Prev. 11:105–111.
2002.PubMed/NCBI
|
|
26
|
McCall MR and Frei B: Can antioxidant
vitamins materially reduce oxidative damage in humans? Free Radic
Biol Med. 26:1034–1053. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ohia SE, Opere CA and Leday AM:
Pharmacological consequences of oxidative stress in ocular tissues.
Mutat Res. 579:22–36. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wood RD, Mitchell M, Sgouros J and Lindahl
T: Human DNA repair genes. Science. 291:1284–1289. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Christmann M, Verbeek B, Roos WP and Kaina
B: O(6)-Methylguanine-DNA methyltransferase (MGMT) in normal
tissues and tumors: Enzyme activity, promoter meth-ylation and
immunohistochemistry. Biochim Biophys Acta. 1816:179–190.
2011.PubMed/NCBI
|
|
30
|
Verbeek B, Southgate TD, Gilham DE and
Margison GP: O6-Methylguanine-DNA methyltransferase
inactivation and chemotherapy. Br Med Bull. 85:17–33. 2008.
View Article : Google Scholar
|
|
31
|
Venkitaraman AR: Cancer susceptibility and
the functions of BRCA1 and BRCA2. Cell. 108:171–182. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu X, Gammon MD, Zhang Y, Bestor TH,
Zeisel SH, Wetmur JG, Wallenstein S, Bradshaw PT, Garbowski G,
Teitelbaum SL, et al: BRCA1 promoter methylation is associated with
increased mortality among women with breast cancer. Breast Cancer
Res Treat. 115:397–404. 2009. View Article : Google Scholar :
|
|
33
|
Bartkova J, Horejsí Z, Sehested M, Nesland
JM, Rajpert-De Meyts E, Skakkebaek NE, Stucki M, Jackson S, Lukas J
and Bartek J: DNA damage response mediators MDC1 and 53BP1:
Constitutive activation and aberrant loss in breast and lung
cancer, but not in testicular germ cell tumours. Oncogene.
26:7414–7422. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gorgoulis VG, Vassiliou LV, Karakaidos P,
Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr,
Kastrinakis NG, Levy B, et al: Activation of the DNA damage
checkpoint and genomic instability in human precancerous lesions.
Nature. 434:907–913. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gou Q, Xie Y, Liu L, Xie K, Wu Y, Wang Q,
Wang Z and Li P: Downregulation of MDC1 and 53BP1 by short hairpin
RNA enhances radiosensitivity in laryngeal carcinoma cells. Oncol
Rep. 34:251–257. 2015.PubMed/NCBI
|
|
36
|
Jackson SP: DNA-dependent protein kinase.
Int J Biochem Cell Biol. 29:935–938. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Belenkov AI, Paiement JP, Panasci LC,
Monia BP and Chow TY: An antisense oligonucleotide targeted to
human Ku86 messenger RNA sensitizes M059K malignant glioma cells to
ionizing radiation, bleomycin, and etoposide but not DNA
cross-linking agents. Cancer Res. 62:5888–5896. 2002.PubMed/NCBI
|
|
38
|
Mi J, Dziegielewski J, Bolesta E,
Brautigan DL and Larner JM: Activation of DNA-PK by ionizing
radiation is mediated by protein phosphatase 6. PLoS One.
4:e43952009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shintani S, Mihara M, Li C, Nakahara Y,
Hino S, Nakashiro K and Hamakawa H: Up-regulation of DNA-dependent
protein kinase correlates with radiation resistance in oral
squamous cell carcinoma. Cancer Sci. 94:894–900. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Matuo R, Sousa FG, Escargueil AE,
Grivicich I, Garcia-Santos D, Chies JA, Saffi J, Larsen AK and
Henriques JA: 5-Fluorouracil and its active metabolite FdUMP cause
DNA damage in human SW620 colon adenocarcinoma cell line. J Appl
Toxicol. 29:308–316. 2009. View Article : Google Scholar
|
|
41
|
Jäämaa S, Af Hällström TM, Sankila A,
Rantanen V, Koistinen H, Stenman UH, Zhang Z, Yang Z, De Marzo AM,
Taari K, et al: DNA damage recognition via activated ATM and p53
pathway in nonproliferating human prostate tissue. Cancer Res.
70:8630–8641. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Brum G, Carbone T, Still E, Correia V,
Szulak K, Calianese D, Best C, Cammarata G, Higgins K, Ji F, et al:
N-acetylcysteine potentiates doxorubicin-induced ATM and p53
activation in ovarian cancer cells. Int J Oncol. 42:211–218.
2013.
|