Introduction
The inventory of cancer research of 2015 shows that
liver cancer is a global high incidence malignant tumor, which is
known as ‘the king of cancer’ (1).
China is the country with high incidence of liver cancer. Chinese
population accounts for 1/5 of the world, while the incidence and
mortality of liver cancer is more than 1/2 of the world. Worlds
Cancer Report of 2014 shows that hepatocellular carcinoma (HCC) has
become the fifth ranked cancer in men (~554,000 new cases each
year) and the ninth cancer in women (~228,000 new cases each year)
(2). The high prevalence and
mortality rate of HCC (the overall mortality rate is 95%) make it
the major health burden in the current society.
Bile acids are the main component of the bile
secreted by liver cells, while conjugated bile acids is the primary
form. Due to the pH environment around the bile, most of which
exists as sodium or potassium salt, called bile salts (sodium salt
combined with bile acids). Glycochenodeoxycholate (Glycine
conjugate of chenodexycholate) is the main ingredient in the bile,
which is considered to be toxic and hydrophobic and may be involved
in mediating apoptosis of liver cells during cholestasis. Previous
studies have shown that toxic bile salt glycochenodeoxycholate
induces hepatocyte apoptosis involving ligand-independent
oligomerization of Fas (3) and by
promoting cytoplasmic transport of Fas to the cell surface
(4). PKC-dependent signaling
pathways play a critical role in bile salt-induced hepatocyte
apoptosis (5,6). Endoplasmic reticulum is involved in
glycochenodeoxycholic acid-induced apoptosis in rat hepatocytes,
although its role may be smaller than mitochondria-mediated pathway
(7). Another study demonstrated
that glycochenodeoxycholate induces apoptosis of liver cells by
modulating the IGF1 system (8).
Although the accumulation of glycochenodeoxycholate induced
hepatocyte apoptosis in cholestasis, many hepatocytes still
survive. The study by Wang et al (9) found that
glycochenodeoxycholate-induced caspase response is reversible,
which may activate anti-apoptotic genes to protect hepatocytes from
apoptosis. Later, another study by Wang et al (10) demonstrated an interesting result: a
low dosage of glycochenodeoxycholate induced hepatocyte apoptosis
to exhibit the biphasic response, which was regulated by the
expression of survivin through NF-κB signaling pathway. Recent
research suggests that PKC-δ activation by glycochenodeoxycholate
stimulates a cytoprotective pathway involving JNK inhibition, Akt
activation and downregulation of BIM (11). Moreover, research has confirmed that
bile salt, especially toxic bile salt, is associated with
carcinogenesis of gastrointenstinal tumors. Bile acids can block
Mcl-1 protein degradation via activation of an EGFR/Raf-1 cascade
resulting in tumorigenesis of cholangiocarcinomas (12). Glycochenodeoxycholic acid activates
the PI3 Kinase/Akt signaling pathway, stimulate cell growth in the
Barretts adenocarcinoma cell line SEG-1 (13) and induces proliferation of a
non-neoplastic Barretts cell line by activation of both ERK and p38
MAPK pathways (14), which suggest
potential mechanisms whereby bile reflux may facilitate the
neoplastic progression of Barretts esophagus. In addition,
long-term effect of oxidative DNA damage caused by
glycochenodeoxycholate may promote carcinogenesis in the biliary
tract (15). The disturbance of
bile circulation metabolism may cause abnormal concentration of
bile salts, which will result in necrosis and apoptosis of liver
cells and become the potential carcinogenesis of hepatocellular
carcinoma (HCC). However, the underlying intracellular signaling
mechanism remains to be further studied.
Bcl-2 (B Cell Leukemia-2) family proteins are key
players in the control of mitochondria-based apoptosis (16,17).
Bcl-2 is the most important anti-apoptotic protein, whose
overexpression and phosphorylation may be involved in regulating
cell proliferation, cell cycle, DNA repair, tumorigenesis and
chemoresistance. There is high level of Bcl-2 protein in many human
tumors, such as liver, prostate, colon, lung, stomach, lymphoma,
neuroblastoma and breast cancer (17,18).
Endogenous Bcl-2 expressed in various cells can be phosphorylated
at multiple sites in the flexible loop domain (FLD), including
Thr69, Ser70 and Ser87, associated with regulation of apoptosis
(19). Studies verified that PKCα,
p44 MAPK/ERK1, p42 MAPK/ERK2 and JNKs are Bcl-2 upstream kinases
(20,21). In recent years, Bcl-2s application
in clinic is quite common. Bcl-2 may play an important role in
mediating the outcome of antidepressant treatment (22). The expression of Bcl-2 may serve as
predictive biomarker of response to induction chemotherapy in HNSCC
(head and neck squamous cell carcinomas) patients (23). The genetic variation in Bcl-2 3-UTR
was associated with a decreased lung cancer risk and better
survival for non-small cell lung cancer in male Chinese (24). Overexpression of Bcl-2 protein
predicts chemoresistance in acute myeloid leukemia (25). Genetic polymorphisms in the Bcl-2
gene may be associated with the risk of endometrial cancer in
Chinese women (26). Some studies
found that inhibitors and microRNA targeting Bcl-2 had effect on
treatment of gliomas (27), myeloma
(28), nasopharyngeal carcinoma
(29), esophageal squamous cell
carcinoma (30), aclcoholic liver
disease (31), colorectal (32), gastric cancer (33) and melanoma (34).
However, studies on the role of Bcl-2 in
hepatocellular carcinoma (HCC) are scarce. The earliest study
demonstrated that normal hepatocytes did not express Bcl-2 and
expression of Bcl-2 by hepatocytes during cholestasis suggested an
adaptive phenomenon to resist apoptosis by toxic bile salts
(35). Hepatocytes in the bile duct
ligation (BDL) rats expressed Bcl-2, which can inhibit the bile
salt-induced hepatocellular apoptosis (36). Related research found that
glycochenodeoxycholate (GCDA) may induce the phosphorylation of
Mcl-1 (one of the anti-apoptotic member in Bcl-2 family) at T163
site, eventually leading to survival and chemoresistance in
hepatocellular carcinoma cells (37). Since GCDA can activate ERK1/2, which
is a physiologic Ser70 site kinase of Bcl-2, in the present study,
we have shown that GCDA induces Bcl-2 phosphorylation at Ser70
through activation of ERK1/2, which leads to enhanced anti-apototic
function in human liver cancer cells.
Materials and methods
Materials
Sodium glycochenodeoxycholate (GCDA, G0759) was
purchased from Sigma-Aldrich (St. Louis, MO, USA). 5-Fluorouracil
was purchased from Shanghai Xudong Haipu Pharmaceutical Co., Ltd.
(Shanghai, China). PD98059 (513001) was obtained from Calbiochem
(San Diego, CA, USA). Bcl-2 (50E3) rabbit (2870) and phospho-Bcl-2
(Ser70) (5H2) rabbit (2827) antibodies were from Cell Signaling
Technology (Danvers, MA, USA). Bcl-2 (N-19) (sc-492), p-Bcl2
(74.Ser70) (sc-135757), ERK1 (K-23) (sc-94), ERK2 (C-14) (sc-154)
and p-ERK (E-4) (sc-7383) antibodies were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Alexa Fluor® 488
goat anti-rabbit IgG (H+L) antibody (A-11008), Alexa
Fluor® 594 goat anti-rabbit IgG (H+L) antibody (A-11012)
and Alexa Fluor® 594 donkey anti-mouse IgG (H+L)
antibody (A-21203) were obtained from Molecular Probes (Eugene, OR,
USA). Monoclonal anti-Flag antibody (F1804) was from Sigma-Aldrich.
The mitochondrial marker (Mito-GFP) and endoplasmic reticulum
marker (ERM-RFP) plasmids were kindly provided by Dr Yuan (Sun
Yat-sen University, Guangzhou, China). All reagents used were
obtained from commercial sources.
Cell lines and culture conditions
Five HCC cell lines HepG2, Bel7402, QGY7703,
SMMC7721 and Huh7 were purchased from the Institute of Biochemistry
and Cell Biology (Shanghai Institutes for Biological Sciences, CAS,
Shanghai, China). HepG2, Bel7402, QGY7703 and SMMC7721 were
maintained in RPMI-1640 supplemented with 10% fetal bovine serum
(FBS). Huh7 was maintained in Dulbeccos modified Eagles medium
(DMEM) supplemented with 10% FBS. All of the cell lines were
cultured at 37°C in a humid incubator with 5% CO2.
siRNA and transfections
For RNA interference, siRNA specific to Bcl-2 (5-AAC
AUC GCC CUG UGG AUG ACU-3), ERK1 (5-GAC CGG AUG UUA ACC UUU ATT-3),
ERK2 (5-CAC CAA CCA UCG AGC AAA UTT-3) and non-specific control
siRNA (5-UUC UCC GAA CGU GUC ACG UTT-3) were purchased from
Shanghai GenePharma, Co., Ltd., (Shanghai, China) siRNAs were
transfected into the cells using Lipofectamine RNAi max
(Invitrogen) according to the manufacturers instruction.
Cell viability assay
Cells treated with GCDA, drugs and inhibitors were
harvested and washed with phosphate-buffered saline (PBS). Cells
were resuspended in binding buffer and stained with Annexin V and
propidium iodide (PI) according to manufacturers instructions (BD
Biosciences Annexin V kit; BD Biosciences, San Diego, CA, USA).
Cell extraction and western
blotting
After the treatment, the cells were washed with cold
PBS and then were removed to 1.5 ml Eppendorf tubes using a cell
scraper. The whole-cell extracts were prepared in lysis buffer [50
mM Tris, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholic
acid sodium salt and cocktail protease inhibitor (Roche)], which
were put on ice for 30 min and then centrifuged at 14,000 rpm for 2
min. The resulting supernatant was collected as the total cell
lysate. Aliquots containing 30 µg of total protein from each sample
were subjected to 10% SDS-PAGE and electrotransferred to
nitrocellulose membranes. The membranes were blocked with 5%
skimmed milk in PBST for 2 h, and then incubated with primary
antibodies (diluted in PBST containing 2.5% skimmed milk) at 4°C
overnight. After the membranes were washed with PBST three times,
they were incubated with horseradish peroxidase-conjugated
anti-rabbit or anti-mouse secondary antibody for 2 h.
Chemiluminescent detection was performed with SuperSignal West Dura
Extended Duration Substrate kit (Thermo Fisher Scientific) and the
membranes were exposed to X-ray film.
Immunofluorescence
QGY7703 cells were seeded on coverslips (10 mm × 10
mm) in a 24-well plate. After 20–24 h, cells were fixed for 15 min
in a 4% formaldehyde solution, washed three times in PBST (0.05%
Tween-20 in PBS) and blocked with 5% BSA in PBST at 4°C overnight.
They were then incubated with Bcl-2, p-Bcl-2-Ser70 or p-ERK
antibodies for 1 h. The cells were washed three times in PBST, and
incubated with the Alexa Fluor® 488 (green) goat
anti-rabbit IgG (H+L) antibody and/or Alexa Fluor® 594
(red) goat anti-rabbit IgG (H+L) antibody and/or Alexa
Fluor® 594 (red) donkey anti-mouse IgG (H+L) antibody
for 1 h. Following triple washing in PBS, cells were labeled with
0.2 µg/ml DAPIs in PBS for 5 min, and then washed three times in
PBS. Cells were mounted in MOWIOL R4-88 reagent (Calbiochem) and
photographed with a Carl Zeiss AxioVision 4 microscope.
Statistical analysis
The data were subject to statistical analysis using
the SPSS software package (version 17.0). All data were expressed
as the means ± SD of three or more independent experiments. The
statistical differences were analyzed by the Students t-test. The
differences were considered to be statistically significant at
P<0.05.
Results
GCDA induces chemoresistance of human
liver cancer cells
5-Fluorouracil (5-FU) is one of the most important
chemotherapy drugs for the advanced liver cancer. However, its
clinical efficacy is not satisfactory due to the chemoresistance.
The underlying mechanism is not clear yet. In order to test whether
bile salt GCDA can induce chemoresistance, five HCC cell lines,
HepG2, Bel7402, QGY7703, SMMC7721 and Huh7 were treated with 5-FU
with respective IC50 concentration (15 µg/ml for HepG2,
30 µg/ml for Bel7402, 1.5 µg/ml for QGY7703, 20 µg/ml for SMMC7721
and 3 µg/ml for Huh7) in the absence or presence of GCDA for 72 h.
Cell viability and apoptosis were determined by analyzing Annexin V
binding on fluorescence-activated cell sorting (FACS). Results
indicate that in all the five cell lines, 5-FU can induce ~50% of
cells to undergo apoptosis and GCDA can significantly attenuate the
apoptotic effect of 5-FU (Fig. 1).
These findings reveal that GCDA may be one reason for
chemoresistance in liver cancer cells. Data represent the mean ± SD
of three determinations. P<0.05 is based on the Students
t-test.
Bcl-2 plays a role in GCDA-induced
chemoresistance of human liver cancer cells
Bcl-2 is a major anti-apoptotic member of Bcl2
family. Consistently, Bcl-2 is widely expressed in human liver
cancer cells. Among them, HepG2, QGY7703 and SMMC7721 expressed
relatively high levels of endogenous Bcl-2, while Bel7402 and Huh7
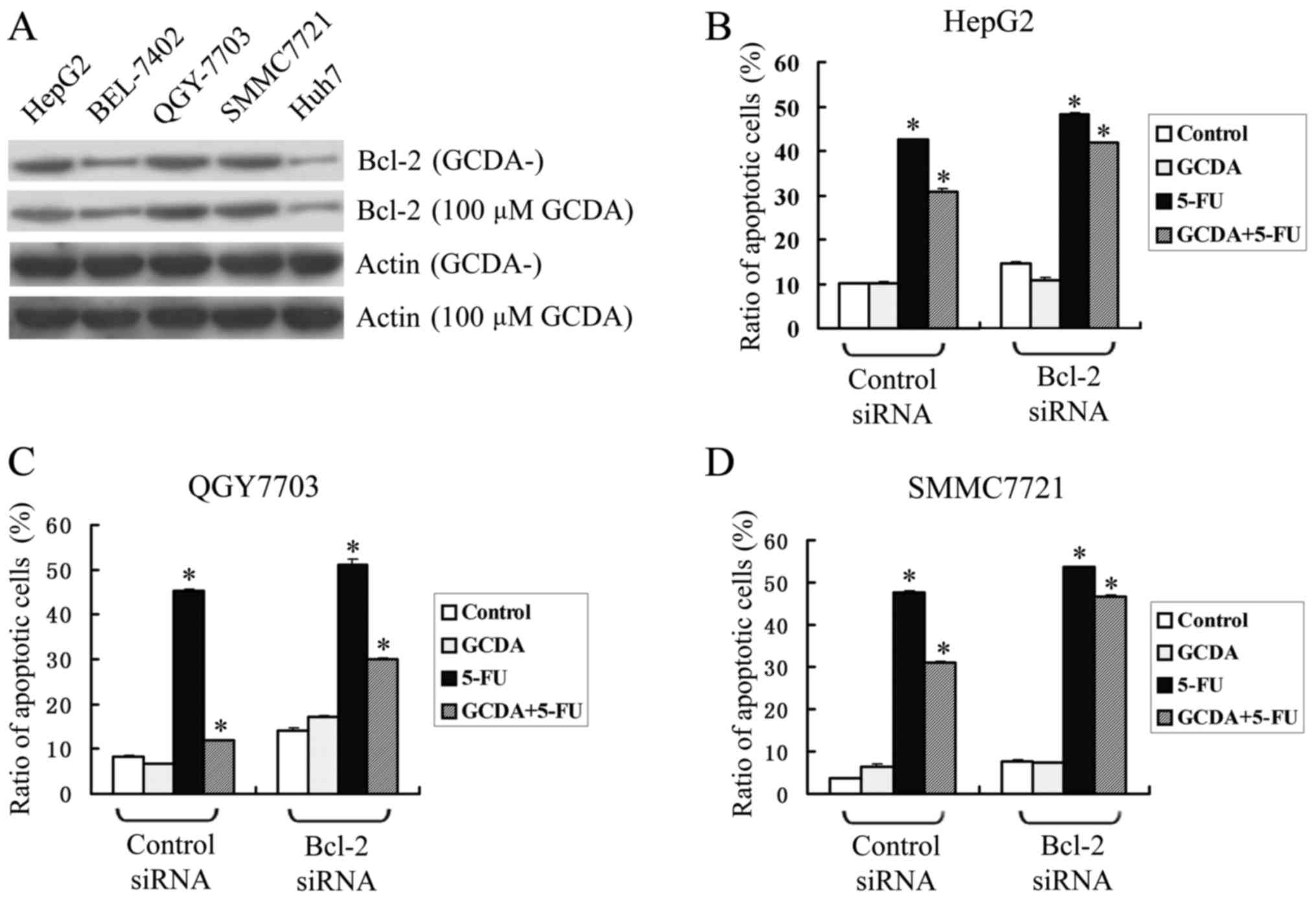
expressed relatively low levels of endogenous Bcl-2 (Fig. 2A). After treating the five types of
cell-line with 100 µM GCDA, the expression levels of endogenous
Bcl-2 changed little (Fig. 2A).
To test whether Bcl-2 plays a role in GCDA-induced
chemoresistance, three HCC cell-lines with high level of endogenous
Bcl-2, HepG2, QGY7703 and SMMC7721 were transfected with siRNA
targeting Bcl-2 in the presence of GCDA (100 µM), 5-FU (15 µg/ml
for HepG2, 1.5 µg/ml for QGY7703 and 20 µg/ml for SMMC7721) or both
GCDA (100 µM) and 5-FU (15 µg/ml for HepG2, 1.5 µg/ml for QGY7703
and 20 µg/ml for SMMC7721) for 72 h. Cell viability and apoptosis
were determined by analyzing Annexin V binding FACS. Results
indicate that in all the three cell-lines, GCDA can attenuate the
apoptotic effect of 5-FU. However, after Bcl-2 was silenced by
siRNA, the ratio of apoptotic cells was upregulated slightly, and
the chemoresistance effect of GCDA was downregulated from 27.23 to
13.41% in HepG2 cells (Fig. 2B),
from 73.86 to 40.93% in QGY7703 cells (Fig. 2C) and 35.20 to 13.13% in SMMC7721
cells (Fig. 2D). Data represent the
mean ± SD of three determinations. P<0.05 is based on the
Students t-test. These findings suggest that Bcl-2 may play a role
in GCDA-induced chemoresistance.
GCDA stimulates Bcl-2 phosphorylation
at Ser70
According to the results above, GCDA did not affect
much the expression of endogenous Bcl-2 (Fig. 2A), but Bcl-2 indeed plays a role in
GCDA-induced chemoresistance (Fig.
2B-D). Consistent with previous studies, in QGY7703 cells Bcl-2
localized at mitochondria and endoplasmic reticulum (Fig. 3A and B), which indicates that Bcl-2
may function both at mitochondria and endoplasmic reticulum
(16). A previous study discovered
that phophorylation of Bcl-2 at the evolutionarily conserved Ser70
site may be required for its full and potent antiapoptotic function
(20,21). Therefore, we supposed that GCDA may
induce the phosphorylation of Bcl-2 at Ser70 and then play an
antiapoptotic effect. To test this, we treated HepG2, Bel7402,
QGY7703, SMMC7721 and Huh7 cells with 100 µM GCDA for 30 min.
Western blot analysis showed that the phosphorylation levels of
Bcl-2 at Ser70, but not the expression levels of Bcl-2, increased
in all the five cell lines after treated with GCDA (Fig. 3C). Furthermore, we treated QGY7703
cells with gradually increasing doses of GCDA for 30 min. Western
blot analysis showed that the phosphorylation levels of Bcl-2 at
Ser70, but not the expression levels of Bcl-2, increased with the
increasing GCDA concentrations (Fig.
3D). Notably, when Bcl-2 was phosphorylated at Ser70, abundant
p-Bcl-2-Ser70 protein clustered in the nucleus, appearing as bigger
spots in vision (Fig. 3E). These
findings indicated that GCDA can induce the phosphorylation of
Bcl-2 at Ser70 site. The p-Bcl-2-Ser70 protein may function at the
nucleus as polymers.
GCDA induces phosphorylation of Bcl-2
at Ser70 through MAPK/ERK1/2 pathway
Previous research identified ERK1/2 kinases as
upstream Bcl-2 kinases (20). ERK1
and ERK2 can colocalize with mitochondrial Bcl-2 and directly
phophorylate Bcl-2 on Ser-70 both in vitro and in
vivo (20). To test whether
GCDA-induced Bcl-2 phosphorylation occurs through MAPK/ERK1/2,
QGY7703 cells were treated with increasing concentrations of GCDA
for 30 min. Phosphorylation of ERK1/2 was analyzed by western blot
analysis using a phospho-specific ERK antibody. Results show that
GCDA induces phosphorylation and activation of ERK1/2 in a
dose-dependent manner (Fig. 4A). To
further test whether inhibition of MAPK/ERK1/2 affects GCDA-induced
Bcl-2 phosphorylation, QGY7703 cells expressing high levels of
endogenous Bcl-2 were treated with GCDA in the absence or presence
of increasing concentrations of PD98059 (MAPK/ERK1/2 inhibitor).
Results suggest that PD98059 suppresses GCDA-stimulated
phosphorylation of Bcl-2 at Ser70 and activation of ERK1/2, and has
no effect on expression levels of endogenous Bcl-2 and ERK1/2
(Fig. 4B and C). QGY7703 cells were
transfected with ERK1/2-siRNA or control-siRNA, phosphorylation of
Bcl-2 at Ser70 and activation of ERK1/2 was analyzed by western
blot analysis using a phospho-specific Bcl-2 Ser70 and ERK
antibody. Results indicate that knockdown of ERK1/2 protein
expression by RNAi significantly decreases phosphorylation of Bcl-2
at Ser70 and activation of ERK1/2 (Fig.
4D). Coimmunofluorescent staining using p-ERK and Bcl-2
antibodies reveals that p-ERK and Bcl-2 colocalized in QGY7703
cells (Fig. 4E), which implies the
direct interaction between p-ERK and Bcl-2. Thus, these findings
suggest that GCDA-induced phosphorylation of Bcl-2 at Ser70 may
occur through activation of ERK1/2.
The MAPK/ERK1/2 inhibitor PD98059
suppresses GCDA-induced survival and chemoresistance
The results above showed that PD98059 can decrease
phosphorylation of Bcl-2 and ERK1/2, then whether PD98059 affects
cell survival was assessed in QGY7703 cells expressing high levels
of endogenous Bcl-2 treated with GCDA in the absence or presence of
increasing concentrations of PD98059 for 30 min. Cell viability and
apoptosis were determined by analyzing Annexin V binding FACS.
Results indicated that PD98059 mediated cell apoptosis in a
dose-dependent manner (Fig. 5A and
C). Next, to test whether PD98059 attenuates GCDA-induced
chemoresistance, QGY7703 cells were treated with GCDA and 5-FU in
the presence or absence of PD98059 for 72 h. Annexin V binding FACS
results showed that GCDA can prolong cell survival following
treatment with 5-FU, and PD98059 significantly attenuated the
chemoresistance of GCDA (Fig. 5B and
D). Data represent the mean ± SD of three determinations.
P<0.05 is based on the Students t-test. These findings suggest
that inhibition of MAPK/ERK1/2 pathway in human liver cancer cells
may be pivotal for suppression of GCDA-induced chemoresistance.
Discussion
Bile acids are considered to be both detergent
molecules that facilitate the solubilization of cheolesterol and
absorption of lipids and regulatory molecules that activate five
distinct receptors (farnesoid X receptor, pregnane X receptor,
constitutive androstane receptor, vitamin D receptor and G
protein-coupled receptor) and cell signaling pathways (c-jun
N-terminal kinase 1/2, AKT and ERK1/2) in cells in the liver and
gastrointestinal tract (38,39).
Hepatocytes synthesize bile acids that secrete into the bile and
are stored in the gallbladder (40). Eating food can stimulate bile acids
to release into the intestinal tract where bile acids act as
detergents to facilitate the solubilization of fatty acids and
absorption of dietary lipids (40),
then, bile acids are efficiently reabsorbed in the ileum and
transported back to the liver via portal blood for resecretion into
the bile. This process is known as enterohepatic circulation of
bile acids (40). Recycling of bile
acids/salts between the liver and intestine occurs 6–10 times each
day and transports 20–40 g bile acids (39). Bile salts and bile acids are
considered to be potential carcinogens. For example, bile acids
play a role in colorectal carcinogenesis through ERKs and PKC
signaling pathway (41);
intrahepatic bile acid accumulation may have cocarcinogenic effects
on the development of cholangiocarcinoma (42). Glycochenodeoxycholate (Glycine
conjugate of chenodexycholate) is the main ingredient in the bile.
Glycochenodeoxycholic acid can stimulate cell growth in Barretts
adenocarcinoma cell line (SEG-1) by activating the PI3 kinase/Akt
signaling pathway (13), and induce
the proliferation of non-neoplastic Barretts cell line by
activation of both ERK and p38 MAPK pathways (14). Hepatocellular carcinoma (HCC) is a
highly malignant tumor that can evolve rapidly to acquire
resistance to conventional chemotherapies (43). Our findings suggest that in all the
five HCC cell lines, 5-FU can induce ~50% of cells to undergo
apoptosis and GCDA can significantly attenuate the apoptotic effect
of 5-FU (Fig. 1). These findings
reveal that GCDA may be one reason of chemoresistance in liver
cancer cells.
Bcl-2 is the most important anti-apoptotic protein.
Specific depletion of Bcl-2 from HepG2, QGY7703 and SMMC7721 cells
by RNAi enhances sensitivity of those liver cancer cells to
chemotherapeutic drug 5-FU, the chemoresistance effect of GCDA is
downregulated (Fig. 2B-D). This
suggests that Bcl-2, which is an essential target for bile
salt-induced chemoresistance in human liver cancer cells, should be
a target for liver cancer treatment. At present, many inhibitors
and microRNAs targeting Bcl-2 have been studied. miR-16 can inhibit
glioma cell growth and invasion through the suppression of Bcl-2
(27). miR-18, a tumor-suppressive
miRNA directly targeting Bcl-2, participated in suppression of cell
proliferation and survival in nasopharyngeal carcinoma (29). MicroRNA-30b functions as a tumor
suppressor in human colorectal cancer by targeting Bcl-2 (32). miR-449a could modulate cell cycle
and apoptosis through regulating Bcl-2 expression in gastric cancer
cell line SGC7901 (33). miR-210
can mediate hypoxia-induced neural apoptosis by targeting Bcl-2
(44). AT-101, a pan-Bcl-2
inhibitor, has high binding specificity for Bcl-2 and Mcl-1 and
potentiates the cytotoxic effects of lenalidomide and dexamethasone
in preclinical models of plasma cell cancers (multiple myeloma and
Waldenstrom macroglobulinaemia) (28). ABT-737, which is an anticancer drug,
is a Bcl-2 Homology 3 (BH-3)-mimetic that induces apoptosis by
inhibiting pro-survival Bcl-2 proteins (45) and enhance the effects of epirubicin
on HepG2 cells by activating autophagy and inducing apoptosis
(46). A BH3 mimetic, ABT-199, has
been developed to selectively bind Bcl-2 and enhances
imatinib-induced cell death in chronic myeloid leukemia progenitors
(47). BDA-366, a small-molecule
Bcl-2-BH4 domain antagonist, can bind BH4 of Bcl-2 with high
affinity and selectivity, and this Bcl-2-BH4 antagonist may provide
a strategy to improve lung cancer outcome (48).
Bcl-2 is an anti-apototic molecule, whose
phosphoylation at Ser70 by growth factor-activated protein kinases
including PKC and the MAPKs (ERK1/2) can positively regulate the
anti-apoptotic function of Bcl-2 (20,49,50).
Our findings indicate that Bcl-2 is expressed in all the five liver
cancer cell lines (Fig. 2A).
Glycochenodeoxycholate (GCDA), the main ingredient in the bile, can
mimic growth factor to stimulate Bcl-2 phosphorylation at Ser70
site in dose-dependent manner (Fig.
3D), which is associated with enhanced chemoresistance of liver
cancer cells (Figs. 1 and 5B). ERK1 and ERK2 are physiologic Bcl-2
kinases that can phosphorylate Bcl-2 at Ser70 site. Intriguingly,
GCDA can stimulate phosphorylation and activation of ERK1/2
(Fig. 4A). Activated ERK1/2
(p-ERK1/2) can colocalize with Bcl-2 in cytoplasm (Fig. 4E). Because both the specific MEK/ERK
inhibitor PD98059 and siRNA targeting ERK1/2 block GCDA-stimulated
phosphorylation of ERK1/2 and Bcl-2 (Fig. 4B-D) and attenuates GCDA-induced
survival and chemoresistance (Fig. 5A
and B), we propose that ERK1 and ERK2 function as
GCDA-activated Bcl-2 kinases to phosphorylate Bcl-2 and regulate
its activity in human liver cancer cells.
Bcl-2 at the ER is required in cancer cells, such
cells will be sensitive to BH4-domain targeting drugs (16). Our results showed that in QGY7703
cells Bcl-2 localized both at mitochondria and endoplasmic
reticulum (Fig. 3A and B), which
indicated that Bcl-2 may prevent apoptosis by disturbing
Bim-mediated Bax/Bak activation or toxic Ca2+ release.
In addition, the function of Bcl-2 at the nucleus is not so clear.
Phosphorylation of Bcl-2 at Ser70 site induced by NNK may
facilitate the direct interaction between Bcl-2 and c-Myc in the
nucleus that enhances the half-life of c-Myc and finally promotes
cell survival and chemoresistance in human lung cancer (51). NNK can induce the accumulation of
Bcl-2 in the nucleus, which disrupts the hMSH2-hMSH6 complex and
suppresses DNA mismatch repair in vitro (52). Besides, the accumulation of Bcl-2 in
the nucleus induced by NNK can also interact with APE1, which
disrupts APE1·XRCC1 complex with suppresion of APE1 endonulease
activity and AP site repair (53).
Our results show that when Bcl-2 was phosphorylated at Ser70,
abundant p-Bcl-2-Ser70 protein cluster in the nucleus, appearing as
bigger spots (Fig. 3E). The
p-Bcl-2-Ser70 protein may function at the nucleus as polymers.
However, the p-Bcl-2-Ser70 protein participation in the nucleus in
transcriptional regulation or DNA repair need to be further
studied.
In summary, the present study found that
glycochenodeoxycholate (GCDA) induces chemoresistance of human
liver cancer cells by activating the anti-apoptotic function of
Bcl-2 via its phosphorylation. GCDA stimulates Bcl-2
phosphorylation at Ser70 site in the flexible loop domain through
activating ERK1/2. GCDA-induced Bcl-2 phosphorylation at Ser70 site
leads to its chemoresistance of human liver cancer cells (Fig. 6). Thus, disruption of the
anti-apoptotic function of Bcl-2 by blocking its Ser70 site
phosphorylation may represent a strategy for the treatment of
GCDA-induce chemoresistance in human liver cancers.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (nos. 81402001, 81272193 and
81302075), the Natural Science Foundation of Hunan Province (no.
2016JJ3177) and the Young Teachers Boost Project of Central South
University (2012QNZT114). We thank Dr Yuan (Sun Yat-sen University)
for kindly providing mitochondrial marker (Mito-GFP) and
endoplasmic reticulum marker (ERM-RFP) plasmids.
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Allemani C, Weir HK, Carreira H, Harewood
R, Spika D, Wang XS, Bannon F, Ahn JV, Johnson CJ, Bonaventure A,
et al: CONCORD Working Group: Global surveillance of cancer
survival 1995–2009: Analysis of individual data for 25,676,887
patients from 279 population-based registries in 67 countries
(CONCORD-2). Lancet. 385:977–1010. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Faubion WA, Guicciardi ME, Miyoshi H,
Bronk SF, Roberts PJ, Svingen PA, Kaufmann SH and Gores GJ: Toxic
bile salts induce rodent hepatocyte apoptosis via direct activation
of Fas. J Clin Invest. 103:137–145. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sodeman T, Bronk SF, Roberts PJ, Miyoshi H
and Gores GJ: Bile salts mediate hepatocyte apoptosis by increasing
cell surface trafficking of Fas. Am J Physiol Gastrointest Liver
Physiol. 278:G992–G999. 2000.PubMed/NCBI
|
|
5
|
Jones BA, Rao YP, Stravitz RT and Gores
GJ: Bile salt-induced apoptosis of hepatocytes involves activation
of protein kinase C. Am J Physiol. 272:G1109–G1115. 1997.PubMed/NCBI
|
|
6
|
Gonzalez B, Fisher C and Rosser BG:
Glycochenodeoxycholic acid (GCDC) induced hepatocyte apoptosis is
associated with early modulation of intracellular PKC activity. Mol
Cell Biochem. 207:19–27. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tsuchiya S, Tsuji M, Morio Y and Oguchi K:
Involvement of endoplasmic reticulum in glycochenodeoxycholic
acid-induced apoptosis in rat hepatocytes. Toxicol Lett.
166:140–149. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Metalli V Drudi, Mancino MG, Mancino A,
Torrice A, Gatto M, Attili AF, Alpini G and Alvaro D: Bile salts
regulate proliferation and apoptosis of liver cells by modulating
the IGF1 system. Dig Liver Dis. 39:654–662. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang K, Brems JJ, Gamelli RL and Ding J:
Reversibility of caspase activation and its role during
glycochenodeoxycholate-induced hepatocyte apoptosis. J Biol Chem.
280:23490–23495. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang K, Brems JJ, Gamelli RL and Holterman
AX: Survivin signaling is regulated through nuclear factor-kappa B
pathway during glycochenodeoxycholate-induced hepatocyte apoptosis.
Biochim Biophys Acta. 1803:1368–1375. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Webster CR, Johnston AN and Anwer MS:
Protein kinase Cδ protects against bile acid apoptosis by
suppressing proapoptotic JNK and BIM pathways in human and rat
hepatocytes. Am J Physiol Gastrointest Liver Physiol.
307:G1207–G1215. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoon JH, Werneburg NW, Higuchi H, Canbay
AE, Kaufmann SH, Akgul C, Edwards SW and Gores GJ: Bile acids
inhibit Mcl-1 protein turnover via an epidermal growth factor
receptor/Raf-1-dependent mechanism. Cancer Res. 62:6500–6505.
2002.PubMed/NCBI
|
|
13
|
Jaiswal K, Tello V, Lopez-Guzman C,
Nwariaku F, Anthony T and Sarosi GA Jr: Bile salt exposure causes
phosphatidyl-inositol-3-kinase-mediated proliferation in a Barretts
adenocarcinoma cell line. Surgery. 136:160–168. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jaiswal K, Lopez-Guzman C, Souza RF,
Spechler SJ and Sarosi GA Jr: Bile salt exposure increases
proliferation through p38 and ERK MAPK pathways in a non-neoplastic
Barretts cell line. Am J Physiol Gastrointest Liver Physiol.
290:G335–G342. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Komichi D, Tazuma S, Nishioka T, Hyogo H
and Chayama K: Glycochenodeoxycholate plays a carcinogenic role in
immortalized mouse cholangiocytes via oxidative DNA damage. Free
Radic Biol Med. 39:1418–1427. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Akl H, Vervloessem T, Kiviluoto S,
Bittremieux M, Parys JB, De Smedt H and Bultynck G: A dual role for
the anti-apoptotic Bcl-2 protein in cancer: Mitochondria versus
endoplasmic reticulum. Biochim Biophys Acta. 1843:2240–2252. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsujimoto Y: Bcl-2 family of proteins:
Life-or-death switch in mitochondria. Biosci Rep. 22:47–58. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cory S and Adams JM: The Bcl2 family:
Regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Deng X, Gao F, Flagg T and May WS Jr:
Mono- and multisite phosphorylation enhances Bcl2s antiapoptotic
function and inhibition of cell cycle entry functions. Proc Natl
Acad Sci USA. 101:153–158. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Deng X, Ruvolo P, Carr B and May WS Jr:
Survival function of ERK1/2 as IL-3-activated,
staurosporine-resistant Bcl2 kinases. Proc Natl Acad Sci USA.
97:1578–1583. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Deng X, Xiao L, Lang W, Gao F, Ruvolo P
and May WS Jr: Novel role for JNK as a stress-activated Bcl2
kinase. J Biol Chem. 276:23681–23688. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang C, Wu Z, Hong W, Wang Z, Peng D,
Chen J, Yuan C, Yu S, Xu L and Fang Y: Influence of BCL2 gene in
major depression susceptibility and antidepressant treatment
outcome. J Affect Disord. 155:288–294. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moreno-Galindo C, Hermsen M,
García-Pedrero JM, Fresno MF, Suárez C and Rodrigo JP: p27 and BCL2
expression predicts response to chemotherapy in head and neck
squamous cell carcinomas. Oral Oncol. 50:128–134. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu P, Liu L, Wang J, Zhang K, Hong X, Deng
Q, Xiang J, Zhang X, He M, Wu T, et al: Genetic variation in BCL2
3-UTR was associated with lung cancer risk and prognosis in male
Chinese population. PLoS One. 8:e721972013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mehta SV, Shukla SN and Vora HH:
Overexpression of Bcl2 protein predicts chemoresistance in acute
myeloid leukemia: Its correlation with FLT3. Neoplasma. 60:666–675.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dorjgochoo T, Xiang YB, Long J, Shi J,
Deming S, Xu WH, Cai H, Cheng J, Cai Q, Zheng W, et al: Association
of genetic markers in the BCL-2 family of apoptosis-related genes
with endometrial cancer risk in a Chinese population. PLoS One.
8:e609152013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang TQ, Luo XJ, Wu TF, Ding DD, Zhao ZH,
Chen GL, Xie XS, Li B, Wei YX, Guo LC, et al: MicroRNA-16 inhibits
glioma cell growth and invasion through suppression of BCL2 and the
nuclear factor-κB1/MMP9 signaling pathway. Cancer Sci. 105:265–271.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Paulus A, Chitta K, Akhtar S, Personett D,
Miller KC, Thompson KJ, Carr J, Kumar S, Roy V, Ansell SM, et al:
AT-101 downregulates BCL2 and MCL1 and potentiates the cytotoxic
effects of lenalidomide and dexamethasone in preclinical models of
multiple myeloma and Waldenström macroglobulinaemia. Br J Haematol.
164:352–365. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhen Y, Liu Z, Yang H, Yu X, Wu Q, Hua S,
Long X, Jiang Q, Song Y, Cheng C, et al: Tumor suppressor PDCD4
modulates miR-184-mediated direct suppression of C-MYC and BCL2
blocking cell growth and survival in nasopharyngeal carcinoma. Cell
Death Dis. 4:e8722013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yao F, Han Q, Zhong C and Zhao H: TRAF6
promoted the tumorigenicity of esophageal squamous cell carcinoma.
Tumour Biol. 34:3201–3207. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Petrasek J, Iracheta-Vellve A, Csak T,
Satishchandran A, Kodys K, Kurt-Jones EA, Fitzgerald KA and Szabo
G: STING-IRF3 pathway links endoplasmic reticulum stress with
hepatocyte apoptosis in early alcoholic liver disease. Proc Natl
Acad Sci USA. 110:16544–16549. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liao WT, Ye YP, Zhang NJ, Li TT, Wang SY,
Cui YM, Qi L, Wu P, Jiao HL, Xie YJ, et al: MicroRNA-30b functions
as a tumour suppressor in human colorectal cancer by targeting
KRAS, PIK3CD and BCL2. J Pathol. 232:415–427. 2013. View Article : Google Scholar
|
|
33
|
Hu J, Fang Y, Cao Y, Qin R and Chen Q:
miR-449a regulates proliferation and chemosensitivity to cisplatin
by targeting cyclin D1 and BCL2 in SGC7901 cells. Dig Dis Sci.
59:336–345. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Drukker L, Margulis A, Chaouat M, Levitzki
R, Maiorenko E and Ben Bassat H: Changes of PI3K/AKT/BCL2 signaling
proteins in congenital Giant Nevi: Melanocytes contribute to their
increased survival and integrity. J Recept Signal Transduct Res.
33:359–366. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kurosawa H, Que FG, Roberts LR, Fesmier PJ
and Gores GJ: Hepatocytes in the bile duct-ligated rat express
Bcl-2. Am J Physiol. 272:G1587–G1593. 1997.PubMed/NCBI
|
|
36
|
Wang J and Zou S: The bcl-2 mRNA
expression in GCDC-induced obstructive jaundice in rats and its
implication in hepatocellular apoptosis. J Huazhong Univ Sci
Technolog Med Sci. 22:34–36. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liao M, Zhao J, Wang T, Duan J, Zhang Y
and Deng X: Role of bile salt in regulating Mcl-1 phosphorylation
and chemoresistance in hepatocellular carcinoma cells. Mol Cancer.
10:442011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hylemon PB, Zhou H, Pandak WM, Ren S, Gil
G and Dent P: Bile acids as regulatory molecules. J Lipid Res.
50:1509–1520. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sipka S and Bruckner G: The
immunomodulatory role of bile acids. Int Arch Allergy Immunol.
165:1–8. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li T and Chiang JY: Bile acid signaling in
metabolic disease and drug therapy. Pharmacol Rev. 66:948–983.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Debruyne PR, Bruyneel EA, Li X, Zimber A,
Gespach C and Mareel MM: The role of bile acids in carcinogenesis.
Mutat Res. 480–481:359–369. 2001. View Article : Google Scholar
|
|
42
|
Lozano E, Sanchez-Vicente L, Monte MJ,
Herraez E, Briz O, Banales JM, Marin JJ and Macias RI:
Cocarcinogenic effects of intrahepatic bile acid accumulation in
cholangiocarcinoma development. Mol Cancer Res. 12:91–100. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang X, Sun D, Tian Y, Ling S and Wang L:
Metformin sensitizes hepatocellular carcinoma to arsenic
trioxide-induced apoptosis by downregulating Bcl2 expression.
Tumour Biol. 36:2957–2964. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chio CC, Lin JW, Cheng HA, Chiu WT, Wang
YH, Wang JJ, Hsing CH and Chen RM: MicroRNA-210 targets
antiapoptotic Bcl-2 expression and mediates hypoxia-induced
apoptosis of neuroblastoma cells. Arch Toxicol. 87:459–468. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rooswinkel RW, van de Kooij B, Verheij M
and Borst J: Bcl-2 is a better ABT-737 target than Bcl-xL or Bcl-w
and only Noxa overcomes resistance mediated by Mcl-1, Bfl-1, or
Bcl-B. Cell Death Dis. 3:e3662012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Du P, Cao H, Wu HR, Zhu BS, Wang HW, Gu
CW, Xing CG and Chen W: Blocking Bcl-2 leads to autophagy
activation and cell death of the HEPG2 liver cancer cell line.
Asian Pac J Cancer Prev. 14:5849–5854. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ko TK, Chuah CT, Huang JW, Ng KP and Ong
ST: The BCL2 inhibitor ABT-199 significantly enhances
imatinib-induced cell death in chronic myeloid leukemia
progenitors. Oncotarget. 5:9033–9038. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Han B, Park D, Li R, Xie M, Owonikoko TK,
Zhang G, Sica GL, Ding C, Zhou J, Magis AT, et al: Small-molecule
Bcl2 BH4 antagonist for lung cancer therapy. Cancer Cell.
27:852–863. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mai H, May WS, Gao F, Jin Z and Deng X: A
functional role for nicotine in Bcl2 phosphorylation and
suppression of apoptosis. J Biol Chem. 278:1886–1891. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ruvolo PP, Deng X, Carr BK and May WS: A
functional role for mitochondrial protein kinase Calpha in Bcl2
phosphorylation and suppression of apoptosis. J Biol Chem.
273:25436–25442. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jin Z, Gao F, Flagg T and Deng X:
Tobacco-specific nitrosamine
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone promotes functional
cooperation of Bcl2 and c-Myc through phosphorylation in regulating
cell survival and proliferation. J Biol Chem. 279:40209–40219.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hou Y, Gao F, Wang Q, Zhao J, Flagg T,
Zhang Y and Deng X: Bcl2 impedes DNA mismatch repair by directly
regulating the hMSH2-hMSH6 heterodimeric complex. J Biol Chem.
282:9279–9287. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhao J, Gao F, Zhang Y, Wei K, Liu Y and
Deng X: Bcl2 inhibits abasic site repair by down-regulating APE1
endonuclease activity. J Biol Chem. 283:9925–9932. 2008. View Article : Google Scholar : PubMed/NCBI
|