Introduction
Cervical cancer is one of the most common
malignancies in women, with an estimated 530,000 new cases and
275,000 deaths each year worldwide (1). Although mounting studies have
demonstrated the importance of human papillomavirus in cervix
tumorigenesis (2,3), the detailed mechanism of cervical
carcinogenesis remains unclear.
Recent studies have shown that many members of the
tripartite motif-containing (TRIM) superfamily are involved in a
broad range of biological processes, including immunity (4,5),
neurological development (6), cell
growth (7), cell differentiation
(8), tumorigenesis (9), and responses to microbial infection
(10). TRIM proteins are
evolutionarily conserved, sharing a common N-terminal really
interesting new gene (RING) finger domain, which can mediate the
conjugation of proteins with ubiquitin (11). Recently, multiple studies have
indicated that some members of the TRIM protein family function as
important regulators for carcinogenesis. For example, TRIM13,
TRIM19, TRIM24, TRIM25, and TRIM59 were shown to be involved in
leukemia, liver, breast, prostate, and gastric cancers,
respectively (11–15), demonstrating the crucial roles of
the TRIM family in tumorigenesis.
TRIM28 also exhibits certain functions in tumor
development (16–18). Yokoe et al (19) found that the expression level of the
TRIM28 gene was significantly higher in gastric cancer
tissues than in non-cancerous tissues. Patients with high
TRIM28 expression showed a higher incidence of peritoneal
carcinomatosis and significantly poorer overall survival compared
to patients with low TRIM28 expression. Addison et al
(20) showed that TRIM28 is
frequently overexpressed in breast tumors at both the mRNA and
protein levels; knockdown of TRIM28 in breast cancer cells led to
the inhibition of cell proliferation, tumor growth, and metastasis.
However, other researchers found that high TRIM28 expression was
correlated with increased overall survival in early-stage lung
tumors, and TRIM28 overexpression reduced cell proliferation in
model lung cancer cell lines (21).
These findings suggest that TRIM28 may play a complex role in human
cancer.
In the present study, we found that the expression
of TRIM28 is greatly increased in cervical cancer tissues and cell
lines. In vitro and in vivo experiments further
demonstrated that TRIM28 overexpression promotes cervical cancer
cell proliferation and xenograft tumor growth, respectively.
Further analyses revealed that TRIM28 induced cervical cancer cell
growth via the mammalian target of rapamycin (mTOR) signaling
pathway.
Materials and methods
Clinical tissue samples
Twenty paired cervical cancer samples and adjacent
normal tissues were obtained from January 2013 to November 2015 at
the Shanghai Eighth People's Hospital (Shanghai, China). None of
the patients had received chemotherapy, immunotherapy, or
radiotherapy before specimen collection. All patients were well
informed and the process was approved by the Ethics Committee of
Eighth People's Hospital affiliated to Jiangsu University,
China.
Cell lines and cell culture
The human cervical cancer cell lines SiHa and CaSki
were purchased from the American Type Culture Collection
(Rockville, MD, USA). The cell lines were cultured in Dulbecco's
modified Eagle's medium (Sigma-Aldrich, St. Louis, MO, USA) with
10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA). The cells
were maintained at 37°C in a humidified atmosphere containing 5%
CO2.
Lentivirus production
The cDNA of TRIM28 and two short hairpin RNA
(shRNA) sequences targeting TRIM28 were separately cloned
into lentiviral expression vectors, as described previously
(11). The lentiviral vectors were
then transfected into 293T cells, along with the lentiviral
packaging mix. At 48 h post-transfection, lentiviral supernatants
were collected to infect the target cell lines, and a further 48 h
later, infected cells were selected with 8 mg/ml puromycin for a
continuous 2-week period to generate the stable transfected cell
line.
Immunohistochemical staining
Paraffin-embedded tissue blocks were deparaffinized,
rehydrated, and subjected to a heat-induced epitope retrieval step
in 0.01 M sodium citrate (pH 6.0). After blocking the endogenous
peroxidase with 3% hydrogen peroxide, the sections were washed with
Tris-buffered saline (TBS) and then incubated with a primary
antibody targeting TRIM28 (Proteintech, Hubei, China) for 1 h at
37°C. Subsequently, the sections were incubated with a horseradish
peroxidase-conjugated secondary antibody for 30 min, developed with
3,3′-diaminobenzidine, and counterstained with hematoxylin.
RNA extraction and quantitative
reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the cervical cancer
samples and cell lines, using TRIzol reagent (Invitrogen). cDNA
synthesis was performed using PrimeScript RT reagent kit (Takara,
Otsu, Japan). qRT-PCR of TRIM28 was conducted using the SYBR
Green PCR Master Mix kit (Takara) and ABI 7900HT Fast Real-Time PCR
system (Applied Biosystems, Foster City, CA, USA). β-actin was used
as the endogenous normalization control. Data were analyzed by the
2−∆∆Ct method. All samples were examined in
triplicate.
Cell viability analysis
To test cell viability, 2×103 cells were
cultured in 96-well plates for 24, 48, 72 and 96 h, and
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT) assays were performed according to the manufacturer's
protocol (Invitrogen). In brief, the cells were stained with 100 µl
MTT (0.5 mg/ml) for 4 h at 37°C, followed by removal of the culture
medium and addition of 150 µl dimethyl sulfoxide. The absorbance
was measured at 490 nm.
Colony formation assay
The cells were plated in a 6-well plate containing
500 cells per well in triplicate. After 14 days, the cells were
stained with 0.005% crystal violet (dissolved in methanol) for 1.5
h, and then counted and photographed.
Cell cycle analysis
Cells (1×106) were harvested and washed
with cold phosphate-buffered saline (PBS), followed by fixation
with 70% ethanol for 24 h at 4°C. After washing with PBS, the cells
were incubated with propidium iodide (Sigma-Aldrich) and DNase-free
RNase for 30 min at room temperature. The cell cycle was measured
using a FACSCalibur flow cyto-meter (BD Biosciences, San Jose, CA,
USA) with CellQuest software.
Bromodeoxyuridine (BrdU) incorporation
assay
The BrdU assay was performed as previously described
(22). In brief, cells grown on
coverslips (Fisher, Pittsburgh, PA, USA) were incubated with BrdU
for 1 h and stained with anti-BrdU antibody (Upstate, Temecula, CA,
USA) according to the manufacturer's instructions. Gray-level
images were acquired under a laser-scanning microscope (Axioskop 2
Plus, Carl Zeiss Co., Ltd., Jena, Germany).
Tumor xenograft experiment
Female BALB/c-nude mice (4–6 weeks old) were
purchased from Shanghai Laboratory Animal Center (Shanghai, China).
All experimental procedures were approved by the Institutional
Animal Care and Use Committee of Eighth People's Hospital
affiliated to Jiangsu University, China. SiHa cells
(2×106 cells/mouse) and CaSki cells (5×106
cells/mouse) were injected subcutaneously into the nude mice (n=5
per group). Tumor volume (V) was monitored every 4 days and
calculated using the formula: V = 0.5 × length × width2.
At the end of the experiment, the mice were sacrificed and tumor
weights were assessed. The proliferation index was determined by
Ki67 immunostaining and calculating the ratio of Ki67-positive
cells among the total number of cells in five randomly selected
fields.
Western blot analysis
The cells were lysed with RIPA buffer supplemented
with protease and phosphatase inhibitors (Roche Diagnostics,
Branchburg, NJ, USA). The protein concentration was measured by the
BCA Protein assay (Bio-Rad Laboratories, Hercules, CA, USA).
Proteins were separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred onto
polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA).
The membranes were blocked with 5% bovine serum albumin in TBS for
1 h at room temperature, and then incubated with the primary
antibodies against TRIM28 (15202–1-AP) and GAPDH (10494-1-AP) (both
from Proteintech) overnight at 4°C. The membranes were then washed
in TBS containing 1% Tween-20, incubated with the horseradish
peroxidase-conjugated secondary antibody for 1 h at room
temperature, and developed by ECL reagent (Pierce, Rockford, IL,
USA).
Statistical analysis
All experiments were performed at least in
triplicate and the data are presented as the means ± standard
deviations. All statistical analyses were carried out using the
SPSS 16.0 statistical software package (SPSS, Inc., Chicago, IL,
USA). The two-sided Student's t-test was performed to assess
differences between groups. P<0.05 was considered statistically
significant.
Results
TRIM28 expression is greatly increased
in cervical cancer tissues and cell lines
To determine the expression of TRIM28 in cervical
cancer, we first analyzed related data in the Oncomine database,
which revealed marked elevation in TRIM28 expression levels in
cervical cancer tissues compared with that in normal tissues
(Fig. 1A). We next tested the
relative expression levels of TRIM28 in 20 cervical cancer
samples and paired adjacent normal tissues by qRT-PCR. The results
indicated that the TRIM28 expression was significantly
upregulated in cervical cancer tissues compared with that in the
adjacent normal tissues (Fig. 1B).
Moreover, the protein level of TRIM28 was increased in the cervical
cancer samples, as shown by immunohistochemistry (Fig. 1C). Consistent with these
observations, both the mRNA and protein levels of TRIM28 were
differentially upregulated in the cervical cancer cell lines SiHa
and CaSki compared with that in the human normal cervical tissues
(Fig. 1D and E), suggesting that
TRIM28 is upregulated in cervical cancer tissues and cell
lines.
TRIM28 promotes proliferation of
cervical cancer cells by accelerating the cell cycle
To examine the biological role of TRIM28 in cervical
cancer, the cervical cancer cell lines SiHa and CaSki were
established to stably express TRIM28 or TRIM28 shRNA,
using a lentivirus-mediated overexpression or knockdown system,
respectively (Fig. 2A). MTT and
colony formation assays showed that ectopic expression of TRIM28
dramatically increased cell growth, whereas depletion of TRIM28
expression reduced the growth rate of cervical cancer cell lines
compared with that of control cells (Fig. 2B and C). Flow cytometry analysis
revealed that the TRIM28-overexpressing cells showed a significant
increase in the percentage of cells in the S peak and a decrease in
the percentage of cells in the G0-G1 peak compared with control
cells, whereas knockdown of TRIM28 had the opposite effect on CaSki
cells (Fig. 2D), indicating that
the effect of TRIM28 on cell proliferation might be due to
regulation of the G1-S phase transition. In addition, BrdU
incorporation was markedly increased by the ectopic expression of
TRIM28 in SiHa cells, and was suppressed by TRIM28 silencing in
CaSki cells (Fig. 2E). These
results further supported the notion that upregulation of TRIM28
regulates the G1-S phase transition of cervical cancer cells.
TRIM28 promotes tumor formation of
cervical cancer cells in vivo
We further evaluated the effect of TRIM28 on the
tumorigenic activity of cervical cancer cells. As shown in Fig. 3A and B, the TRIM28-overexpressing
tumors grew at a much higher rate, as determined by size and
weight, than the control tumors, whereas the tumors formed by
TRIM28-silenced cells were smaller and had lower tumor weights than
those formed from shRNA-vector control cells. Immunohistochemistry
analysis revealed that TRIM28-overexpressing tumors displayed a
higher Ki67 proliferation index, whereas the TRIM28-silenced tumors
showed reduced numbers of Ki67-positive cells (Fig. 3C). Protein expression levels of
TRIM28 in xenografts were further examined by western blotting.
TRIM28 was robustly upregulated in tumors formed by
TRIM28-overexpressing SiHa cells, but was downregulated in tumors
formed by TRIM28-silencing CaSki cells (Fig. 3D). Taken together, these results
suggest that TRIM28 promotes the tumorigenicity of cervical cancer
cells in vivo.
TRIM28 stimulates cervical cancer cell
proliferation via the mTOR signaling pathway
As mTOR is a central regulator of the cell cycle,
cell growth, and proliferation (23,24),
we investigated whether the TRIM28-induced cervical cancer cell
proliferation was mediated through the mTOR signaling pathway.
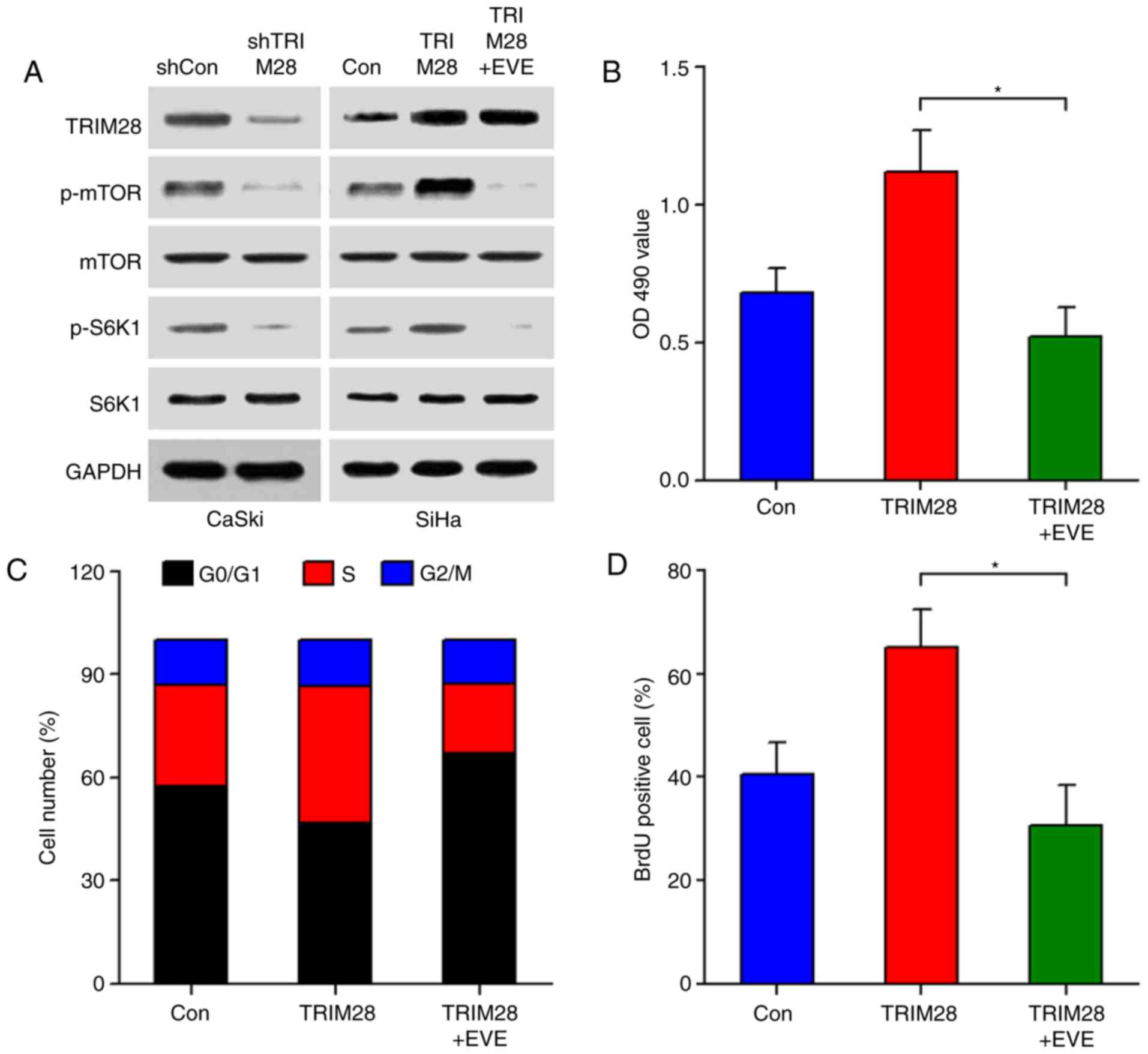
Western blot analyses showed that downregulation of TRIM28
significantly inhibited the phosphorylation of mTOR and its
downstream molecule S6K1, while upregulation of TRIM28 increased
the activity of mTOR/S6K1 signaling. However, modulation of TRIM28
did not alter the total protein levels of mTOR and S6K1 (Fig. 4A). TRIM28-modulated activation of
mTOR was also confirmed in tumor xenografts (Fig. 3D). We next examined the effect of
TRIM28 upregulation on the mTOR signaling pathway in the presence
of everolimus, a specific mTOR inhibitor. As shown in Fig. 4A, everolimus significantly abrogated
the TRIM28-induced phosphorylation of mTOR and S6K1. In line with
this, TRIM28-induced cell proliferation was also abolished by
everolimus, as determined by MTT, cell cycle, and BrdU
incorporation assays (Fig. 4B-D).
Collectively, our results indicate that TRIM28 promotes cervical
cancer cell proliferation through activating the mTOR signaling
pathway.
Discussion
Many members of the TRIM family have been shown to
play pivotal roles in tumorigenesis. TRIM28 is one of 60 members of
the TRIM family. To date, the biological functions and molecular
mechanisms of TRIM28 in cervical cancer remain unclear. In this
study, we found that the expression of TRIM28 is significantly
upregulated in cervical cancer tissues and cell lines compared with
that in their normal counterparts. Moreover, TRIM28 promoted
cervical cancer cell proliferation by activating the mTOR signaling
pathway. Thus, our findings may provide a novel target for the
therapeutic intervention of cervical cancer.
TRIM28, also known as Krüppel-associated box
(KRAB)-associated protein 1 (KAP1) or transcription intermediary
factor 1 (TIF1β), is a universal co-repressor for KRAB family zinc
finger proteins (KRAB-ZNF) (25).
TRIM28 has multifaceted roles in many organismal processes. Mouse
embryos deficient in TRIM28 develop normally to the blastocyst
stage and can implant but fail to gastrulate, indicating that
TRIM28 is essential for early post-implantation mouse development
(26). TRIM28 is also critical for
maintenance of the pluripotent state of embryonic stem cells
(27). Recently, TRIM28 was
reported to regulate autophagy, a stress-induced process that
degrades subcellular constituents (18). Some researchers have demonstrated
that TRIM28 is significantly upregulated in various types of
tumors, including breast, liver, brain, and ovarian cancers
(28,29), and contributes to tumor progression.
However, the underlying cellular functions of TRIM28 and the
related mechanisms involved in its carcinogenicity remain largely
unexplored. Lin et al (30)
identified TRIM28 as a potential marker of cervical cancer
metastasis; increase in TRIM28 expression enhanced the migration
and invasion of cervical cancer cells both in vitro and
in vivo. In the present study, we also detected the elevated
expression of TRIM28 in cervical cancer tissues relative to that in
matched normal tissues. Importantly, TRIM28 overexpression promoted
the proliferation, clone formation, and cell cycle progression of
cervical cancer cell lines, as well as the growth of xenograft
tumors in nude mice. Conversely, knockdown of TRIM28 reduced cell
proliferation and tumor growth. The present results clearly
indicate that TRIM28 is involved in cervical cancer cell
proliferation. However, further investigations are needed to
explore the underlying mechanisms involved in these TRIM28-mediated
effects.
The conserved serine/threonine kinase mTOR is a key
downstream effector of several signaling pathways involved in
cancer progression, including the phosphoinositide 3-kinase,
mitogen-activated protein kinase, and AMP-activated protein kinase
pathways (24,31). Abnormal activation of mTOR signaling
occurs in different types of tumors, including cervical cancer
(32). For example, through the
post-transcriptional regulation of mTOR expression, La-related
protein 1 (LARP1) contributes to cervical cancer progression and
adverse prognosis (33). Inhibition
of mTOR signaling with rapamycin decreased the
progranulin-stimulated protein synthesis, transformation, and
proliferation of cervical cells in vitro, as well as tumor
formation and growth in vivo, suggesting that the activated
mTOR pathway is critical for cervical cancer cell growth,
proliferation, and survival (34).
Therefore, we investigated whether mTOR signaling is involved in
the TRIM28-induced carcinogenesis of cervical cancer. We found that
upregulation of TRIM28 significantly increased the phosphorylation
of mTOR and its downstream molecule S6K1, while downregulation of
TRIM28 inhibited the activity of mTOR/S6K1 signaling.
TRIM28-modulated activation of mTOR was also confirmed in tumor
xenografts. Furthermore, we evaluated the effects of everolimus on
TRIM28-overexpressing cervical cancer cells. The results showed
that treatment with 20 nM everolimus significantly abrogated the
TRIM28-induced phosphorylation of mTOR/S6K1 and cervical cancer
cell proliferation, suggesting that mTOR may be the main downstream
effector of TRIM28 in promoting cervical cancer growth.
In conclusion, this study revealed that TRIM28 plays
an essential role in the progression of cervical cancer, and thus
might represent a new therapeutic target for cervical cancer.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Walboomers JM, Jacobs MV, Manos MM, Bosch
FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ and Muñoz N:
Human papillomavirus is a necessary cause of invasive cervical
cancer worldwide. J Pathol. 189:12–19. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Garland SM, Hernandez-Avila M, Wheeler CM,
Perez G, Harper DM, Leodolter S, Tang GW, Ferris DG, Steben M,
Bryan J, et al Females United to Unilaterally Reduce
Endo/Ectocervical Disease (FUTURE) I Investigators, : Quadrivalent
vaccine against human papillomavirus to prevent anogenital
diseases. N Engl J Med. 356:1928–1943. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ozato K, Shin DM, Chang TH and Morse HC
III: TRIM family proteins and their emerging roles in innate
immunity. Nat Rev Immunol. 8:849–860. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gallouet AS, Ferri F, Petit V, Parcelier
A, Lewandowski D, Gault N, Barroca V, Le Gras S, Soler E, Grosveld
F, et al: Macrophage production and activation are dependent on
TRIM33. Oncotarget. 8:5111–5122. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Song S, Ge Q, Wang J, Chen H, Tang S, Bi
J, Li X, Xie Q and Huang X: TRIM-9 functions in the UNC-6/UNC-40
pathway to regulate ventral guidance. J Genet Genomics. 38:1–11.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Raheja R, Liu Y, Hukkelhoven E, Yeh N and
Koff A: The ability of TRIM3 to induce growth arrest depends on
RING-dependent E3 ligase activity. Biochem J. 458:537–545. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schwamborn JC, Berezikov E and Knoblich
JA: The TRIM-NHL protein TRIM32 activates microRNAs and prevents
self-renewal in mouse neural progenitors. Cell. 136:913–925. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yin J, Kim TH, Park N, Shin D, Choi HI,
Cho S, Park JB and Kim JH: TRIM71 suppresses tumorigenesis via
modulation of Lin28B-let-7-HMGA2 signaling. Oncotarget.
7:79854–79868. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang Y, Yu Y, Yang Y, Yang M, Zhou L,
Huang X and Qin Q: Fish TRIM8 exerts antiviral roles through
regulation of the proinflammatory factors and interferon signaling.
Fish Shellfish Immunol. 54:435–444. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou Z, Ji Z, Wang Y, Li J, Cao H, Zhu HH
and Gao WQ: TRIM59 is up-regulated in gastric tumors, promoting
ubiquitination and degradation of p53. Gastroenterology.
147:1043–1054. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tyybakinoja A, Vilpo J and Knuutila S:
High-resolution oligonucleotide array-CGH pinpoints genes involved
in cryptic losses in chronic lymphocytic leukemia. Cytogenet Genome
Res. 118:8–12. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chung YL and Wu ML: Promyelocytic
leukaemia protein links DNA damage response and repair to hepatitis
B virus-related hepatocarcinogenesis. J Pathol. 230:377–387. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsai WW, Wang Z, Yiu TT, Akdemir KC, Xia
W, Winter S, Tsai CY, Shi X, Schwarzer D, Plunkett W, et al: TRIM24
links a non-canonical histone signature to breast cancer. Nature.
468:927–932. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang S, Kollipara RK, Humphries CG, Ma SH,
Hutchinson R, Li R, Siddiqui J, Tomlins SA, Raj GV and Kittler R:
The ubiquitin ligase TRIM25 targets ERG for degradation in prostate
cancer. Oncotarget. 7:64921–64931. 2016.PubMed/NCBI
|
|
16
|
Herquel B, Ouararhni K, Khetchoumian K,
Ignat M, Teletin M, Mark M, Béchade G, Van Dorsselaer A,
Sanglier-Cianférani S, Hamiche A, et al: Transcription cofactors
TRIM24, TRIM28, and TRIM33 associate to form regulatory complexes
that suppress murine hepatocellular carcinoma. Proc Natl Acad Sci
USA. 108:pp. 8212–8217. 2011; View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim WJ, Wittner BS, Amzallag A, Brannigan
BW, Ting DT, Ramaswamy S, Maheswaran S and Haber DA: The WTX tumor
suppressor interacts with the transcriptional corepressor TRIM28. J
Biol Chem. 290:14381–14390. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pineda CT and Potts PR: Oncogenic
MAGEA-TRIM28 ubiquitin ligase downregulates autophagy by
ubiquitinating and degrading AMPK in cancer. Autophagy. 11:844–846.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yokoe T, Toiyama Y, Okugawa Y, Tanaka K,
Ohi M, Inoue Y, Mohri Y, Miki C and Kusunoki M: KAP1 is associated
with peritoneal carcinomatosis in gastric cancer. Ann Surg Oncol.
17:821–828. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Addison JB, Koontz C, Fugett JH, Creighton
CJ, Chen D, Farrugia MK, Padon RR, Voronkova MA, McLaughlin SL,
Livengood RH, et al: KAP1 promotes proliferation and metastatic
progression of breast cancer cells. Cancer Res. 75:344–355. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen L, Chen DT, Kurtyka C, Rawal B, Fulp
WJ, Haura EB and Cress WD: Tripartite motif containing 28 (Trim28)
can regulate cell proliferation by bridging HDAC1/E2F interactions.
J Biol Chem. 287:40106–40118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin C, Wu Z, Lin X, Yu C, Shi T, Zeng Y,
Wang X, Li J and Song L: Knockdown of FLOT1 impairs cell
proliferation and tumorigenicity in breast cancer through
upregulation of FOXO3a. Clin Cancer Res. 17:3089–3099. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fingar DC, Richardson CJ, Tee AR, Cheatham
L, Tsou C and Blenis J: mTOR controls cell cycle progression
through its cell growth effectors S6K1 and 4E-BP1/eukaryotic
translation initiation factor 4E. Mol Cell Biol. 24:200–216. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pópulo H, Lopes JM and Soares P: The mTOR
signalling pathway in human cancer. Int J Mol Sci. 13:1886–1918.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Czerwińska P, Shah PK, Tomczak K, Klimczak
M, Mazurek S, Sozańska B, Biecek P, Korski K, Filas V, Mackiewicz
A, et al: TRIM28 multi-domain protein regulates cancer stem cell
population in breast tumor development. Oncotarget. 8:863–882.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cammas F, Mark M, Dollé P, Dierich A,
Chambon P and Losson R: Mice lacking the transcriptional
corepressor TIF1beta are defective in early postimplantation
development. Development. 127:2955–2963. 2000.PubMed/NCBI
|
|
27
|
Seki Y, Kurisaki A, Watanabe-Susaki K,
Nakajima Y, Nakanishi M, Arai Y, Shiota K, Sugino H and Asashima M:
TIF1beta regulates the pluripotency of embryonic stem cells in a
phosphorylation-dependent manner. Proc Natl Acad Sci USA. 107:pp.
10926–10931. 2010; View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wei C, Cheng J, Zhou B, Zhu L, Khan MA, He
T, Zhou S, He J, Lu X, Chen H, et al: Tripartite motif containing
28 (TRIM28) promotes breast cancer metastasis by stabilizing TWIST1
protein. Sci Rep. 6:298222016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Y, Jiang J, Li Q, Ma H, Xu Z and Gao
Y: KAP1 is overexpressed in hepatocellular carcinoma and its
clinical significance. Int J Clin Oncol. 21:927–933. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin LF, Li CF, Wang WJ, Yang WM, Wang DD,
Chang WC, Lee WH and Wang JM: Loss of ZBRK1 contributes to the
increase of KAP1 and promotes KAP1-mediated metastasis and invasion
in cervical cancer. PLoS One. 8:e730332013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pineda CT, Ramanathan S, Fon Tacer K, Weon
JL, Potts MB, Ou YH, White MA and Potts PR: Degradation of AMPK by
a cancer-specific ubiquitin ligase. Cell. 160:715–728. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Molinolo AA, Marsh C, El Dinali M, Gangane
N, Jennison K, Hewitt S, Patel V, Seiwert TY and Gutkind JS: mTOR
as a molecular target in HPV-associated oral and cervical squamous
carcinomas. Clin Cancer Res. 18:2558–2568. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mura M, Hopkins TG, Michael T, Abd-Latip
N, Weir J, Aboagye E, Mauri F, Jameson C, Sturge J, Gabra H, et al:
LARP1 post-transcriptionally regulates mTOR and contributes to
cancer progression. Oncogene. 34:5025–5036. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Feng T, Zheng L, Liu F, Xu X, Mao S, Wang
X, Liu J, Lu Y, Zhao W, Yu X, et al: Growth factor progranulin
promotes tumorigenesis of cervical cancer via PI3K/Akt/mTOR
signaling pathway. Oncotarget. 7:58381–58395. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Biewenga P, Buist MR, Moerland PD, Ver
Loren van Themaat E, van Kampen AH, ten Kate FJ and Baas F: Gene
expression in early stage cervical cancer. Gynecol Oncol.
108:520–526. 2008. View Article : Google Scholar : PubMed/NCBI
|