Introduction
Cholangiocarcinoma (CCA) is a highly malignant
disease with a poor prognosis, and comprises approximately 3% of
all gastrointestinal malignant tumors (1,2). The
etiology of this malignancy is mostly unknown, while the incidence
and mortality rate of the disease are increasing in many countries
(3). Due to limited diagnostic
methodologies, most patients are diagnosed at an advanced stage and
are ineligible for surgical resection. As a result, the 5-year
survival rate of CCA has remained at 10% for many years (2). Chemotherapy (cisplatin plus
gemcitabine) has been the exclusive therapy for a significant
percentage of CCA patients (4), but
chemoresistance attenuates the efficacy of conventional
chemotherapy, thus making it essential to identify novel curative
targets for the treatment of this disease.
Metformin, a biguanide, has been commonly prescribed
for decades as an anti-hyperglycemic agent in the treatment of type
II diabetes mellitus (5). Although
metformin has been extensively used as an anti-diabetic for 40
years, the first report indicating its antitumor effect in mammals
was in 2001 (6), and the first
study discussing the association between a reduced incidence of
cancer in patients with type II diabetes and the use of metformin
was published only about 10 years ago (7). Since then, more and more evidence has
shown that metformin has antitumor properties and can be used as a
chemosensitizer (8–11). However, the antitumor and
chemopreventive mechanisms of metformin have not yet been fully
elucidated.
Tumor cells preferentially use glycolysis for energy
production even in the presence of oxygen, which is the so-called
Warburg effect (12). This
metabolic alteration accumulates enough lactate and glycolytic
intermediates to support tumor growth and invasion. Therefore,
reversal of the Warburg effect is a potential therapeutic
methodology for the treatment of cancer (13,14).
Epigenetic changes, including histone modifications,
have been reported to play a crucial role in malignant disease
(15). Histone deacetylases (HDACs)
have emerged as new therapeutic targets in many cancers, as they
can remove acetyl groups from histone to decrease gene
transcription. HDACs can be divided into 4 classes: Class-I (HDAC1,
2, 3 and 8), Class-II (HDAC4, 5, 6, 7, 9 and 10), Class-III
(SIRT1-7) and Class-IV (HDAC11) (16). Recent studies suggest that Class-I
HDACs are upregulated in many malignancies, and that they inhibit
the expression of specific tumor-suppressor genes through
epigenetic modulation (17–19). Our previous studies demonstrated
that high levels of HDAC3 expression and activity play a critical
role in CCA, and that the inhibition of HDAC3 could induce
apoptosis in CCA cells (20).
Several HDAC inhibitors have been developed in
clinical trials for cancer treatment, and SAHA, as well as
romidepsin, have been approved by the US Food and Drug
Administration for the treatment of cutaneous T-cell lymphoma
(21). Novel Class I HDAC
inhibitors (4SC202, BG45 and SBHA) have shown efficacy in cancer
cells with near-marginal toxicity (15,16,22).
BG45, a selective HDAC3 inhibitor, has been demonstrated to be
effective in the treatment of leukemia (16). However, it remains unknown as to
whether BG45 can be used as a new treatment for CCA.
In the present study, we found that metformin could
reverse the Warburg effect by downregulating the protein levels of
LDHA, which was overexpressed in CCA; this could, in turn, make CCA
cells vulnerable. Therefore, combining metformin with BG45 markedly
inhibited the growth of cholangiocarcinoma via the induction of
cellular apoptosis. Our findings strongly suggest that metformin
combined with the HDAC3 inhibitor BG45 can be used as a therapeutic
strategy for the treatment of CCA.
Materials and methods
Ethics, consent and permissions
The protocol for the animal experiments was reviewed
and approved by the Ethics Committee of Medical Research, Nanjing
Drum Tower Hospital, Affiliated Hospital of Nanjing University
Medical School (Nanjing, China). Donors provided consent for any
use of human samples for research and the study protocol was
approved by the Ethics Committee of Medical Research, Nanjing Drum
Tower Hospital, Affiliated Hospital of Nanjing University Medical
School.
Bioinformatic analysis
The differentially expressed genes (DEGs) in CCA
were analyzed using data from The Cancer Genome Atlas (TCGA). CCA
RNA-Seq data were downloaded from the TCGA database using the GDC
Data Portal (https://gdc-portal.nci.nih.gov/), which consisted of 9
normal samples and 9 paired CCA samples. KEGG pathway enrichment
analysis was performed to detect the potential biological functions
and pathways of these genes in CCA. The heat map was drawn using
the gplots package in Bioconductor software (Bioconductor, org.,
version 3.6).
Immunohistochemistry
We purchased the tissue microarray slides from
Shanghai Outdo Biotech Co., Ltd. (Shanghai, China). Staining
intensity was graded as follows: absence of staining, 0; weak, 1;
moderate, 2 and strong, 3. The scoring approach in the assessment
of staining was as follows: 0 (no positive cells), 1 (<25%
positive cells), 2 (25–50% positive cells), 3 (>50-75% positive
cells), and 4 (>75% positive cells). The score for each tissue
was calculated by multiplication of these two grades, and the range
of this calculation was 0–12 (23).
Cell culture and reagents
HuCCT1 (JCRB, Osaka, Japan) and RBE (The Institute
of Biochemistry and Cell Biology, Shanghai Institutes for
Biological Sciences, Chinese Academy of Sciences, Shanghai, China)
cells were cultured in RPMI-1640 medium (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine
serum (Biological Industries, Cromwell, CT, USA), penicillin (100
U/ml; Invitrogen; Thermo Fisher Scientific) and streptomycin (100
U/ml; Invitrogen; Thermo Fisher Scientific, Inc.). Cells were all
maintained at 37°C with 5% CO2. Metformin was purchased
from Sangon Biotech Co., Ltd. (Shanghai, China) and BG45 was
purchased from MedChem Express (Monmouth Junction, NJ, USA).
Cell transfection
HuCCT1 and RBE cells were transfected using
Lipofectamine RNAiMax reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), following the manufacturer's protocol. HDAC3
siRNA was produced as described previously (20). Briefly, 50 pmol siRNA and 0.5 ml
Opti-MEM I Medium (Invitrogen; Thermo Fisher Scientific, Inc.) were
mixed, and then 5.5 µl RNAiMax reagent was added to each well of a
6-well plate and incubated for 15 min. Next, the mixed reagent was
added to a cell suspension containing 25×104 cells in
1.5 ml RPMI-1640 with 10% FBS.
Western blotting
Cells were lysed with ice-cold RIPA buffer (50 mM
Tris-HCl at pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium
deoxycholate, 0.1% SDS, 1% NP-40 and 1 mM EDTA), mixed with a
protease and phosphatase inhibitor cocktail (Roche Diagnostics
GmbH, Mannheim, Germany) and phenylmethylsulfonyl fluoride (PMSF)
(Biosharp, Hefei, China) for 15 min on ice. Extracted proteins were
supplemented with loading buffer containing 5% 2-mercaptoethanol
and then denatured at 100°C for 10 min. The protein samples were
separated by 8–12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred to polyvinylidene
fluoride (PVDF) membranes (EMD Millipore, Billerica, MA, USA). The
membranes were blocked in Tris-buffered saline containing 0.1%
Tween-20 with 5% non-fat milk for 2 h at room temperature.
Subsequently, membranes were incubated with specific primary
antibodies overnight at 4°C. All the primary antibodies were
diluted in Tris-buffered saline containing 0.1% Tween-20 with 5%
bovine serum albumin (BSA; Biosharp, Hefei, China). The dilution
ratio for β-actin and GAPDH was 1:5,000, and all the other
antibodies were in 1:1,000. Next, the membranes were treated with
the appropriate horseradish peroxidase (HRP)-conjugated secondary
antibodies (1:3,000 dilution). The blots were visualized with ECL
western blotting reagents (EMD Millipore), following the
manufacturer's instructions. HRP-conjugated anti-mouse IgG (7076;
Cell Signaling Technology, Inc.) and anti-rabbit antibodies (7074;
Cell Signaling Technology, Inc.) were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). The following antibodies were
used: HDAC3 (ab32369; Abcam, Cambridge, UK), cleaved caspase 3
(9664s; Cell Signaling Technology, Inc.), cleaved PARP (5625s; Cell
Signaling Technology, Inc.), PKM2 (4053s; Cell Signaling
Technology, Inc.), PDHA1 (ab92696; Abcam), LDHA (3582s; Cell
Signaling Technology, Inc.), GLUT1 (12939; Cell Signaling
Technology, Inc.), GAPDH (ab128915; Abcam) and β-actin (A5441;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany).
Apoptosis assay
Cellular apoptosis was detected with an Annexin
V-fluorescein isothiocyanate (FITC) apoptosis detection kit
(556547; BD Biosciences, Franklin Lakes, NJ, USA), following the
manufacturer's instructions. Briefly, HuCCT1 and RBE cells were
seeded in 6-well plates for 24 h. Then cells were cultured in
normal medium or treated with 10 mM metformin, 10 µM BG45, or the
combination of both drugs. Next, the control and treated cells were
collected and resuspended in 100 µl Annexin V binding buffer, and
incubated with 5 µl FITC-conjugated Annexin V and 5 µl propidium
iodide (PI) for 15 min in the dark. Annexin V binding buffer (400
µl) was then added to each tube. Samples were analyzed using a BD
FACSCanto II flow cytometer (BD Biosciences) with CellQuest (BD
Biosciences) version 3.3 software within 1 h.
Cell viability assay
Cell viability was measured using a Cell Counting
Kit-8 assay (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto,
Japan) in 96-well plates (2×103 cells/well). Cells were
seeded in 96-well plates. At 24 h, drugs were added and cells were
cultured for 24, 48 or 72 h at 37°C with 5% CO2. After
treatment, the medium was removed and 100 µl CCK-8 reagent was
added to each well, according to the manufacturer's instructions.
Then, the cells were incubated for 90 min at 37°C with 5%
CO2. The absorbance of the samples at 450 nm was
detected using a spectrophotometer (BioTek Instruments, Inc.,
Winooski, VT, USA). Relative cell viability (%) = (absorbance at
450 nm of the treated group-absorbance at 450 nm of the
blank)/(absorbance at 450 nm of the control group-absorbance at 450
nm of the blank) × 100%.
Lactate production
HuCCT1 and RBE cells were seeded in 6-well plates at
2.5×105 cells/well. At confluence, the cells were
treated with or without 10 mM metformin for 24 h. Then the media
and cells were collected separately for lactate production
detection. The lactate production was detected with a lactate assay
kit (K627; BioVision, Inc., Milpitas, CA, USA), according to the
manufacturer's instructions. The absorbance values were measured at
450 nm with a spectrophotometer (BioTek Instruments, Inc.). The
values were normalized to the cell number.
Mitochondrial oxidative
phosphorylation analysis
The oxygen consumption rate (OCR) and extracellular
acidification rate (ECAR) were detected in real time with an XF96
Extracellular Flux Analyzer from Seahorse Bioscience, Inc. (North
Billerica, MA, USA), following the manufacturer's instructions.
HuCCT1 and RBE cells were plated in 96-well XF cell culture
microplates at 1×104 cells/well and incubated for 24 h
at 37°C. Then, 1 mM metformin was added into each well of the
plates. Before measurement, the medium was replaced with 175
µl/well XF-96 running medium (supplemented with RPMI-1640 without
serum) and pre-incubated at 37°C for 20 min in the absence of
CO2. For each analysis, different compounds that
modulate mitochondrial respiration were injected into each well,
according to standard protocols: for OCR, oligomycin,
carbonylcyanide p-trifluoromethoxy-phenylhydrazone, rotenone and
antimycin A; for ECAR, glucose, oligomycin and 2-deoxy-D-glucose.
The cell number was used for data normalization. OCR is expressed
as pmol/min (picomoles/minute). ECAR is expressed as mpH/min
[milli-pH units (mpH) per minute].
CCA cancer xenograft model
Nude mice were purchased from the Department of
Laboratory Animal Science, Nanjing Drum Tower Hospital. HuCCT1
cells (3×106) in serum-free RPMI-1640 medium were
subcutaneously injected into the right flank of the mice. Once
palpable tumors were exhibited, mice were randomly assigned into 4
groups: control (100 µl natural saline, NS), metformin (200 mg/kg
diluted in 100 µl NS), BG45 (20 mg/kg diluted in 100 µl NS), and a
combination of both drugs (metformin at 200 mg/kg and BG45 at 20
mg/kg in 100 µl NS). Each group received an intraperitoneal
injection 3 times/week for 4 weeks. Tumor volume (V) was calculated
using the following formula: V = length × width2/2. All
the experiments involving animals were reviewed and approved by the
Animal Welfare Committee of Nanjing Drum Tower Hospital.
Statistical analysis
Data are expressed as the mean ± standard error of
the mean (SEM). A one-way analysis of variance (ANOVA) was
performed with Dunnett's multiple comparisons test (SPSS 17.0;
SPSS, Inc., Chicago, IL, USA). The Chi-squared test and unpaired
Student's t-test were performed for comparisons between two groups.
Overall survival time was calculated by the Kaplan-Meier analysis.
Staining scores were analyzed by the log-rank test. P<0.05 was
considered to indicate a statistically significant difference. In
figures, *P<0.05 and **P<0.01.
Results
Metabolic abnormalities are important
features of CCA
Reprogramming of cellular metabolism is common in
tumor cells, and is regulated by the expression of multiple genes,
thus accelerating the malignant behavior of tumor cells (24). Therefore, we analyzed the
differentially expressed genes (DEGs) in CCA using data from The
Cancer Genome Atlas (TCGA). We confirmed the DEGs and detected the
potential biological functions and pathways of these genes in CCA
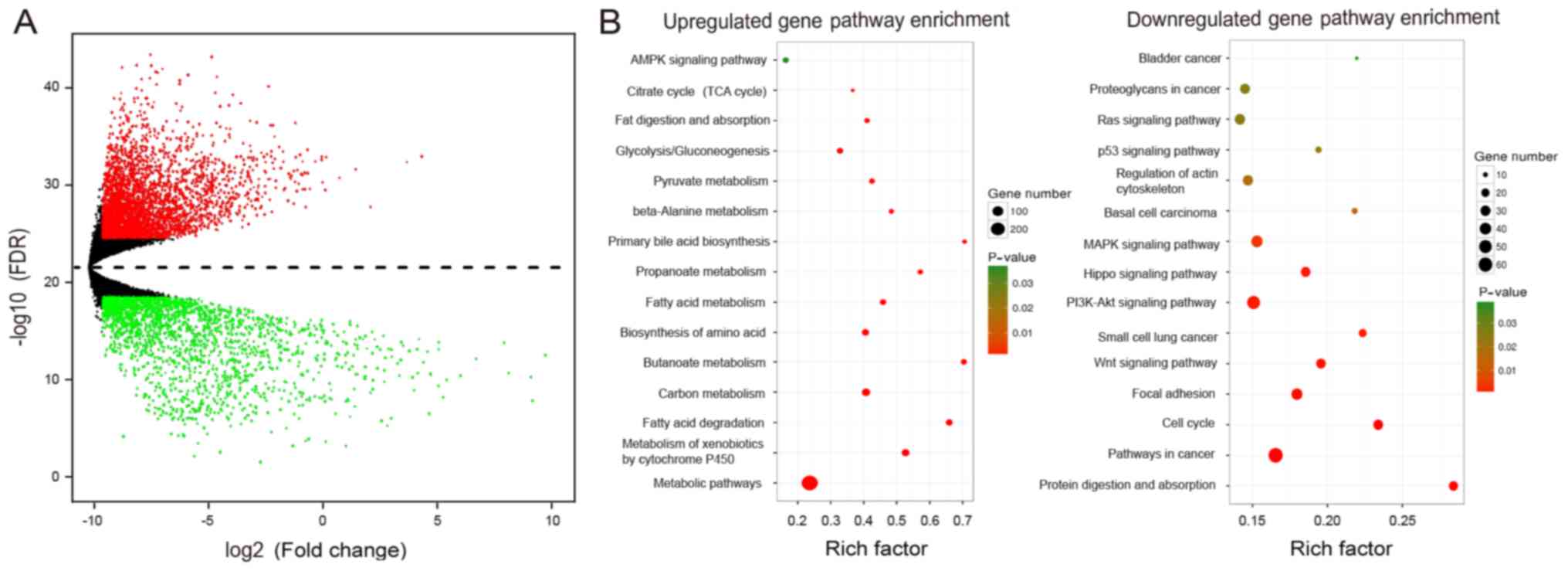
through KEGG pathway analysis (Fig.
1A). We observed that the most relevant pathways in CCA were
metabolic and tumor growth pathways (Fig. 1B). Together, these results indicate
that metabolic reprogramming is an important feature of CCA;
moreover, these results underscore the complex and unclear
regulatory mechanisms.
Metformin suppresses the Warburg
effect in CCA cells by decreasing LDHA
Metformin is a widely accepted first-line drug for
the treatment of type 2 diabetes (5). Many studies have shown that metformin
could serve as a potential cancer therapy, although little is known
concerning the mechanism of its antitumor functionality (25,26).
It has been reported that the antitumor effects of metformin are
partially caused by altering cellular metabolism (14). We then investigated the effects of
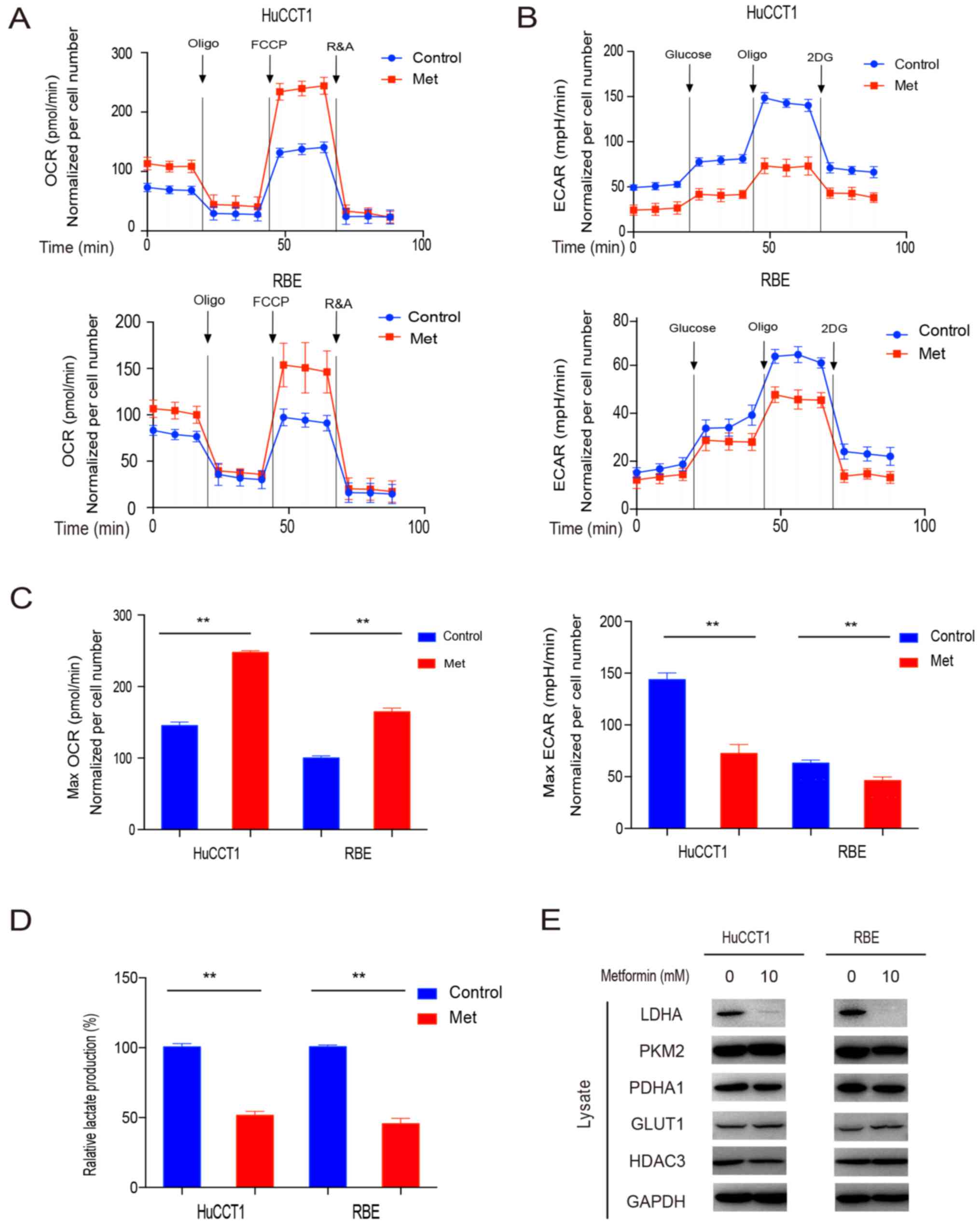
metformin on the cellular metabolic status. After employing the
Seahorse bio-energy analyzer, we found that OCR was notably
increased, while ECAR was markedly decreased after the use of
metformin (Fig. 2A-C). This
indicated the metabolic shift from glycolysis to oxidative
phosphorylation in CCA cells following treatment with
metformin.
To further validate the anti-Warburg effect
properties of metformin, we measured the level of lactate
production after metformin. We found a significant decrease in the
lactate production of both CCA cell lines after the treatment,
which confirmed the effect of metformin on reversing the Warburg
effect (Fig. 2D).
To clarify the mechanisms underlying the fact that
metformin suppressed the Warburg effect, we detected a number of
candidate proteins that may be involved in the Warburg effect. We
found that the expression of LDHA was notably decreased following
the use of metformin, although this result was not observed with
the other possible proteins PKM2, PDHA1, GLUT1 and HDAC3 (Fig. 2E). Collectively, these data
suggested that metformin reversed the Warburg effect by decreasing
LDHA in CCA cells.
Metformin marginally affects the
growth of CCA cells
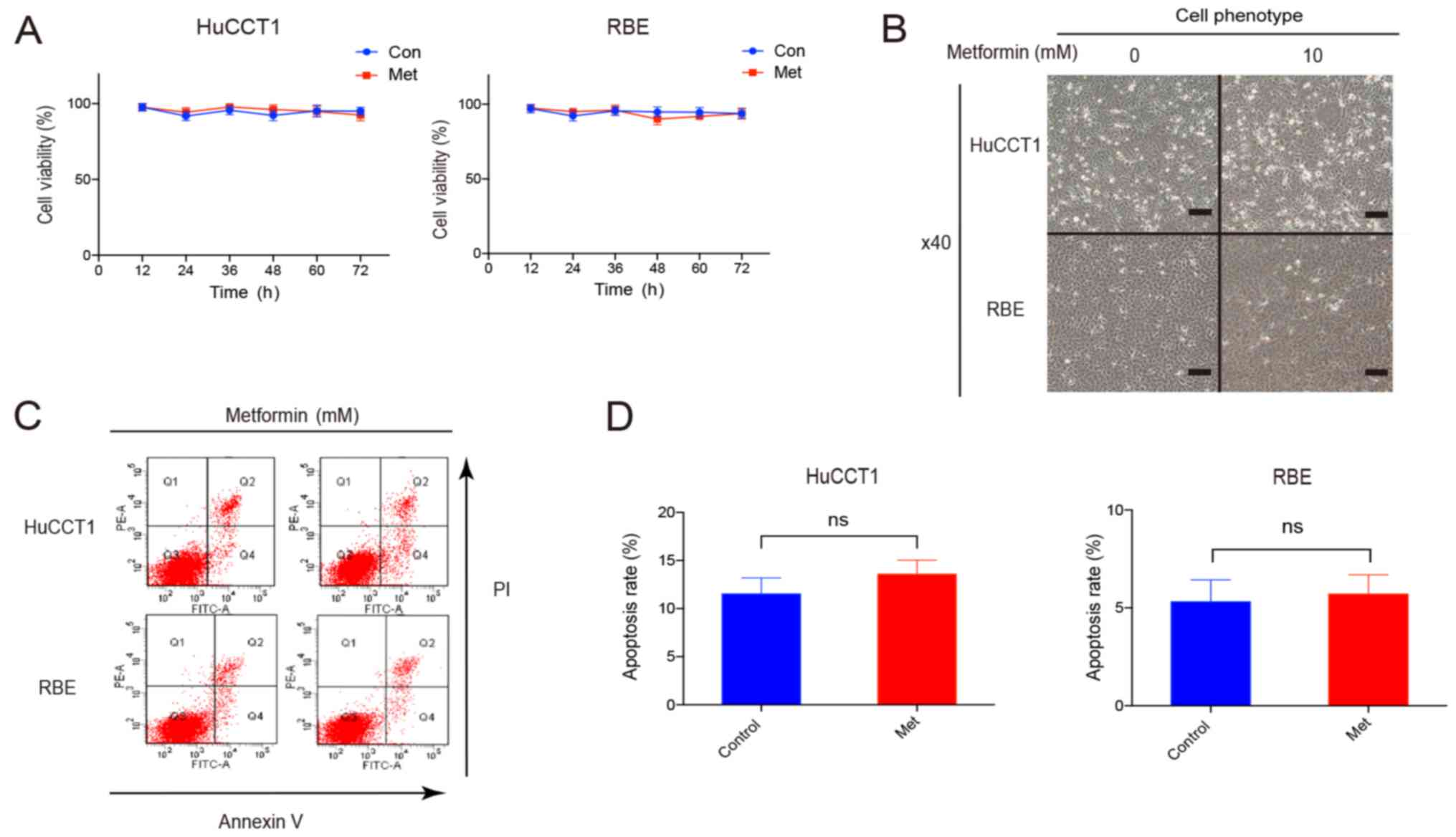
To explore the function of metformin in CCA, we
detected the proliferation and apoptosis of CCA cells. Notably, we
observed that a low concentration of metformin did not inhibit cell
proliferation or induce cellular apoptosis, although the Warburg
effect was suppressed (Fig. 3A-D).
This suggested that metformin alone may only slightly affect the
proliferation or apoptosis of CCA cells, although it suppresses the
Warburg effect.
Metformin facilitates BG45-induced
apoptosis
Several studies have reported that metabolic
abnormalities can accelerate malignant behaviors, increase
chemoresistance and inhibit tumor cell apoptosis (12,27).
As we mentioned above, the present study demonstrated that
metformin could regulate the energy utilization of CCA by
decreasing the expression of LDHA. It is possible that reversing
the Warburg effect in CCA cells with metformin could then increase
tumor cell fragility and render them more susceptible to
chemotherapy (14,28). Recently, we found that HDAC3
inhibitors are promising chemotherapeutic agents in the treatment
of CCA (20). BG45, a novel HDAC3
selective inhibitor, has been validated as a therapeutic agent in
multiple myeloma (16). However,
our data showed that in CCA, the antitumor properties of BG45 were
not significantly effective (Fig.
4A). As a result, we tested the combination treatment of
metformin and BG45, and found that this combination inhibited cell
viability (Fig. 4A and B). We
further explored the mechanism of this combination-induced cell
viability inhibition by performing flow cytometry. The results
revealed that combined metformin and BG45 led to a significant
increase in the apoptosis of CCA cells, compared with single drug
use alone (Fig. 4C and D).
Consistent with the flow cytometry results, higher levels of
cleaved caspase 3 and cleaved PARP were found in CCA cells treated
with the combined treatment, compared to single drug treatment
(Fig. 4E). We further inhibited
HDAC3 using siRNA (Fig. 4F), and
found that the expression levels of cleaved caspase 3 and cleaved
PARP were significantly increased. However, the levels of these two
apoptotic markers were not promoted following treatment with the
combination of both drugs after HDAC3 inhibition (Fig. 4G).
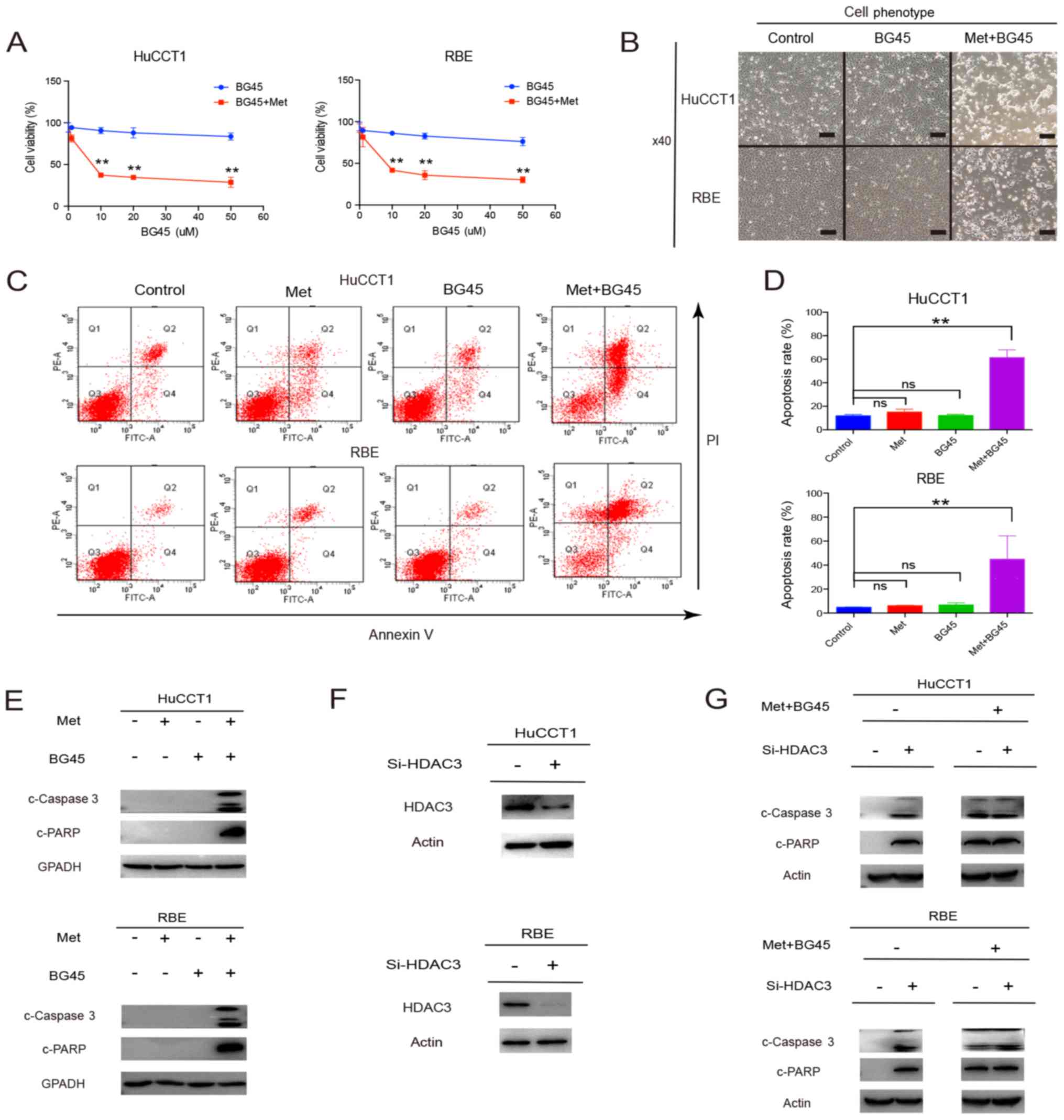 | Figure 4.Metformin facilitates BG45-induced
apoptosis. (A) Cholangiocarcinoma (CCA) cells were treated with
BG45 0–60 µM alone or a combination of BG45 0–60 µM and metformin
10 mM for 24 h, then cell proliferation was quantified via CCK-8
assay. (B) CCA cells were treated with BG45 10 µM or a combination
of BG45 10 µM and metformin 10 mM for 24 h, and cellular phenotype
changes were observed. Scale bars, 100 µm. (C and D) After
treatment with metformin 10 mM, BG45 10 µM, or a combination of
both, CCA cells were analyzed using Annexin V/PI staining, and then
apoptosis was measured by flow cytometry (left). The percentages of
early and late apoptotic cells were quantified (right). (E) Cells
were treated with 10 mM metformin, 10 µM BG45, or the combination
of both drugs for 48 h, and then lysates were collected and
apoptosis-associated markers were examined via western blot
analysis. (F) HDAC3 was inhibited with siRNA, and then protein
samples were collected and HDAC3 was detected via western blotting.
(G) HDAC3 was inhibited with siRNA, cells were treated with 10 mM
metformin and 10 µM BG45, protein samples were collected, and
apoptotic markers were detected via western blotting. ns, not
significant; **P<0.01. |
Collectively, these results demonstrated that
combined treatment using metformin and BG45 could significantly
inhibit the proliferation of CCA cells by inducing apoptosis.
Although metformin alone hardly induced cellular apoptosis at low
concentrations, it did nonetheless facilitate HDAC3 inhibitor
BG45-induced apoptosis.
Effects of metformin and BG45 on tumor
xenografts
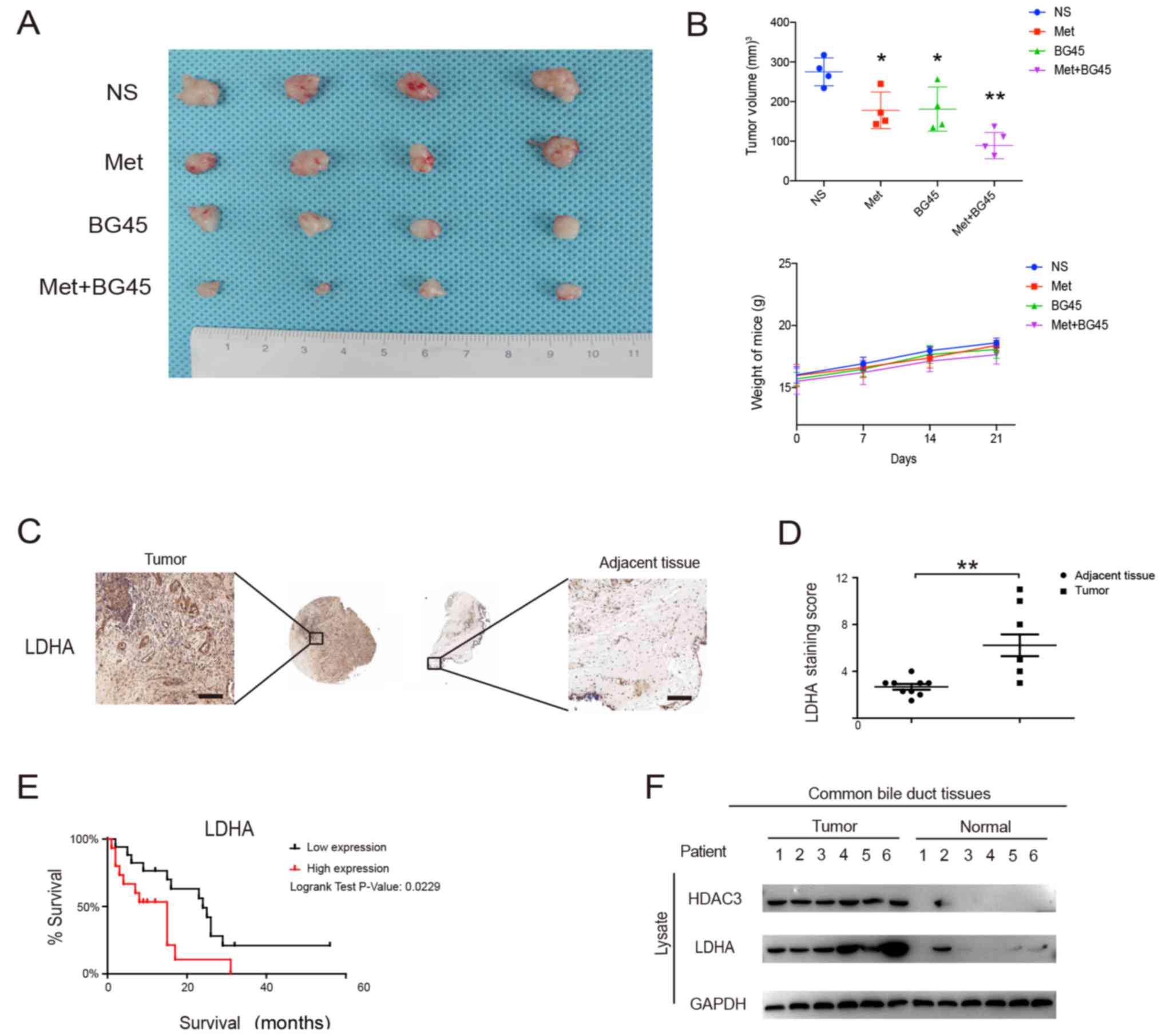
To further validate our findings in vitro, we
evaluated the antitumor effect of the combined treatment in
vivo using a CCA cell tumor xenograft model. We observed that
the combined treatment group significantly inhibited tumor growth
compared to the monotherapy groups (Fig. 5A and B). The weight loss of the mice
was not found to be significant, which indicated that the
combination therapy was safe in vivo (Fig. 5B). Altogether, these data revealed
that the combination of metformin and BG45 could significantly
induce cellular apoptosis and inhibit proliferation in
vivo.
LDHA expression is upregulated in CCA
tissues and indicates poor prognosis
By evaluating the expression of LDHA on the tissue
microarrays from Shanghai Outdo Biotech Co., Ltd., we found that
LDHA was significantly upregulated in tumor tissues compared to
that noted in adjacent tissues (Fig. 5C
and D). Furthermore, we evaluated the clinical data of the
tissue microarrays and found that LDHA protein was overexpressed in
68/127 cases (54.5%), and was associated with tumor size (Table I). Employing the 33 follow-up cases,
we found that high LDHA protein expression in CCA reduced patient
survival (P<0.001, log-rank test) (Fig. 5E). Next, we assessed the expression
of HDAC3 and LDHA in fresh tissues, and found that HDAC3 and LDHA
were markedly upregulated in tumor tissues compared to levels noted
in normal tissues (Fig. 5F).
Collectively, our data suggest that LDHA is overexpressed in CCA
tissues and is associated with a worse prognosis.
 | Table I.Clinical characteristics and
metabolic protein levels in patients with cholangiocarcinoma. |
Table I.
Clinical characteristics and
metabolic protein levels in patients with cholangiocarcinoma.
|
|
| LDHA
expression |
|
|---|
|
|
|
|
|
|---|
|
| N | Low, n (%) | High, n (%) | P-value |
|---|
| Sex |
|
|
| 0.499 |
|
Male | 80 | 39 (49) | 41 (51) |
|
|
Female | 47 | 20 (43) | 27 (57) |
|
| Age (years) |
|
|
| 0.393 |
|
≤70 | 99 | 44 (44) | 55 (56) |
|
|
>70 | 28 | 15 (54) | 13 (46) |
|
| Size (mm) |
|
|
| 0.012a |
| ≤7 | 71 | 40 (56) | 31 (44) |
|
|
>7 | 56 | 19 (34) | 37 (66) |
|
|
Differentiation |
|
|
| 0.186 |
|
Well | 107 | 47 (44) | 60 (56) |
|
|
Poor | 20 | 12 (60) | 8 (40) |
|
| T stage |
|
|
| 0.052 |
|
T1-T2 | 113 | 56 (50) | 57 (50) |
|
| T3 | 14 | 3 (21) | 11 (79) |
|
| Lymph node
metastasis |
|
|
| 0.535 |
|
Negative | 102 | 46 (45) | 56 (55) |
|
|
Positive | 25 | 13 (52) | 12 (48) |
|
| Venous
invasion |
|
|
| 0.542 |
|
Negative | 114 | 54 (47) | 60 (53) |
|
|
Positive | 13 | 5 (38) | 8 (62) |
|
| Nerve invasion |
|
|
| 0.982 |
|
Negative | 114 | 53 (46) | 61 (54) |
|
|
Positive | 13 | 6 (46) | 7 (54) |
|
Discussion
CCA is a highly lethal disease, with an increasing
incidence and mortality rate worldwide (1). Due to the lack of effective diagnostic
methods, most patients with CCA are diagnosed at an advanced stage,
and are thus ineligible for surgical resection (1). Although these patients can and do
receive palliative chemotherapy (cisplatin and gemcitabine), the
efficacy is limited due to drug resistance (2,4).
Therefore, there remains an urgent need to develop new potential
treatments for this malignancy.
In the present study, we showed that metformin could
suppress the Warburg effect in CCA, which decreases aerobic
glycolysis and promotes oxidative phosphorylation, thus making CCA
cells vulnerable to chemotherapy. Moreover, we found that LDHA is
more susceptible to metformin than other indicated proteins.
Furthermore, we demonstrated that the combination of metformin and
the HDAC3 inhibitor BG45 can be used as a novel curative
therapeutic strategy in the treatment of CCA.
Through bioinformatic analysis, we found that
metabolic and tumor proliferation pathways were most relevant in
CCA. The Warburg effect, also known as aerobic glycolysis, refers
to the phenomenon whereby cancer cells display a high level of
glucose uptake and metabolism by glycolysis, even in the presence
of normal oxygen levels (12). This
leads to metabolic abnormalities in cancer cells and thereby
promotes malignant behaviors, increases chemoresistance, and
inhibits tumor cell apoptosis.
Metformin is a widely adopted therapy for type 2
diabetes. Recent studies have confirmed its antitumor properties,
but the mechanisms require further elaboration (25,26).
We found that low concentrations of metformin could change the
metabolic status of tumor cells and reverse the Warburg effect
through the inhibition of LDHA, which was overexpressed in CCA
tissues and indicated a shorter survival time.
Notably, we found that metformin alone could hardly
induce cellular apoptosis in CCA cells. It is widely accepted that
multiple key pathways, as well as genes, converge to change
cellular metabolism in order to support tumor growth and
development (29). Therefore, it is
difficult to induce cellular apoptosis under single-target
inhibition.
Since metabolic reprogramming in tumor cells could
possibly endow the cells with resistance to chemotherapies
(30,31), we thought that low concentrations of
metformin could alter the metabolic abnormalities of tumor cells,
causing the cells to become fragile and sensitive to chemotherapy.
Our previous data showed that HDAC3 is a potential chemotherapeutic
target (20). However, the present
study found that the new selective HDAC3 inhibitor, BG45, could
hardly induce CCA cellular apoptosis. As a result, we utilized a
combination of low-concentration metformin and BG45, and found that
cell viability, as well as proliferation, was inhibited, and that
the apoptosis rate was increased dramatically. These results were
validated both in vitro and in vivo. In summary, we
revealed that in CCA, metformin could be adopted as a chemotherapy
sensitizer, which could enhance the antitumor properties of HDAC3
inhibitors.
Our findings suggest that metformin could reverse
the Warburg effect through inhibition of LDHA. This sensitizes
cells to the antitumor effects of HDAC3 inhibitors and induces
cellular apoptosis both in vitro and in vivo. Our
study provides support for the use of metformin with BG45 as a
novel therapeutic strategy in CCA treatment.
Acknowledgements
The authors would like to thank the Zhao laboratory
for offering their help.
Funding
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 81672935, 81472756
and 81602076), the Jiangsu Clinical Medical Center of Digestive
Disease (grant no. BL2012001), the Natural Science Foundation from
the Department of Science & Technology of Jiangsu Province
(grant no. BK20160113), the Fundamental Research Funds for the
Central Universities (grant no. 021414380244), and the Foundation
of Jiangsu Provincial Commission of Health and Family Planning
(grant no. Q201611).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
MZ, LW, XZ designed the study; DT, LX, QZ, YL and YP
did the cell experiments; YL, YY, YW, LZ and DT collected the
tissue samples; QZ, DT and YL performed the protein analysis; LZ,
DT, LX, MZ, RGD drafted the manuscript and performed the
immunohistochemistry experiment; RGD did the language editing; MZ,
LW and XZ supported the study. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The protocol for the animal experiments was reviewed
and approved by the Ethics Committee of Medical Research, Nanjing
Drum Tower Hospital, Affiliated Hospital of Nanjing University
Medical School (Nanjing, China). Donors provided consent for any
use of human samples for research and the study protocol was
approved by the Ethics Committee of Medical Research, Nanjing Drum
Tower Hospital, Affiliated Hospital of Nanjing University Medical
School.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Razumilava N and Gores GJ:
Cholangiocarcinoma. Lancet. 383:2168–2179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rizvi S and Gores GJ: Pathogenesis,
diagnosis, and management of cholangiocarcinoma. Gastroenterology.
145:1215–1229. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bridgewater J, Galle PR, Khan SA, Llovet
JM, Park JW, Patel T, Pawlik TM and Gores GJ: Guidelines for the
diagnosis and management of intrahepatic cholangiocarcinoma. J
Hepatol. 60:1268–1289. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Valle JW, Lamarca A, Goyal L, Barriuso J
and Zhu AX: New horizons for precision medicine in biliary tract
cancers. Cancer Discov. 7:943–962. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Inzucchi SE, Lipska KJ, Mayo H, Bailey CJ
and McGuire DK: Metformin in patients with type 2 diabetes and
kidney disease: A systematic review. Jama. 312:2668–2675. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schneider MB, Matsuzaki H, Haorah J,
Ulrich A, Standop J, Ding XZ, Adrian TE and Pour PM: Prevention of
pancreatic cancer induction in hamsters by metformin.
Gastroenterology. 120:1263–1270. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Camacho L, Dasgupta A and Jiralerspong S:
Metformin in breast cancer-an evolving mystery. Breast Cancer Res.
17:882015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ezewuiro O, Grushko TA, Kocherginsky M,
Habis M, Hurteau JA, Mills KA, Hunn J, Olopade OI, Fleming GF and
Romero IL: Association of Metformin Use with outcomes in advanced
endometrial cancer treated with chemotherapy. PLoS One.
11:e01471452016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Griss T, Vincent EE, Egnatchik R, Chen J,
Ma EH, Faubert B, Viollet B, DeBerardinis RJ and Jones RG:
Metformin antagonizes cancer cell proliferation by suppressing
mitochondrial-dependent biosynthesis. PLoS Biol. 13:e10023092015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ling S, Xie H, Yang F, Shan Q, Dai H, Zhuo
J, Wei X, Song P, Zhou L, Xu X and Zheng S: Metformin potentiates
the effect of arsenic trioxide suppressing intrahepatic
cholangiocarcinoma: Roles of p38 MAPK, ERK3, and mTORC1. J Hematol
Oncol. 10:592017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Green DR, Galluzzi L and Kroemer G: Cell
biology. Metabolic control of cell death. Science. 345:12502562014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu X, Romero IL, Litchfield LM, Lengyel E
and Locasale JW: Metformin targets central carbon metabolism and
reveals mitochondrial requirements in human cancers. Cell Metab.
24:728–739. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhijun H, Shusheng W, Han M, Jianping L,
Li-sen Q and Dechun L: Pre-clinical characterization of 4SC-202, a
novel class I HDAC inhibitor, against colorectal cancer cells.
Tumor Biol. 37:10257–10267. 2016. View Article : Google Scholar
|
|
16
|
Minami J, Suzuki R, Mazitschek R, Gorgun
G, Ghosh B, Cirstea D, Hu Y, Mimura N, Ohguchi H, Cottini F, et al:
Histone deacetylase 3 as a novel therapeutic target in multiple
myeloma. Leukemia. 28:680–689. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lakshmaiah KC, Jacob LA, Aparna S,
Lokanatha D and Saldanha SC: Epigenetic therapy of cancer with
histone deacetylase inhibitors. J Cancer Res Ther. 10:469–478.
2014.PubMed/NCBI
|
|
18
|
Harting K and Knöll B: SIRT2-mediated
protein deacetylation: An emerging key regulator in brain
physiology and pathology. Eur J Cell Biol. 89:262–269. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang LT, Liou JP, Li YH, Liu YM, Pan SL
and Teng CM: A novel class I HDAC inhibitor, MPT0G030, induces cell
apoptosis and differentiation in human colorectal cancer cells via
HDAC1/PKCδ and E-cadherin. Oncotarget. 5:5651–5662. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yin Y, Zhang M, Dorfman RG, Li Y, Zhao Z,
Pan Y, Zhou Q, Huang S, Zhao S, Yao Y and Zou X: Histone
deacetylase 3 overexpression in human cholangiocarcinoma and
promotion of cell growth via apoptosis inhibition. Cell Death Dis.
8:e28562017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang QL, Wang L, Zhang YW, Jiang XX, Yang
F, Wu WL, Janin A, Chen Z, Shen ZX, Chen SJ and Zhao WL: The
proteasome inhibitor bortezomib interacts synergistically with the
histone deacetylase inhibitor suberoylanilide hydroxamic acid to
induce T-leukemia/lymphoma cells apoptosis. Leukemia. 23:1507–1514.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Richon VM, Emiliani S, Verdin E, Webb Y,
Breslow R, Rifkind RA and Marks PA: A class of hybrid polar
inducers of transformed cell differentiation inhibits histone
deacetylases. Proc Natl Acad Sci USA. 95:pp. 3003–3007. 1998;
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xiao Y, Wang J, Qin Y, Xuan Y, Jia Y, Hu
W, Yu W, Dai M, Li Z, Yi C, et al: Ku80 cooperates with CBP to
promote COX-2 expression and tumor growth. Oncotarget. 6:8046–8061.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shibata T and Aburatani H: Exploration of
liver cancer genomes. Nat Rev Gastroenterol Hepatol. 11:340–349.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Alimova IN, Liu B, Fan Z, Edgerton SM,
Dillon T, Lind SE and Thor AD: Metformin inhibits breast cancer
cell growth, colony formation and induces cell cycle arrest in
vitro. Cell Cycle. 8:909–915. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ben Sahra I, Laurent K, Loubat A,
Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le
Marchand-Brustel Y and Bost F: The antidiabetic drug metformin
exerts an antitumoral effect in vitro and in vivo through a
decrease of cyclin D1 level. Oncogene. 27:3576–3586. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou Y, Tozzi F, Chen J, Fan F, Xia L,
Wang J, Gao G, Zhang A, Xia X, Brasher H, et al: Intracellular ATP
levels are a pivotal determinant of chemoresistance in colon cancer
cells. Cancer Res. 72:304–314. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Iliopoulos D, Hirsch HA and Struhl K:
Metformin decreases the dose of chemotherapy for prolonging tumor
remission in mouse xenografts involving multiple cancer cell types.
Cancer Res. 71:3196–3201. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vander Heiden MG: Targeting cancer
metabolism: A therapeutic window opens. Nat Rev Drug Discov.
10:671–684. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shukla SK, Purohit V, Mehla K, Gunda V,
Chaika NV, Vernucci E, King RJ, Abrego J, Goode GD, Dasgupta A, et
al: MUC1 and HIF-1alpha signaling crosstalk induces anabolic
glucose metabolism to impart gemcitabine resistance to pancreatic
cancer. Cancer Cell. 32:71–87.e7. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Herranz D, Ambesi-Impiombato A, Sudderth
J, Sánchez-Martín M, Belver L, Tosello V, Xu L, Wendorff AA,
Castillo M, Haydu JE, et al: Metabolic reprogramming induces
resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic
leukemia. Nat Med. 21:1182–1189. 2015. View Article : Google Scholar : PubMed/NCBI
|