Introduction
Colorectal cancer (CRC) remains a critical health
concern worldwide, with an estimated 1.4 million cases occurring in
2012 (1). It has become the fourth
most commonly diagnosed cancer and is the fifth leading cause of
cancer-related death in China (2).
Although much progress has been made, the biological mechanism and
potential biomarkers of CRC remain to be clarified. Therefore, more
studies on the molecular mechanism of CRC are required to
facilitate the early diagnosis and treatment of CRC.
Ultraconserved regions (UCRs) are 481 DNA segments
of more than 200 bp in length that are absolutely conserved (100%)
in the human, rat and mouse genomes, most of which are also 95–99%
conserved in chickens and dogs. They are always classified into
three types, namely, exonic, non-exonic, and possibly exonic
(3). Most UCRs can be transcribed
and their transcriptional products, called T-UCRs, represent an
important category of long non-coding RNAs (lncRNAs) (4). T-UCRs have been reported to be
involved in various cancer processes (5–9). As a
member of the T-UCR family, uc.338 has been reported to promote
tumorigenesis in hepatocellular carcinoma (9–11),
cervical cancer (12) and lung
carcinoma (13). In CRC, uc.338 was
reported to promote the invasion and metastasis of HCT116 and SW480
cells (14). However, the exact
role of uc.338 in CRC proliferation remains unknown.
Therefore, in the present study, we detected the
role and potential mechanism of uc.338 in CRC proliferation. Our
results indicated that uc.338 upregulation is vital for the
progression of CRC, highlighting the potential of uc.338 as a
therapeutic target in CRC.
Materials and methods
Human tissues
Our study was approved by the Ethics Committee of
the First Affiliated Hospital of Nanjing Medical University. From
July 2010 to December 2012, we collected 100 pairs of human CRC
tissues and adjacent normal tissues from patients after they had
signed an informed consent form. Of the patients, 64 were males and
36 were females. Ages of patients at the time of surgery ranged
from 27 to 82 years (60.14±11.72 years). The tumor tissues and
adjacent normal tissues were collected in liquid nitrogen within 5
min, and then immediately transferred to a −70°C freezer for
long-term storage. The tumor-node-metastasis (TNM) stage was
classified according to the National Comprehensive Cancer Network
(NCCN). In this study, none of the patients received any
preoperative treatments.
Cell lines and cell culture
CRC cell lines LoVo, HCT116, DLD-1, and human colon
epithelial mucosal cell line NCM460 were maintained in our
laboratory. All cell lines were cultured with Dulbecco's modified
Eagle's medium (DMEM; Winsent, Quebec, Canada) supplemented with
10% fetal bovine serum (Winsent), 100 U/ml penicillin, and 100
µg/ml streptomycin in a moist incubator (stabilized at 5%
CO2 and 37°C).
Quantitative real-time PCR (qPCR)
Total RNA was extracted from CRC tissues and
adjacent normal tissues using Invitrogen™ TRIzol reagent (Thermo
Fisher Scientific, Inc., Waltham, MA, USA) according to the
manufacturer's instructions. The RNA was converted to cDNA using
the PrimeScript RT reagent kit (Takara Biotechnology, Co., Ltd.,
Dalian, China). The qPCR experiment was performed using a SYBR
Green PCR kit (Roche Diagnostics, Indianapolis, IN, USA) in a
volume of 20 µl, and then the final reaction was carried using athe
Applied Biosystems Step-One Plus Real-Time PCR System (Thermo
Fisher Scientific, Inc.). The qPCR cycling was performed as
follows: Hot-start DNA polymerase activation at 95°C for 10 min; 40
cycles of 95°C for 15 sec, and 60°C for 1 min; followed by 1 cycle
of melting curve analysis of 95°C for 15 sec, 60°C for 1 min, and
95°C for 15 sec. The primer sequences for uc.338 were as follows:
5′-AGCGACAGTGCGAGCTTT-3′ (forward) and 5′-TTCCGAGTGAGTTAGGAAGG-3′
(reverse). The primer sequences for glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) were as follows:
5′-GTGGACATCCGCAAAGAC-3′ (forward) and 5′-AAAGGGTGTAACGCAACTA-3′
(reverse).
siRNA and plasmid transfection
Small interfering RNA (siRNA) targeting uc.338 and a
negative control (NC) siRNA were designed by Genepharma Corp.
(Shanghai, China) and transferred into LoVo and HCT116 cells. The
target sequence was as follows: siuc. 338,
5′-CCACAGGACAGGUACAGCA-3′. For uc.338 overexpression, 5 µg per-well
(6-well plate) of the plasmids expressing uc.338 and negative
control sequence designed by GeneCopoeia, Inc. (Rockville, MD, USA)
were transferred into DLD-1 cells. All the above siRNA sequences
and plasmids were transfected using Invitrogen™ Lipofectamine 3000
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocols. Transfection efficiency was analyzed after 48 h.
Additionally, a lentivirus expressing a small hairpin RNA against
uc.338 (shuc.338) for an in vivo study was designed
according to the sequence of siuc.338. The
phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) inhibitor
LY294002 was purchased from Cell Signaling Technology (Danvers, MA,
USA).
Cell proliferation assay
Cell proliferation was measured using a Cell
Counting Kit-8 assay (CCK-8; Dojindo Laboratories, Tokyo, Japan)
according to the manufacturer's protocol. After 24, 48, 72 and 96
h, 10 µl CCK-8 assay solution mixed with 90 µl of serum-free medium
was added in each well containing the cells to be tested. The
absorbance was measured 2 h later using a microplate reader at a
test wavelength of 450 nm and a reference wavelength of 630 nm.
5-Ethynyl-2′-deoxyuridine (EdU)
assay
The EdU assay kit (RiboBio, China) was used to
detect the number of cells that were in the DNA replication period,
which could indirectly reveal the cell proliferation rate. The
steps were as follows. Before the addition of EdU (50 µM), the
cells were cultured with DMEM (10% FBS) for 24 h in 24-well plates
(2×104 cells/well). The cells were then incubated for 2
h at 37°C, fixed in 4% formaldehyde for 30 min, and permeabilized
with 0.5% Triton X-100 for 10 min at room temperature. After
washing with phosphate-buffered saline (PBS) for 5 min, 400 µl of
1X ApolloR reaction cocktail was added to react with the EdU for 30
min. Finally, 400 µl of Hoechst 33342 was added for 30 min to
visualize the cell nuclei. The cells were then observed under a
Nikon microscope (Nikon TI-DH; Nikon, Tokyo, Japan).
Plate colony formation assay
Five hundred cells were treated with siuc.338 and NC
and cultured in wells of a 6-well plate to perform the colony
formation assay. All cells were cultured with DMEM + 10% serum +
100 U/ml penicillin and 100 µg/ml streptomycin under the same
conditions. One week later, the cells were washed with cold PBS
three times at room temperature. The cells were immediately fixed
with ethyl alcohol for 30 sec, and stained with crystal violet dye
for 20 min. Cell colonies (≥50 cells/colony) were observed and
imaged using a digital camera (Canon DS126211; Canon, Tokyo, Japan)
in each plate after washing with PBS.
Cell cycle analysis
All the CRC cells were collected and saved in 75%
ethyl alcohol at 4°C overnight. Before detection, the treated cells
were fixed with 500 µl of propidium iodide (PI) staining solution
and incubated for 30 min in the dark. The analysis of cell cycles
was performed using a fluorescence-activated cell sorting (FACS)
Calibur flow cytometer with BD CellQuest software (version 3.0; BD
Biosciences, Franklin Lakes, NJ, USA).
In vivo tumor xenograft model
Our animal experiments were approved by the Animal
Ethics Committee of Nanjing medical university (NJMU). Fifteen male
BALB/c nude mice (aged 3–4 weeks, 13–15 g weight) were purchased
from the Animal Center of NJMU and were randomly divided to two
groups (the LoVo cell group and the HCT116 cell group). Then,
2×106 of CRC cells mixed with 200 µl PBS were
subcutaneously injected into the anesthetized mice. Each mouse was
randomly injected with cells treated in two different ways
(shuc.338 and NC) in their right and left armpits. The mice were
maintained under the following conditions: room temperature,
20–26°C; relative humidity, 40–70%; light/dark cycle, 12 h; food
and water, 5 g food and 100 ml water per 100 g body weight per day.
Four weeks after injection, all mice were sacrificed by cervical
dislocation and all the tumors were surgically removed. The maximum
tumor size was as allowable by IACUC guidelines (diameter, 1.5 cm;
area, 1.8 cm2; and volume 1.8 cm3).
Western blotting
Protein was extracted from CRC cells by using a
Radioimmunoprecipitation assay (RIPA) kit (Beyotime Institute of
Biotechnology, Shanghai, China) according to the manufacturer's
protocols. The concentration was determined using the Bicinchoninic
Acid Protein Assay kit (BCA). Proteins (40 µg) with different
molecular weights were separated on 10% SDS-PAGE gels in running
buffer and transferred to polyvinylidene fluoride (PVDF) membranes
(Millipore, Bedford, MA, USA) in transfer buffer. The membranes
were then blocked in 5% non-fat milk at room temperature for over 2
h and incubated in specific primary antibodies at 4°C overnight.
After washing in TBST three times (10 min each), the membranes were
incubated with secondary antibodies (anti-rabbit or anti-mouse) at
room temperature for 2 h. Immunoreactive protein bands were
visualized using ECL Plus (Millipore, Billerica, MA, USA) with a
Bio-Imaging System. The specific primary antibodies recognized the
following proteins: p-PI3K (diluted 1:1,000; cat. no. ab182651;
Abcam), PI3K (diluted 1:1,000; cat. no. ab86714; Abcam), p-AKT
(diluted 1:500; cat. no. ab38449; Abcam), AKT (diluted 1:500; cat.
no. ab8805; Abcam), p21 (diluted 1:5,000; cat. no. ab109520; Abcam)
and cyclin D1 (diluted 1:10,000; cat. no. ab134175; Abcam). GAPDH
(diluted 1:5,000; cat. no. ab8245; Abcam) was used as an internal
control. The secondary antibody was horseradish peroxidase
(HRP)-goat anti-rabbit (diluted 1:5,000; cat. no. GAB007; Hangzhou
Multi Sciences Biotech, Co., Ltd., Hangzhou, China) or anti-mouse
(diluted 1:5,000; cat. no. GAM007; Hangzhou Multi Sciences Biotech,
Co., Ltd., Hangzhou, China) IgG.
Statistical analysis
SPSS (version 15.0) (SPSS, Inc., Chicago, IL, USA)
and Graphpad Prism5 (GraphPad Software, Inc., La Jolla, CA, USA)
were used for the statistical analysis. Data were derived from at
least three repeated experiments. The statistical analyses were
performed using t-tests, Pearson's χ2 tests and ANOVA.
Differences were considered to be statistically significant at
P≤0.05. The Kaplan-Meier method was used to perform the cumulative
survival analysis.
Results
uc.338 is upregulated in CRC
tissues
To reveal the role of uc.338 in CRC progression, we
first detected its expression level in 100 pairs of CRC tissues and
adjacent normal tissues using qPCR. The results showed that uc.338
expression was significantly upregulated in CRC tissues compared
with that in normal tissues (Fig.
1A). According to the expression level of uc.338 in CRC
tissues, we further verified that higher uc.338 expression
predicted a larger tumor size, deeper tumor invasion, and increased
lymph node metastasis (P<0.05, Table
I). However, we did not find any association between uc.338
expression and other clinical features, including sex, age, tumor
stage, liver metastasis, location and CEA. We then examined whether
the expression level of uc.338 could predict the overall survival
(OS) of patients diagnosed with CRC. Using Kaplan-Meier curves, we
found that higher uc.338 expression predicted poorer overall
survival (OS) in patients with CRC (Fig. 1B). The P-value was 0.012
(P<0.05).
 | Table I.Expression of uc.338 in colorectal
carcinoma and adjacent normal tissues. |
Table I.
Expression of uc.338 in colorectal
carcinoma and adjacent normal tissues.
|
| uc.338
expression |
|---|
|
|
|
|---|
| Clinical
features | N | High | Low | P-value |
|---|
| Sex |
| 50 | 50 | 0.096 |
|
Male | 64 | 36 | 28 |
|
|
Female | 36 | 14 | 22 |
|
| Age (year) |
|
|
| 0.288 |
|
>60 | 67 | 31 | 36 |
|
|
≤60 | 33 | 19 | 14 |
|
| Tumor size
(cm) |
|
|
| 0.002a |
|
>5 | 41 | 28 | 13 |
|
| ≤5 | 59 | 22 | 37 |
|
| Depth of
invasion |
|
|
| 0.032a |
|
T1/T2 | 32 | 11 | 21 |
|
|
T3/T4 | 68 | 39 | 29 |
|
| Lymph node
metastasis |
|
|
| 0.005a |
|
Absent | 42 | 14 | 28 |
|
|
Present | 58 | 36 | 22 |
|
| Tumor stage |
|
|
| 0.539 |
|
I/II | 39 | 18 | 21 |
|
|
III/IV | 61 | 32 | 29 |
|
| Liver
metastasis |
|
|
| 0.275 |
|
Yes | 16 | 10 | 6 |
|
| No | 84 | 40 | 44 |
|
| Location |
|
|
| 0.546 |
|
Rectal | 55 | 26 | 29 |
|
|
Colon | 45 | 24 | 21 |
|
| CEA (ng/ml) |
|
|
| 0.155 |
|
>4.7 | 41 | 17 | 24 |
|
|
≤4.7 | 59 | 33 | 26 |
|
Uc.338 is upregulated in CRC cell
lines and was inhibited by siRNA
To detect whether uc.338 is upregulated in CRC cell
lines, we detected the mRNA expression level of uc.338 using qPCR.
Compared with that in the human colon epithelial mucosa cell line
NCM460, the mRNA expression of uc.338 was upregulated in LoVo,
HCT116 and DLD-1 cells (Fig. 1C).
To knockdown uc.338 expression, LoVo and HCT116 cells were
transfected with a siRNA designed to inhibit uc.338 expression
(siuc.338). The result revealed that the mRNA expression of uc.338
was significantly inhibited by siuc.338 in both LoVo and HCT116
cells (Fig. 1D and E). The
interference efficiency of siuc.338 was 71 and 79% in LoVo and
HCT116 cells, respectively (P<0.05).
Knockdown of uc.338 inhibits cell
proliferation in LoVo and HCT116 cells
To detect the effect of uc.338 on cell
proliferation, we performed a CCK-8 assay, an EdU assay, and a
plate colony formation assay in succession. In the CCK-8 assay, we
found that the viability of LoVo and HCT116 cells was significantly
decreased after knockdown of uc.338 (Fig. 2A and B). On the fourth day, the
viability of LoVo and HCT116 cells had decreased by 33.9 and 41.1%,
respectively (P<0.05). In the EdU assay, compared with the
control cells, we found that the proportion of cells in the DNA
replication phase decreased by 19.8 and 21.9% in the LoVo and
HCT116 cells, respectively, after knockdown of uc.338 (Fig. 2C and D), which revealed that
knockdown of uc.338 could inhibit DNA synthesis in CRC cells. In
the plate colony formation assay, the result revealed that
knockdown of uc.338 significantly inhibited the formation of cell
colonies in the LoVo and HCT116 cells (Fig. 3A and B). After 7 days, we found that
the number of cell colonies decreased by 46 and 42% in LoVo and
HCT116 cells, respectively, after knockdown of uc.338 (P<0.05).
According to the results of three assays, knockdown of uc.338
inhibited the cell proliferation in CRC LoVo and HCT116 cells.
Knockdown of uc.338 results in cell
cycle arrest in G1/S phases in LoVo and HCT116 cells
To detect the role of uc.338 in the cell cycle of
CRC cells, we performed cell cycle analysis using flow cytometry.
After knockdown of uc.338, we found that the cell number in the G1
phase increased by 15.5 and 23.6% in LoVo and HCT116 cells,
respectively, whereas the cell number in the S phase decreased by
11.4 and 15.5%, respectively, in these two cell lines (Fig. 3C and D). Our result indicated that
knockdown of uc.338 caused marked cell cycle arrest in the G1/S
phases in the LoVo and HCT116 cells.
Knockdown of uc.338 suppresses tumor
growth in nude mice
To detect the role of uc.338 in the tumor growth in
nude mice, LoVo and HCT116 cells were transfected with uc.338
inhibitor lentivirus (shuc.338) (Fig.
4B). Then, 2×106 cells transfected with shuc.338 and
a negative control sequence (control) were separately injected
subcutaneously into the nude mice (Fig.
4A). After four weeks, we found that the size and weight of the
tumors were significantly decreased in the nude mice transfected
with the cells with knockdown of uc.338 (Fig. 4C and D). Compared with the tumors
from the control groups, the weight of tumors formed by the
uc.338-knockdown LoVo and HCT116 cells was decreased by 61.1 and
54.9% in the shuc.338 groups (P<0.05). Therefore, knockdown of
uc.338 was able to suppress tumor growth in nude mice.
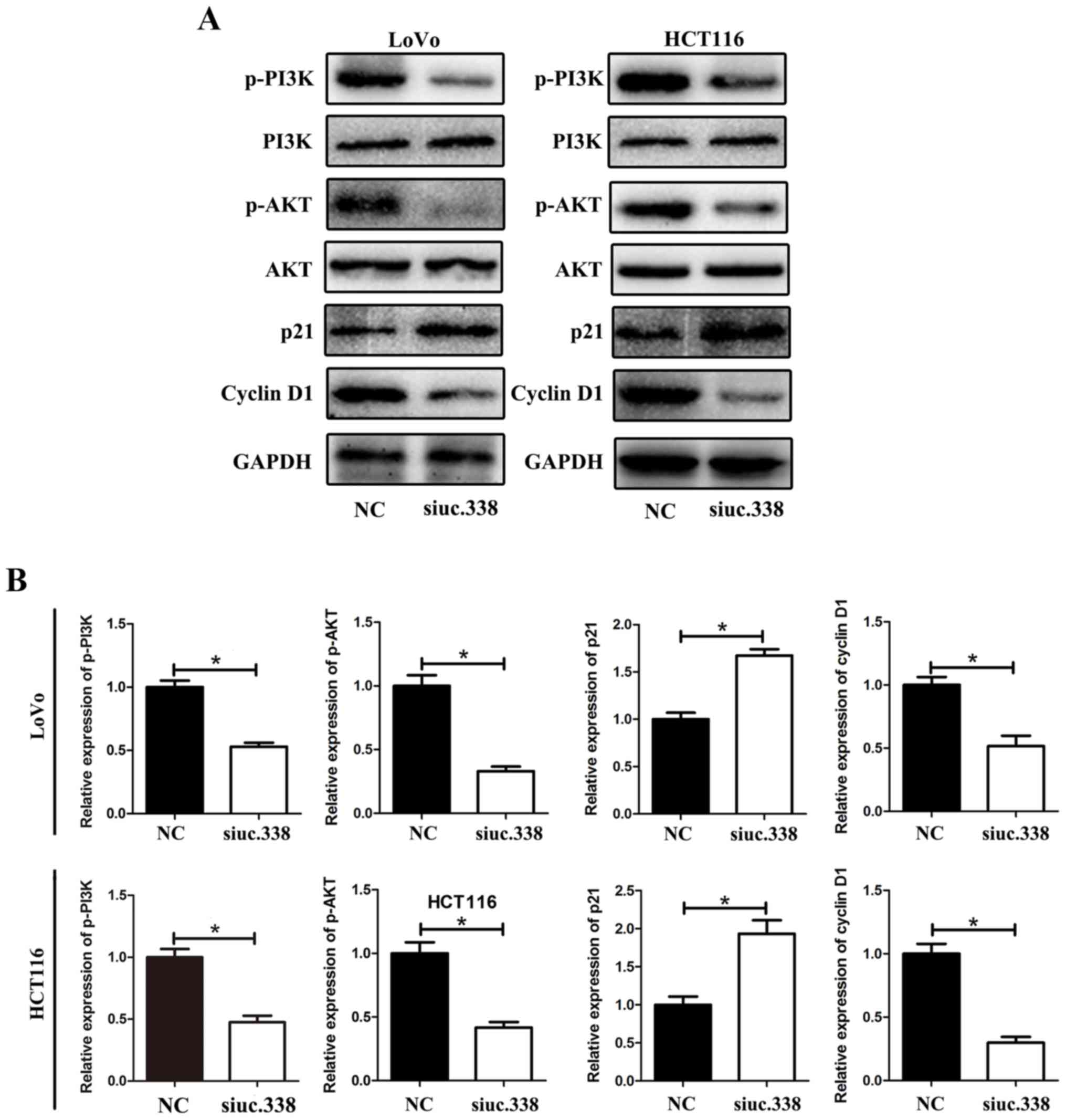
Downregulation of uc.338 inhibits the
expression of cyclin D1 and promotes the expression of p21,
possibly via the PI3K/AKT pathway
We revealed that uc.338 could cause cell cycle
arrest in the G1/S phase in CRC cells. To further detect the
potential mechanism of cell cycle arrest, we detected a series of
cell cycle-related proteins that could regulate the G1/S phase in
LoVo and HCT116 cells. The results showed that the level of cyclin
D1 was significantly downregulated and the level of p21 was
upregulated by uc.338 knockdown. Further investigation revealed
that the levels of p-PI3K and p-AKT were downregulated by uc.338
knockdown, whereas no significant change was detected in the levels
of total PI3K and AKT (Fig. 5A and
B).
To further investigate the role of the PI3K/AKT
pathway in uc.338-induced cell proliferation in CRC, a specific
inhibitor of PI3K (LY294002) was used. uc.338 was overexpressed in
the DLD-1 cell line (Fig. 6A),
which expresses a relatively low level of uc.338, and then the
cells were treated with LY294002. By western blotting, we found
that the PI3K/AKT pathway was significantly inhibited by LY294002
(Fig. 6B and C). The colony
formation assay showed that overexpression of uc.338 promoted cell
proliferation in DLD-1 cells; however, this promotion of cell
proliferation was inhibited by LY294002 (Fig. 6D and E). Taken together, the results
demonstrated that knockdown of uc.338 inhibited the expression of
cyclin D1 and promoted the expression of p21, possibly via the
PI3K/AKT pathway.
Discussion
Their high degree of conservation means that
ultraconserved regions (UCRs) are considered to have fundamental
functional importance for the ontogeny and phylogeny of mammals and
other vertebrates (3). Since Calin
et al first detected the role of T-UCRs in human cancers
(6), increasing evidence has shown
that aberrant expression of T-UCRs is involved in tumor
pathogenesis. Simultaneously, single nucleotide polymorphisms
(SNPs) within UCRs were also verified to be closely related to
clinical features in various carcinomas (15–17).
These observations revealed the vital importance of T-UCRs in human
carcinomas. uc.338 is an exonic type T-UCR that overlaps with the
mRNA of a known human protein-coding gene (6,18).
Previous studies have shown that uc.338 is involved in the
progression of various tumors. For example, Bo et al found
that uc.338 promoted HCC cell proliferation and induced cell cycle
progression through an association with BMI1 in hepatocellular
carcinoma (10). Gao et al
found that uc.338 could promote proliferation and metastasis by
cyclin B1 and EMT activation in lung carcinoma (13). In CRC, Wang et al first
detected that uc.338 could promote CRC cell migration and invasion
by targeting the TIMP1 (14); however, the role of uc.338 in CRC
cell proliferation was not completely clear.
In the present study, we first detected that the
expression level of uc.338 mRNA was upregulated in CRC tissues. On
this basis, we further analyzed the clinical features and prognosis
of patients with CRC. The results indicated that a higher
expression level of uc.338 mRNA was associated with larger tumors,
deeper tumor invasion, increased lymph node metastasis, and poorer
prognosis. Our results not only confirmed the results of Wang et
al, but also suggested a possible role of uc.388 in CRC
proliferation and prognosis. To further examine the role of uc.338
in promoting cell proliferation, we performed a series of cell
proliferation experiments in vivo and in vitro. In
vitro, we found that uc.338 was significantly upregulated in
CRC cell lines (LoVo, HCT116 and DLD-1) compared with its level in
normal colon epithelial cells (NCM460). Further experiments
revealed that the cell viability, DNA replication, and colony
formation were significantly inhibited by uc.338 knockdown in LoVo
and HCT116 cells. In vivo, we transplanted CRC cells into
nude mice, which revealed that the speed of tumor growth from cells
with uc.338 knockdown was significantly inhibited, which supported
our hypothesis that uc.338 could promote cell proliferation in CRC.
Flow cytometry showed that the cell cycle in the G1/S phase was
arrested by uc.338 inhibition, suggesting that uc.338 could promote
cell cycle transition. However, no difference was found in the rate
of cell apoptosis in CRC cells, indicating that uc.338 is not
involved in cell apoptosis control (data not shown). These results
indicated that uc.338 promotes tumor proliferation possibly via
cell cycle G1/S promotion in CRC cells.
Numerous components of the cell cycle kinases or
kinase inhibitors are involved in mediating the G1/S transition
(19,20). Protein p21 is generally recognized
as a negative regulator in the G1/S transition (21), and downregulation of p21 is involved
in tumor promotion in various cancers (22,23).
Conversely, cyclin D1 is widely accepted as an oncogene and has
been implicated in many activities, such as cell cycle promotion,
chromosomal instability, mitochondrial function, and cellular
senescence (24–26). To better understand the mechanisms
that promote the G1/S cell cycle transition mediated by uc.338, the
expression levels of certain cell cycle-related proteins were
determined using western blotting. Our results indicated that
arrest of the cell cycle at the G1/S phase in CRC cells might be
facilitated by uc.338-mediated p21 upregulation and cyclin D1
downregulation. To further understand the mechanism of
uc.338-mediated regulation of p21 and cyclin D1, we detected the
protein levels of key members of the PI3K/AKT pathway. The PI3K/AKT
pathway is recognized as a classic signaling mediator that
modulates cell proliferation and apoptosis, and is involved in the
progression of various tumors (26–28).
In this process, p21 and cyclin D1 are two common downstream
regulatory genes of the PI3K/AKT pathway in a variety of tumors,
including CRC (29,30). Western blotting showed that the
levels of p-PI3K and p-AKT were significantly decreased by uc.338
knockdown in CRC cells, whereas no significant difference was found
in the total PI3K and AKT levels. Moreover, after treatment with a
specific inhibitor of PI3K (LY294002), the oncogenic role of uc.338
in cell proliferation was distinctly inhibited. Hence, we
hypothesized that uc.338 could promote p21 downregulation and
cyclin D1 upregulation, possibly via PI3K/AKT pathway
activation.
p53, usually called as ‘guardian of the genome’, is
an important tumor-suppressor gene that controls response to
oncogene activation such as cell cycle arrest, apoptosis, and
senescence. In the mammalian genome, p53 is also the most
frequently mutated gene in human cancer (31–33).
It has been reported that elevated PI3K/Akt activity is observed in
cells harboring high levels of p53 mutant protein (34,35),
and p53 could bind to p21 to promote cell death or replicative
senescence (36,37). To date, no direct evidence has shown
that uc.338 is related to p53 status. To further investigate
whether uc.338 is associated with p53, we detected the mRNA
expression of p53 by uc.338 knockdown, but our results showed no
significant relationship between uc.338 andp53 expression. Even so,
it remains unclear whether uc.338 is related to the p53 mutation or
in other states, and a more in-depth study on the relationship
between uc.338 and p53 should be performed in our next study.
Meanwhile, the aberrant regulation of T-UCR expression consistently
plays a role in cancer by altered interactions with miRNAs
(18). It is uncertain whether
uc.338 interacts with certain miRNAs, and we cannot rule out the
possibility that other signaling pathways might also be affected by
uc.338 in regards to CRC proliferation. Furthermore, the exact
association between uc.338, p21, cyclin D1 and the PI3K/AKT pathway
requires further investigation.
In conclusion, we demonstrated that uc.338 was
significantly upregulated in CRC tissues. We proposed that uc.338
expression is associated with tumor size, invasion depth, and
prognosis in patients with CRC. Further study provided evidence
supporting our hypothesis that uc.338 promotes CRC cell
proliferation by facilitating cell cycle G1/S transition. Finally,
we indicated that uc.338 might target p21 and cyclin D1 to promote
cell cycle G1/S transition, possibly by activation of the PI3K/Akt
pathway.
Acknowledgments
We would like to thank Dr Hao Fan for the excellent
technical assistance. We also would like to thank Dr Yuanguangyan
Zhang for the great statistical guidance.
Funding
The present study was supported by the Jiangsu Key
Medical Discipline (General Surgery) (grant no. ZDXKA2016005).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
YS, ZF and YZ contributed to the design of this
study. ShijiaW, WQ and ZZ contributed to the experimental work. YZ,
BJ and SenW contributed to the data collection and analysis. YZ, QW
and DJ contributed to the interpretation of data and drafting the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The present study using human tissues was approved
by the Ethics Committee of the First Affiliated Hospital of Nanjing
Medical University. Our animal experiments were approved by the
Animal Ethics Committee of Nanjing Medical University (NJMU).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CRC
|
colorectal cancer
|
|
UCRs
|
ultraconserved regions
|
|
T-UCRs
|
transcribed ultraconserved regions
|
|
NCCN
|
National Comprehensive Cancer
Network
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
PBS
|
phosphate-buffered saline
|
|
qPCR
|
quantitative real-time PCR
|
|
siRNA
|
small interfering RNA
|
|
CCK-8
|
Cell Counting Kit-8
|
|
EdU
|
5-ethynyl-2′-deoxyuridine
|
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in china,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bejerano G, Pheasant M, Makunin I, Stephen
S, Kent WJ, Mattick JS and Haussler D: Ultraconserved elements in
the human genome. Science. 304:1321–1325. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kung JT, Colognori D and Lee JT: Long
noncoding RNAs: Past, present, and future. Genetics. 193:651–669.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mestdagh P, Fredlund E, Pattyn F, Rihani
A, Van Maerken T, Vermeulen J, Kumps C, Menten B, De Preter K,
Schramm A, et al: An integrative genomics screen uncovers ncRNA
T-UCR functions in neuroblastoma tumours. Oncogene. 29:3583–3592.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Calin GA, Liu CG, Ferracin M, Hyslop T,
Spizzo R, Sevignani C, Fabbri M, Cimmino A, Lee EJ, Wojcik SE, et
al: Ultraconserved regions encoding ncRNAs are altered in human
leukemias and carcinomas. Cancer Cell. 12:215–229. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sana J, Hankeova S, Svoboda M, Kiss I,
Vyzula R and Slaby O: Expression levels of transcribed
ultraconserved regions uc.73 and uc.388 are altered in colorectal
cancer. Oncology. 82:114–118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hudson RS, Yi M, Volfovsky N, Prueitt RL,
Esposito D, Volinia S, Liu CG, Schetter AJ, Van Roosbroeck K,
Stephens RM, et al: Transcription signatures encoded by
ultraconserved genomic regions in human prostate cancer. Mol
Cancer. 12:132013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Braconi C, Valeri N, Kogure T, Gasparini
P, Huang N, Nuovo GJ, Terracciano L, Croce CM and Patel T:
Expression and functional role of a transcribed noncoding RNA with
an ultraconserved element in hepatocellular carcinoma. Proc Natl
Acad Sci USA. 108:786–791. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bo C, Li N, Li X, Liang X and An Y: Long
noncoding RNA uc.338 promotes cell proliferation through
association with BMI1 in hepatocellular carcinoma. Hum Cell.
29:141–147. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jin W, Chen L, Cai X, Zhang Y, Zhang J, Ma
D, Cai X, Fu T, Yu Z, Yu F and Chen G: Long non-coding RNA TUC338
is functionally involved in sorafenib-sensitized hepatocarcinoma
cells by targeting RASAL1. Oncol Rep. 37:273–280. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Q, Shen F and Wang C: TUC338 promotes
cell migration and invasion by targeting TIMP1 in cervical cancer.
Oncol Lett. 13:4526–4532. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gao X, Gao X, Li C, Zhang Y and Gao L:
Knockdown of long noncoding RNA uc.338 by siRNA inhibits cellular
migration and invasion in human lung cancer cells. Oncol Res.
24:337–343. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang C, Wang Z, Zhou J, Liu S, Wu C, Huang
C and Ding Y: TUC.338 promotes invasion and metastasis in
colorectal cancer. Int J Cancer. 140:1457–1464. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Catucci I, Verderio P, Pizzamiglio S,
Manoukian S, Peissel B, Barile M, Tizzoni L, Bernard L, Ravagnani
F, Galastri L, et al: SNPs in ultraconserved elements and familial
breast cancer risk. Carcinogenesis. 30:544–545; author reply 546.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bao BY, Lin VC, Yu CC, Yin HL, Chang TY,
Lu TL, Lee HZ, Pao JB, Huang CY and Huang SP: Genetic variants in
ultraconserved regions associate with prostate cancer recurrence
and survival. Sci Rep. 6:221242016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shen H, Lu C, Jiang Y, Tang J, Chen W,
Zhang H, Zhang Q, Wang J, Liang J, Hu Z and Shen H: Genetic
variants in ultraconserved elements and risk of breast cancer in
chinese population. Breast Cancer Res Treat. 128:855–861. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peng JC, Shen J and Ran ZH: Transcribed
ultraconserved region in human cancers. RNA Biol. 10:1771–1777.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Motokura T and Arnold A: Cyclin D and
oncogenesis. Curr Opin Genet Dev. 3:5–10. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pines J: Cyclins: Wheels within wheels.
Cell Growth Differ. 2:305–310. 1991.PubMed/NCBI
|
|
21
|
Niculescu AB III, Chen X, Smeets M, Hengst
L, Prives C and Reed SI: Effects of p21(Cip1/Waf1) at both the G1/S
and the G2/M cell cycle transitions: PRb is a critical determinant
in blocking DNA replication and in preventing endoreduplication.
Mol Cell Biol. 18:629–643. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nie FQ, Sun M, Yang JS, Xie M, Xu TP, Xia
R, Liu YW, Liu XH, Zhang EB, Lu KH and Shu YQ: Long noncoding RNA
ANRIL promotes non-small cell lung cancer cell proliferation and
inhibits apoptosis by silencing KLF2 and P21 expression. Mol Cancer
Ther. 14:268–277. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Casimiro MC and Pestell RG: Cyclin d1
induces chromosomal instability. Oncotarget. 3:224–225.
2012.PubMed/NCBI
|
|
25
|
Leontieva OV, Lenzo F, Demidenko ZN and
Blagosklonny MV: Hyper-mitogenic drive coexists with mitotic
incompetence in senescent cells. Cell Cycle. 11:4642–4649. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang C, Li Z, Lu Y, Du R, Katiyar S, Yang
J, Fu M, Leader JE, Quong A, Novikoff PM and Pestell RG: Cyclin D1
repression of nuclear respiratory factor 1 integrates nuclear DNA
synthesis and mitochondrial function. Proc Natl Acad Sci USA.
103:11567–11572. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen J, Zhao KN, Li R, Shao R and Chen C:
Activation of PI3K/Akt/mTOR pathway and dual inhibitors of PI3K and
mTOR in endometrial cancer. Curr Med Chem. 21:3070–3080. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee JJ, Loh K and Yap YS: PI3K/Akt/mTOR
inhibitors in breast cancer. Cancer Biol Med. 12:342–354.
2015.PubMed/NCBI
|
|
29
|
Wang L, Cao XX, Chen Q, Zhu TF, Zhu HG and
Zheng L: DIXDC1 targets p21 and cyclin D1 via PI3K pathway
activation to promote colon cancer cell proliferation. Cancer Sci.
100:1801–1808. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yan L and Shi G: Effect of IFN-α on
hepatic cancer SMCC-7721 cell via PI3K/Akt signaling pathway and
related mechanism research. Zhonghua Yi Xue Za Zhi. 95:2960–2963.
2015.(In Chinese). PubMed/NCBI
|
|
31
|
Liu J, Zhang C, Hu W and Feng Z: Tumor
suppressor p53 and its mutants in cancer metabolism. Cancer Lett.
356:197–203. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu J, Zhang C and Feng Z: Tumor
suppressor p53 and its gain-of-function mutants in cancer. Acta
Biochim Biophys Sin (Shanghai). 46:170–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Duffy MJ, Synnott NC, McGowan PM, Crown J,
O'Connor D and Gallagher WM: p53 as a target for the treatment of
cancer. Cancer Treat Rev. 40:1153–1160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Muller PA, Caswell PT, Doyle B, Iwanicki
MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL,
Gosselin P, et al: Mutant p53 drives invasion by promoting integrin
recycling. Cell. 139:1327–1341. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hanel W, Marchenko N, Xu S, Yu SX, Weng W
and Moll U: Two hot spot mutant p53 mouse models display
differential gain of function in tumorigenesis. Cell Death Differ.
20:898–909. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Riley T, Sontag E, Chen P and Levine A:
Transcriptional control of human p53-regulated genes. Nat Rev Mol
Cell Biol. 9:402–412. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Haupt S, Berger M, Goldberg Z and Haupt Y:
Apoptosis-the p53 network. J Cell Sci. 116:4077–4085. 2003.
View Article : Google Scholar : PubMed/NCBI
|