Introduction
Colorectal cancer (CRC) is the third most common
cancer worldwide (1). In recent
years, >1.2 million patients have been diagnosed annually and
~600,000 patients succumb to CRC every year (2). Surgical resection is the main method
for treatment of early-stage CRC with a favorable prognosis.
However, most patients are diagnosed with metastatic cancer, which
is not suitable for resection. Traditional chemotherapy is the main
approach for metastatic CRC (mCRC) and the overall survival can
reach 15–19 months (3). With the
advances in therapies, targeted drugs, such as cetuximab and
bevacizumab, can prolong the overall survival to 28.7–33.1 months
in mCRC patients (4,5). Meanwhile, small EGFR tyrosine kinase
inhibitors (TKIs), such as gefitinib and erlotinib, have been
commonly used in non-small cell lung cancer and have improved
progression-free survival, exhibiting great application potential.
However, EGFR-inhibitor resistance has restricted their
application, creating an urgent need to discover novel treatment
strategies.
Yes-associated protein (YAP), a downstream effector
of the Hippo pathway, is involved in tissue overgrowth and tumor
formation (6). A study revealed
that YAP could act as a transcriptional co-activator to promote the
expression of genes involved in cell proliferation and apoptosis
(7). A recent study revealed that
YAP may be useful for identifying resistance to cetuximab (8). However, the role of YAP in cetuximab
resistance remains elusive in CRC.
In the present study, we observed that the
expression of YAP was negatively associated with cetuximab
sensitivity in CRC cell lines, independent of KRAS mutation status,
and YAP knockdown enhanced the cytotoxicity of cetuximab.
Simvastatin, a 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA)
reductase inhibitor, has been reported to inhibit YAP bioactivity
in multiple types of cancer cell lines (9,10). We
determined that simvastatin increased the sensitivity of CRC cells
to cetuximab both in vitro and in vivo. These
findings indicated that YAP may be useful in identifying cetuximab
resistance in CRC cancer.
Materials and methods
Cell culture and agents
The CRC cell lines COLO 320, HCT 116, HT 29, SW 48,
SW 480 and SW 1116 were purchased from the American Type Culture
Collection (ATCC; Manassas, VA, USA). All cell lines were
maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco
Laboratories; Thermo Fisher Scientific, Inc., Grand Island, NY,
USA) supplemented with 10% heat-inactivated fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc., Gaithersburg, MD,
USA). Cell cultures were maintained with 100 U/ml of penicillin G
sodium and 100 µg/ml streptomycin sulfate (Sigma-Aldrich; Merck
KGaA, St. Louis, MO, USA) at 37°C in a humidified 5%
CO2, 95% air incubator. Simvastatin was purchased from
J&K Scientific Ltd. (Beijing, China). Cetuximab was purchased
from Merck & Co., Inc. (Whitehouse Station, NJ, USA) and
YM53601 was purchased from Cayman Chemical Company (Ann Arbor, MI,
USA). Gefitinib, FTI277 and GGTI298 were purchased from Selleck
Chemicals (Houston, TX, USA) and EGF and GGPP were purchased from
Sigma-Aldrich; Merck KGaA.
Transfection
siRNAs against YAP and control siRNAs were designed
and synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China).
We used two independent siRNAs to target YAP. The siRNA sequence
for YAP-1 was 5′-GACAUCUUCUGGUCAGAGA-3′, the siRNA sequence for
YAP-2 was 5′-CUGGUCAGAGAUACUUCUU-3′ and the control siRNA sequence
was 5′-AAUUCUCCGAACGUGUCACGUUU-3′. Cells were transfected with
control siRNA and YAP siRNA for 72 h or at the indicated
time-points using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) according to the manufacturer's
instructions.
Western blot analysis
Cells were lysed in ice-cold RIPA buffer containing
protease inhibitor cocktail (Sigma-Aldrich; Merck KGaA). Cell
lysates from mouse xenograft tumors (four groups) were homogenized
and lysed in glass homogenizers in RIPA buffer. Subsequently, cell
lysates were quantified with a BCA protein assay (Pierce Chemical
Co., Dallas, TX, USA). Approximately 20 µg of total protein was
loaded onto a 10 or 12% SDS-PAGE gel, separated electrophoretically
and then transferred to an NC membrane (EMD Millipore, Bedford, MA,
USA). The membranes were blocked with 5% non-fat milk at room
temperature for ~30 min and then were washed three times with
phosphate-buffered saline (PBS). Then, the membranes were incubated
with the horseradish peroxidase conjugated anti-mouse (cat. no.
HA1022) or anti-rabbit (cat. no. HA1001) secondary antibodies
(Huabio, Hangzhou, China) with dilution of 1:5,000–1:10,000 at room
temperature for 3 h. Finally, the protein expression was detected
with an Odyssey Infrared Imaging System (LI-COR Biosciences,
Lincoln, NE, USA). Protein expression was quantified using ImageJ
1.48 (National institute of Health, Bethesda, MA, USA). Primary
antibodies targeting the following proteins were used in the
present study: pEGFR (cat. no. 3777), cyclin D1 (cat. no. 2922),
YAP (cat. no. 4912), CYR61 (cat. no. 14479), PTEN (cat. no. 9552),
GAPDH (cat. no. 97166) and Lamin B (cat. no. 13435), were purchased
from Cell Signaling Technology (Danvers, MA, USA); EGFR (cat. no.
sc-03-G) was purchased from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA); and pAKT (cat. no. EP2109Y) and AKT (cat. no. EPR16798)
were purchased from Abcam (Cambridge, UK). All the primary
antibodies above were added at a 1:1,000 dilution at 4°C
overnight.
Cell proliferation assay
Cells were seeded (4×103 cells/well in
100 µl DMEM with 10% FBS) in a 96-well flat-bottomed plate (Corning
Inc., Corning, NY, USA) 24 h before treatment. Following
incubation, cell growth was determined with a Cell Counting Kit-8
(CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan)
assay following the manufacturer's instructions.
Hoechst 33342 staining
Cells were seeded (4×103 cells/well in
100 µl DMEM with 10% FBS) in a 96-well flat-bottomed plate 24 h
before treatment. Apoptotic cells were determined using Hoechst
33342 (Sigma-Aldrich; Merck KGaA) DNA staining according to the
manufacturer's protocol. Apoptotic cells were detected by
fluorescence microscopy (Nikon, Tokyo, Japan), which revealed
nuclear condensation and DNA fragmentation.
Quantitative real-time PCR
Cells were collected in TRIzol (Invitrogen) for
total RNA extraction according to the manufacturer's protocol.
Retrotranscription was performed with Reverse Transcriptase M-MLV
(Takara Biotechnology, Co., Ltd., Dalian, China). Real-time PCR
reactions were performed with a SYBR Premix Ex Taq™ kit (Takara
Biotechnology) on an iQ5 Real-Time PCR Detection system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The mixtures were incubated
at 95°C for 10 min, followed by 40 amplification cycles of 95°C for
20 sec, 55–65°C for 20 sec and 72°C for 30 sec. The primers used
were as follows: YAP sense, 5′-GGTGCCACTGTTAAGGAAAGG-3′ and
antisense, 5′-GTGAGGCCACAGGAGTTAGC-3′; CTGF sense,
5′-TGGTGCAGCCAGAAAGCTC-3′ and antisense, 5′-CCAATGACAACGCCTCCTG-3′;
CYR61 sense, 5′-TTCTTTCACAAGGCGGCACTC-3′ and antisense,
5′-AGCCTCGCATCCTATACAACC-3′; GAPDH sense, 5′-CAAGGCCAACCGCGAGAA-3′
and antisense, 5′-CCCTCGTAGATGGGCACAGT-3′. The data were analyzed
with the 2−ΔΔCq method (11).
Nuclear and cytoplasmic protein
extraction
After the addition of reagents as indicated, nuclear
and cytoplasmic proteins from cells and mouse xenograft tumors were
extracted with a Nuclear and Cytoplasmic Extraction kit (Thermo
Fisher Scientific, Inc., Waltham, MA, USA) according to the
manufacturer's instructions.
Immunofluorescence
Approximately 4×103 cells were seeded on
a 24-well culture dish. After the addition of reagents, the cells
were fixed with PBS-buffered 4% paraformaldehyde for 10 min
followed by permeabilization with 0.3% Triton X-100 for 10 min and
blocking with 3% BSA for 30 min. The primary antibody (YAP) was
added at a 1:75 dilution in PBS overnight at 4°C, followed by three
PBS washes. The secondary FITC-labeled antibody (cat. no. 31568)
(Invitrogen; Thermo Fisher Scientific, Inc.) at a 1:75 dilution in
PBS was then incubated with cells for 2 h at 37°C. After three PBS
washes, immunofluorescence was viewed with a fluorescence
microscope (Eclipse TE300; Nikon, Tokyo, Japan). All procedures
were performed at room temperature unless otherwise specified.
In vivo assay and ethical
standards
Four-week-old female BALB/c nude mice obtained from
Beijing HFK Biotechnology Co., Ltd. (Beijing, China) were
maintained under specific pathogen-free conditions. Mice were
injected subcutaneously with SW 480 cells. When the xenografts
reached a volume of 80–100 mm3, the mice (five mice per
group) were assigned into four groups (control, cetuximab,
simvastatin and cetuximab + simvastatin). The mice were treated
with intraperitoneal injection of 20 mg/kg cetuximab in dimethyl
sulfoxide (DMSO) once every 3–4 days and/or orally with 6 mg/kg
simvastatin in DMSO once daily. Tumor diameters were determined
with a digital caliper every three days following treatment. All
animal experimental procedures used in the present study were
approved by the Experimental Animal Manage Committee of West China
Hospital, Sichuan University under contract 2016003A and were
performed strictly according to the guidelines of the Animal Ethics
Committee Guidelines of the Animal Facility of West China Hospital
and the Animal Care and Use Committee of Sichuan University.
Statistical analysis
Values are presented as the means ± standard error
of the mean (SEM). All statistical analyses were performed with
SPSS 14.0 software for Windows (SPSS, Inc., Chicago, IL, USA).
Correlation analysis between the expression of YAP and
proliferation inhibition rate was performed by Spearman's
correlation test as previously described (12). Student's t-test was performed when
two groups were compared; one-way ANOVA and post hoc Tukey's test
were performed when multiple comparisons were conducted. P<0.05
was considered to indicate a statistically significant result.
Results
YAP expression is negatively
associated with the sensitivity of CRC cells to cetuximab
First, we examined YAP expression in five CRC cell
lines (SW 1116, HCT 116, SW 480, COLO 320 and SW 48). Since KRAS
mutation status is reported as an independent biomarker for
cetuximab efficacy (13), we
divided the cancer cells into two groups, wild-type KRAS CRC cell
lines and mutant KRAS CRC cell lines. Subsequently, we investigated
whether YAP participated in cetuximab resistance in vitro.
We assessed the relative expression of YAP compared with the
expression of GAPDH in the five cell lines. The relative YAP
expression of the mutant KRAS CRC cell lines (SW 1116, HCT 116 and
SW 480) were 0.64, 0.56 and 0.42, respectively; and that of the
wild-type KRAS CRC cell lines (COLO 320 and SW 48) were 0.96 and
0.24, respectively (Fig. 1A and B).
Subsequently, we determined the relative proliferation rate of each
cell line treated with cetuximab compared with each untreated cell
line, separately. We observed that SW 1116 and COLO 320, which had
the highest YAP expression in each group, demonstrated the lowest
sensitivity to cetuximab (Fig. 1C).
SW 480 and SW 48 cell lines had the lowest YAP levels and the
highest sensitivity to cetuximab in each group (Fig. 1C). The relative proliferation rate
of the mutant KRAS CRC cell lines was 0.96 vs. 0.85 for SW 1116 vs.
SW 480, and 0.79 vs. 0.60 for the wild-type KRAS CRC cell lines
COLO 320 vs. SW 48. We then conducted a correlation analysis to
determine statistical relationships involving YAP expression and
rate of proliferation inhibition against cetuximab. The correlation
coefficients of the mutant KRAS CRC cell lines and wild-type KRAS
cell lines were 0.83 and 0.89 (P<0.05), respectively.
Collectively, these results indicated that cetuximab sensitivity
was negatively correlated with YAP expression independent of KRAS
mutation status.
YAP knockdown enhances the sensitivity
of CRC cell lines to cetuximab
To further investigate the effect of YAP on the
sensitivity of cancer cell lines to cetuximab, we transfected cells
with two independent RNA interference constructs (siYAP-1 and
siYAP-2) (Fig. 2A and B, E and F).
We selected two cell lines with primary cetuximab resistance,
namely, HCT 116 and SW 480 (Fig.
1C), to conduct the following study. The results revealed that
the combination of cetuximab and siYAP-1 significantly reduced cell
proliferation compared with cetuximab or siYAP-1 alone (HCT 116,
0.7 µM cetuximab vs. 0.7 µM cetuximab + siYAP-1, mean relative cell
proliferation rate: 83.5 vs. 49.9%, P<0.01; SW 480, 0.7 µM
cetuximab vs. 0.7 µM cetuximab + siYAP-1, mean relative cell
proliferation rate: 78.3 vs. 57.9%, P<0.01) (Fig. 2C and D). In addition, results
involving siYAP-2 revealed a similar trend (Fig. 2G and H). Furthermore, colony
formation assays revealed that knockdown of YAP-1 promoted colony
inhibition caused by cetuximab (Fig. 2I
and J). These results indicated that YAP may be related to
primary resistance to cetuximab.
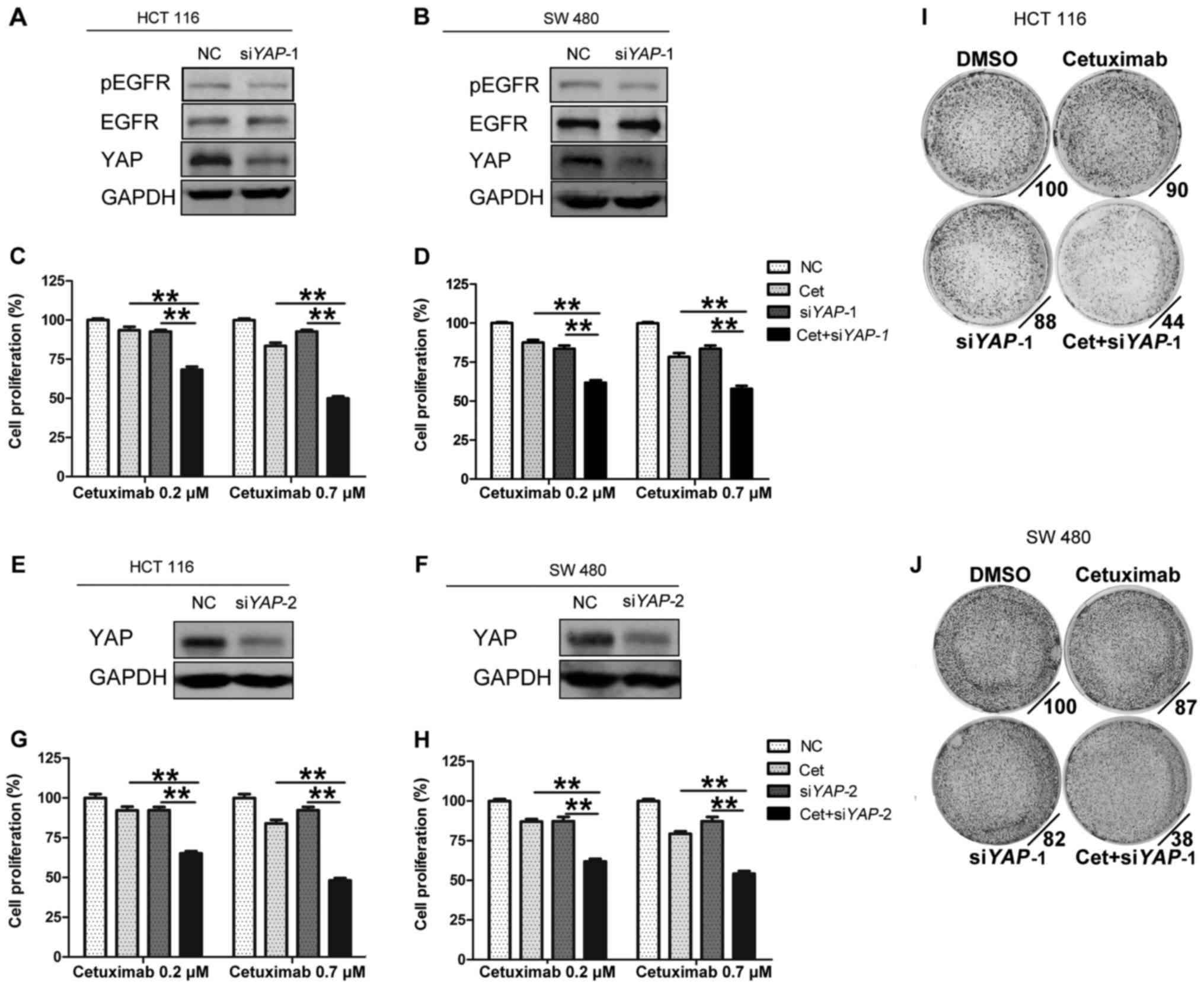 | Figure 2.YAP knockdown enhances the
sensitivity of CRC cell lines to cetuximab. (A, B, E and F) HCT 116
and SW 480 cells were transfected with NC or siYAP (siYAP-1 and
siYAP-2) for 72 h. Immunoblotting analyses were then performed with
the indicated antibodies. (C, D, G and H) Effects of YAP knockdown
on sensitivity to cetuximab in HCT 116 and SW 480 cells. Cell
proliferation was determined with CCK-8 assays following YAP
knockdown for 72 h and cetuximab treatment for 48 h. (I and J)
Colony formation of HCT 116 and SW 480 cells treated with DMSO, 0.2
µM cetuximab, siYAP-1, or a combination of 0.2 µM cetuximab and
siYAP-1 for 7 days in 0.5% FBS. Cells were stained with 0.1%
crystal violet. All the data are presented as the means ± SEM; n=3
biological replicates. One-way ANOVA and post hoc Tukey's test were
performed. **P<0.01. YAP, yes-associated protein; NC, control;
Cet, cetuximab. |
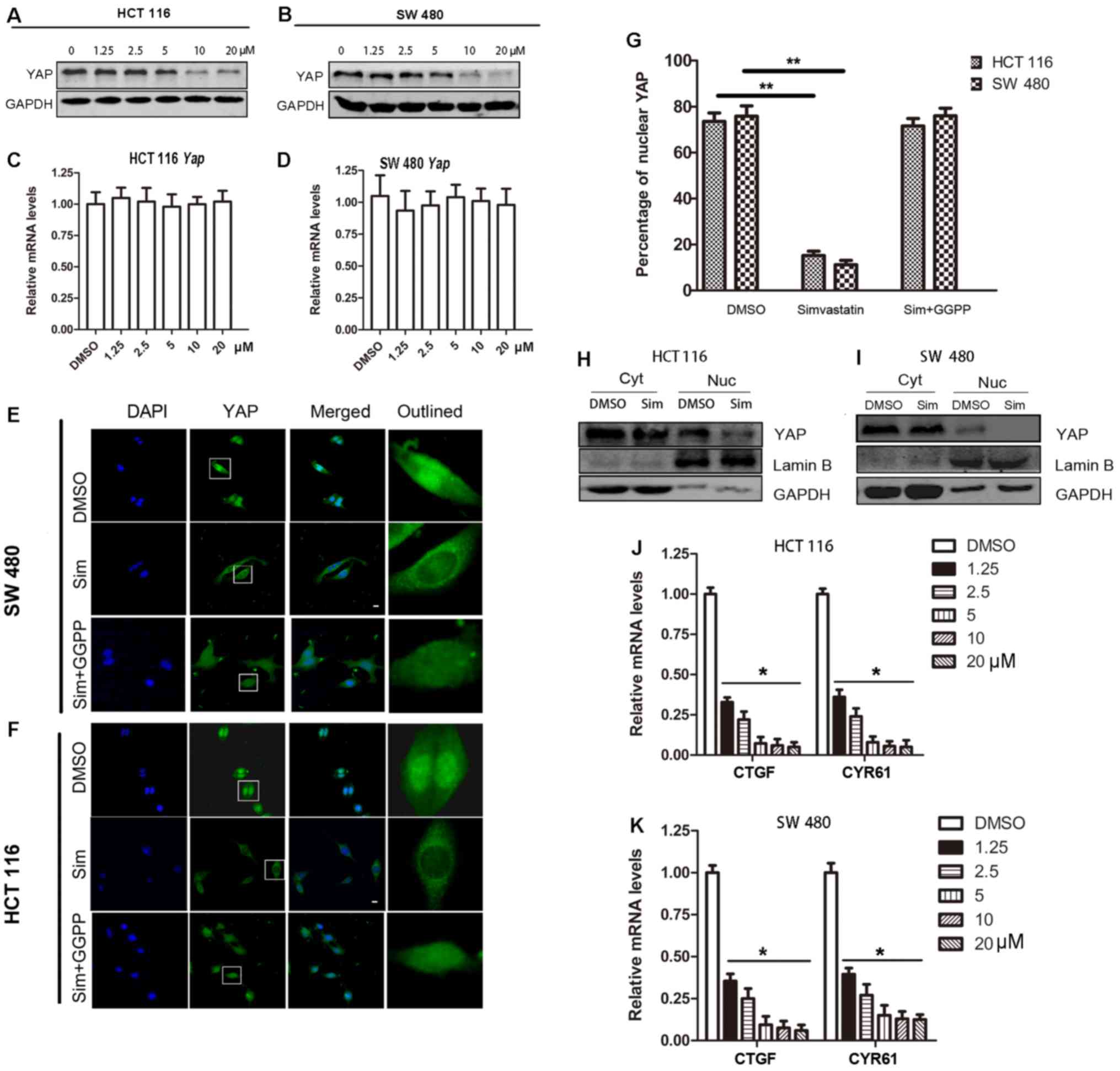
Simvastatin inhibits YAP bioactivity
through total YAP expression and nuclear translocation
Simvastatin, an HMG-CoA reductase inhibitor, is
widely used to lower cellular cholesterol levels in patients with
hypercholesterolemia (10). To
investigate the effects of simvastatin on YAP bioactivity, we first
detected the total YAP protein expression. We observed that
simvastatin treatment caused the total YAP protein level to
decrease in a dose-dependent manner (Fig. 3A and B). Furthermore, real-time PCR
results revealed that simvastatin treatment did not change the
YAP mRNA level (Fig. 3C and
D). YAP subcellular localization is also important for its
bioactivity, as only nuclear YAP can interact with TEAD1-4
transcription factors and execute its function as a transcriptional
co-activator. Thus, we analyzed the effects of simvastatin on YAP
localization via immunofluorescence, and the results indicated that
simvastatin treatment caused a significant decrease in YAP presence
in the nucleus (HCT 116, DMSO vs. simvastatin, mean percentage of
nuclear YAP was 73.6 vs. 15.2%, P<0.01; SW 480, DMSO vs.
simvastatin, mean percentage of nuclear YAP was 75.8 vs. 11.2%,
P<0.01) (Fig. 3E, F and G).
Following the addition of geranylgeranyl pyrophosphate (GGPP), the
effects of simvastatin on nuclear YAP were partly reversed
(Fig. 3E, F and G). Western blot
analysis also revealed that the nuclear YAP level significantly
decreased following simvastatin treatment (Fig. 3H and I). In addition, experiments
revealed that simvastatin inhibited the downstream targets of YAP,
namely, CYR61 and CTGF, at the mRNA level (Fig. 3J and K). Collectively, these results
indicated that simvastatin could act as a YAP inhibitor through the
inhibition of the expression of YAP and nuclear translocation.
Simvastatin increases the antitumor
activity of EGFR inhibitors in CRC cell lines
To investigate the antitumor effects of simvastatin
and (or) cetuximab, cell proliferation was assessed. Compared with
single agents, cetuximab + simvastatin demonstrated more
significant proliferation inhibition (HCT 116, 0.7 µM cetuximab vs.
0.7 µM cetuximab + 3 µM simvastatin, mean relative cell
proliferation rate = 83.4 vs. 61.8%, P<0.01; SW 480, 0.7 µM
cetuximab vs. 0.7 µM cetuximab + 3 µM simvastatin, mean relative
cell proliferation rate = 78.4 vs. 52.5%, P<0.01) (Fig. 4A). Similar results were observed in
COLO 320 and HT 29 cell lines (Fig.
4B). Additionally, colony formation assays demonstrated that
the combination of simvastatin and cetuximab achieved greater
colony inhibition than single agents (Fig. 4C). Subsequently, Hoechst staining
indicated that cetuximab + simvastatin induced apoptosis more
strongly than each agent alone (Fig. 4F
and G). Collectively, these results revealed that simvastatin
increased the cytotoxicity of cetuximab against CRC cells. To
investigate whether simvastatin could enhance the cytotoxicity of
gefitinib, a further combination of simvastatin with gefitinib was
examined, and the results revealed greater cytotoxicity and colony
inhibition compared with each drug alone (HCT 116, 6 µM gefitinib
vs. 6 µM gefitinib + 1 µM simvastatin, mean relative cell
proliferation rate: 40.3 vs. 19.0%, P<0.01; SW 480, 12 µM
gefitinib vs. 12 µM gefitinib + 1 µM simvastatin, mean relative
cell proliferation rate: 48.7 vs. 28.1%, P<0.01) (Fig. 4D and E). These results indicated
that simvastatin could increase the cytotoxicity of EGFR inhibitors
(cetuximab and gefitinib) against CRC cells. To further investigate
the effect of the combined strategy on tumor growth, we implanted
SW 480 cells in nude mice and divided these mice into four groups
(control, cetuximab, simvastatin and cetuximab + simvastatin).
Consistent with the in vitro results, the combination of
cetuximab and simvastatin caused significant inhibition of tumor
growth compared with cetuximab (cetuximab vs. cetuximab +
simvastatin, and the mean tumor volumes [Volume = (L ×
W2)/2, L=length and W=width] were 648.1±143.3
mm3 (mean ± SEM) vs. 310.3±67.7 mm3,
P<0.01) (Fig. 5C and D).
Simvastatin had no obvious impact on the daily movements or the
weights of mice. In addition, we did not observe any severe side
effects in the mice.
 | Figure 4.Simvastatin increases the antitumor
activity of EGFR inhibitors in CRC cells. (A, B and D) Cell
proliferation was determined with CCK-8 assays of CRC cells treated
with the indicated agents (concentration indicated) for 48 h.
One-way ANOVA and post hoc Tukey's tests were performed. (C) Colony
formation of HCT 116 and SW 480 cells treated with DMSO, 0.2 µM
cetuximab, 3 µM simvastatin, or a combination of 0.2 µM cetuximab
and 3 µM simvastatin for 7 days in 0.5% FBS and stained with 0.1%
crystal violet. (E) Colony formation of HCT 116 and SW 480 cells
treated with DMSO, 3 µM (HCT 116) or 6 µM (SW 480) gefitinib, 1 µM
simvastatin, or a combination of gefitinib and 1 µM simvastatin for
5 days in 10% FBS. Cells were stained with 0.1% crystal violet. (F
and G) Cell apoptosis assessed by Hoechst staining of HCT 116 and
SW 480 cells treated with DMSO, 0.2 µM cetuximab, 3 µM simvastatin,
or a combination of 0.2 µM cetuximab and 3 µM simvastatin for 48 h
in 0.5% FBS. Representative immunofluorescence images are shown.
Apoptoticc cells (white arrows). (G) At least 100 cells were scored
in every replicate. All the data are shown as the means ± SEM
except when otherwise indicated; n=3 biological replicates.
*P<0.05, **P<0.01. CRC, colorectal cancer; DMSO, dimethyl
sulfoxide; Cet, cetuximab; Gef, gefitinib; Sim, simvastatin. |
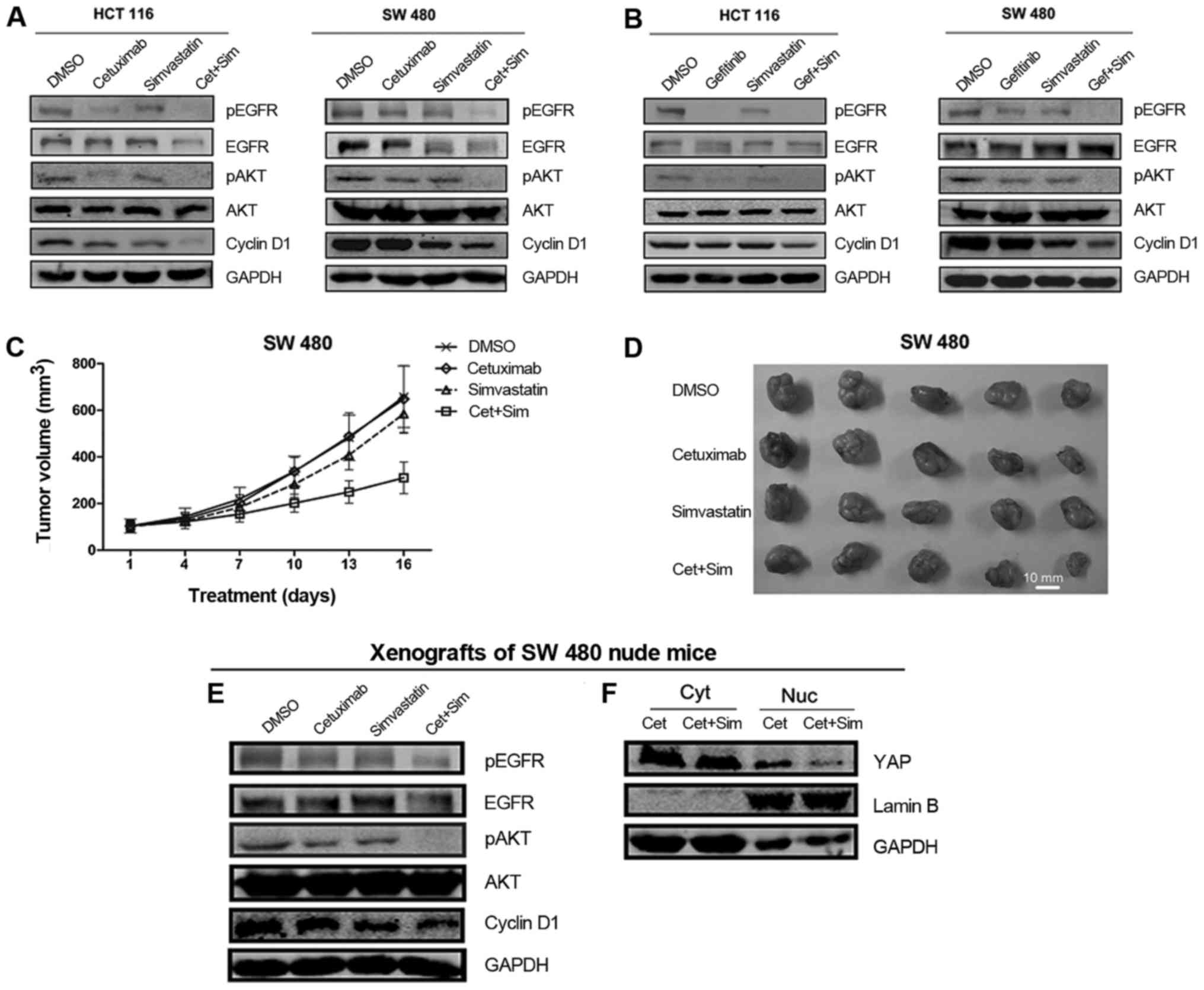 | Figure 5.Simvastatin increases the antitumor
activity of cetuximab and gefitinib. (A and B) Effect of
simvastatin, cetuximab (or gefitinib), or a combination of
simvastatin and cetuximab (or gefitinib) on the EGFR signaling
pathway and cyclin D1 were determined by immunoblotting analysis.
Cells were treated with DMSO, 0.2 µM cetuximab (or 10 µM
gefitinib), 10 µM simvastatin, or a combination of 0.2 µM cetuximab
(or 10 µM gefitinib) and 10 µM simvastatin for 48 h. (C) Graphical
representation of SW 480 cell-derived tumor volumes on different
days following treatment (four groups). The error bars represent
the 95% confidence intervals of the mean volume. Two-tailed
Student's t-test was performed. (D) Representative SW 480 ×enograft
tumors were resected on day 16 after treatment with the indicated
agents. Agent concentrations are shown in the Materials and methods
section. Scale bar, 10 mm. (E) Effects of simvastatin, cetuximab,
or the combination of simvastatin and cetuximab on the EGFR
signaling pathway and cyclin D1 in vivo were determined by
immunoblotting analysis. (F) BALB/c nude mice were treated with
cetuximab or a combination of simvastatin and cetuximab. Levels of
YAP in the cytoplasm or nucleus of SW 480 tumor cells were
determined via immunoblotting analysis. GAPDH and Lamin B were used
as loading controls for the cytoplasm and nucleus, respectively.
All the data are shown as the means ± SEM; n=3 biological
replicates. Cyt, cytoplasm; Nuc, nucleus; Cet, cetuximab; Sim,
simvastatin. |
Effects of simvastatin and EGFR
inhibitors on EGFR signaling and cyclin D1
We then detected the effects of simvastatin and EGFR
inhibitors on EGFR pathway signaling. Western blot analysis
revealed that simvastatin treatment inhibited pEGFR and pAKT. The
combination of cetuximab with simvastatin caused additional
inhibition of pEGFR and pAKT compared with cetuximab alone
(Fig. 5A). Additionally, the
combination of simvastatin and gefitinib revealed a similar trend
in pEGFR and pAKT inhibition (Fig.
5B). Subsequently, we analyzed the expression levels of cyclin
D1, a protein related to proliferation. The results revealed that
the combination of simvastatin and EGFR inhibitors caused an
additional decrease in cyclin D1 compared with single agents
(Fig. 5A and B). Consistent with
the in vitro results, we observed a similar trend in the
expression of pEGFR, pAKT and cyclin D1 in SW 480 ×enograft tumors
(Fig. 5E). Cetuximab + simvastatin
treatment markedly reduced nuclear YAP levels compared with
cetuximab alone in vivo (Fig.
5F).
Simvastatin inhibition of EGFR-AKT
signaling may be mediated through GGPP
To determine the internal mechanism of pAKT
inhibition caused by simvastatin, we hypothesized that simvastatin
could inhibit the expression of pAKT through EGFR, which acts
upstream of AKT and (or) acceleration of phosphatase and tensin
homolog (PTEN), which negatively regulates the PI3K/AKT signaling
pathway. We observed that EGF-activated pEGFR and pAKT signaling
was markedly decreased following simvastatin treatment compared
with the EGF control group, while the expression of PTEN was nearly
unchanged (Fig. 6A and B). GGPP, a
key product of the mevalonate pathway, reversed the decrease in
pEGFR and pAKT caused by simvastatin (Fig. 6C and D, lanes 5 and 8). Since
simvastatin is an inhibitor of the mevalonate pathway and
simvastatin treatment inhibits synthesis of GGPP (9,10),
these results indicated that the simvastatin-induced inhibition of
the EGFR/AKT pathway may be mediated through the mevalonate pathway
product GGPP.
 | Figure 6.Simvastatin inhibits EGF-induced AKT
phosphorylation and YAP activation through EGFR. (A and B)
Simvastatin inhibited EGF-induced AKT phosphorylation through EGFR.
HCT 116 and SW 480 cells were cultured in serum-free medium for 24
h. Then, 10 µM simvastatin was added to treat the cells for 48 h.
Before protein extraction, the cultures were exposed to 30 ng EGF
for 30 min, as indicated. (C and D) Simvastatin inhibited the
EGFR/AKT pathway through GGPP. HCT 116 and SW 480 cells were
cultured in serum-free medium for 24 h. Then, the cells were
treated with 10 µM simvastatin and (or) 10 µM GGPP for 48 h. Before
protein extraction, cultures were exposed to 30 ng EGF for 30 min,
as indicated. (E) EGF promoted YAP activity. SW 48 (left) and HCT
116 (right) cells were cultured in serum-free medium for 24 h.
Subsequently, the cells were treated with 0–30 ng EGF for 2 h. (F)
Combination of cetuximab and simvastatin inhibited EGF-promoted YAP
activity more thoroughly than single agents. SW 48 cells were
cultured in serum-free medium for 24 h. Subsequently, cells were
treated with 10 µM simvastatin and (or) 0.2 µM cetuximab for 48 h.
Prior to protein extraction, cultures were exposed to 30 ng EGF for
30 min, as indicated. n=3 biological replicates. DMSO was used as
the control. GAPDH was used as the loading control. GGPP,
geranylgeranyl pyrophosphate; YAP, yes-associated protein; DMSO,
dimethyl sulfoxide; Sim, simvastatin. |
EGF promotes YAP bioactivity in some
CRC cells
Recent studies have revealed that crosstalk between
the Hippo pathway and other signaling pathways plays a vital role
in carcinogenesis and drug resistance (13,14).
In the present study, we particularly studied the interaction
between YAP, an effector of the hippo pathway, and EGFR in CRC
cells, and the data indicated that YAP knockdown nearly had no
effect on the expression of pEGFR (Fig.
2A and B). Notably, EGF promoted YAP expression and that of its
direct downstream target CYR61 in a dose-dependent manner in SW 48
and HCT 116 cells (Fig. 6E). When
we blocked EGFR signaling with cetuximab, we observed that
cetuximab reversed the EGF-induced increase in YAP and CYR61
(Fig. 6F, lanes 1, 2 and 5).
Furthermore, when we combined cetuximab and simvastatin, YAP and
CYR61 were inhibited more markedly than with cetuximab alone
following EGF activation (Fig. 6F,
lanes 5 and 8). These results indicated that activation of EGFR
signaling can promote YAP signaling in some CRC cells, and a
combination of cetuximab and simvastatin can inhibit YAP signaling
more thoroughly than single treatment.
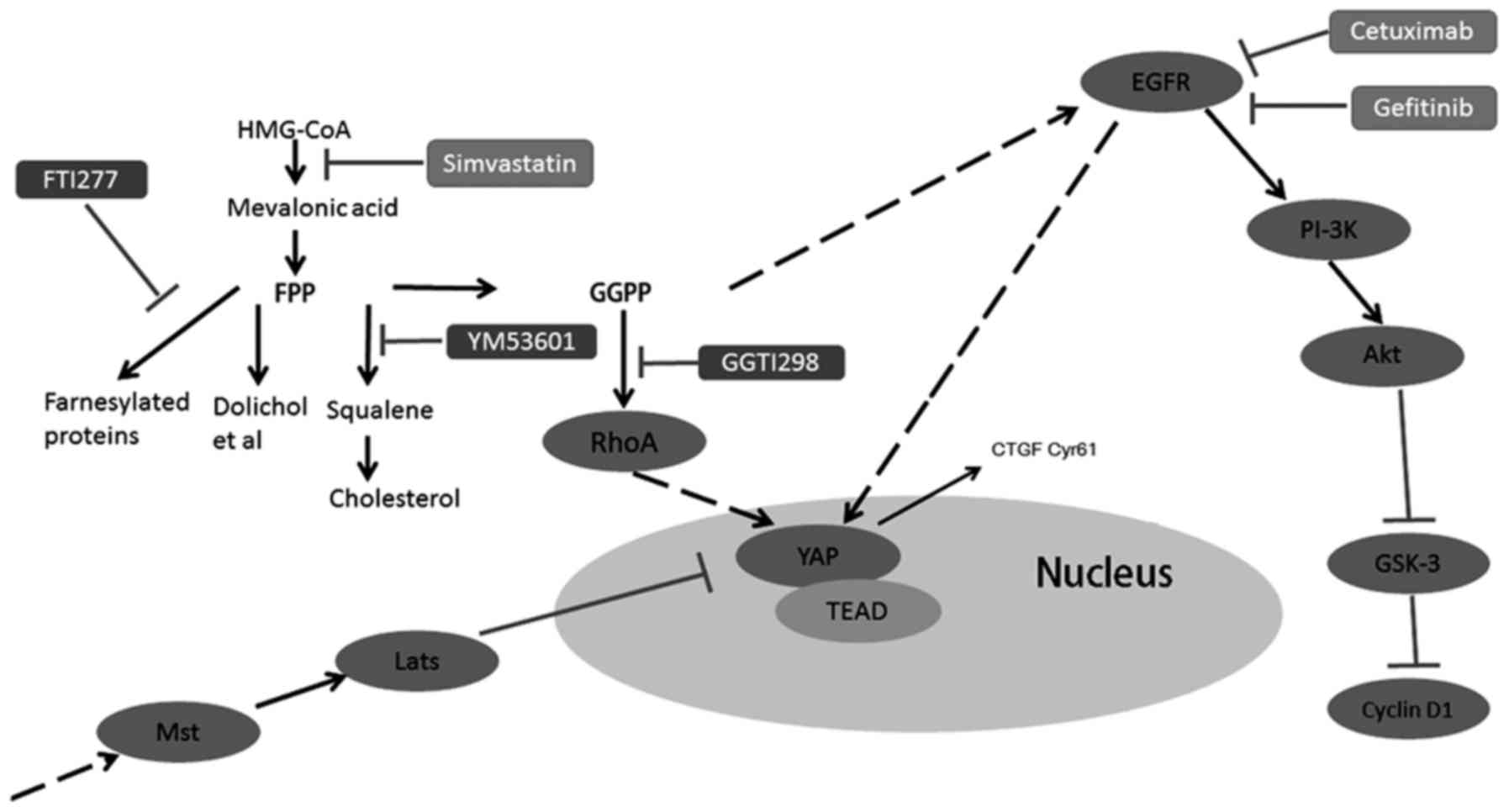
The GGPP pathway mainly mediates the
combined effect of simvastatin and EGFR inhibitors
The mevalonate pathway plays an important role in
the tumorigenesis of many cancers (14). To determine detailed information
about which signaling branch of the mevalonate pathway, primarily
mediates the combined therapy effect, we used three different
inhibitors, FTI277, YM53601 and GGTI298, to inhibit farnesyl
transferase, squalene synthase and geranylgeranyl transferase,
respectively (Fig. 7A). The
combination of FTI277 or YM53601 with EGFR inhibitors had no
significant effect on cell proliferation compared with EGFR
inhibitors alone (Fig. 7C, E, H and
J). Adding back squalene, the product of squalene synthase,
could not reverse the cell proliferation inhibition mediated by
simvastatin and EGFR inhibitors (Fig.
7B, D, G and I), while a combination of GGTI298 and cetuximab
(or gefitinib), achieved approximately the same effect as the
combination of simvastatin and cetuximab (or gefitinib) (Fig. 7C, E, H and J). In addition, after
adding back GGPP, the expression of YAP exhibited no obvious
change, but the expression of CYR61 was markedly increased, which
indicated that YAP bioactivity was recovered (Fig. 7F and K, lanes 4 and 8), consistent
with this, the inhibition of cyclin D1 and cell proliferation
caused by the combination of simvastatin and EGFR inhibitors was
rescued (Fig. 7F and K, lanes 5 and
9; lanes 6 and 10; Fig. 7B and D; G
and I). Collectively, the aforementioned data indicated that the
simvastatin-induced inhibition with EGFR inhibitors was mainly
mediated through the GGPP pathway but not through the farnesyl
pyrophosphate (FPP) pathway or squalene pathway (Fig. 7A).
 | Figure 7.The GGPP pathway mainly mediates the
combined effect of simvastatin and EGFR inhibitors. (A) Overview of
the mevalonate pathway. (B, C, G and H) Cell proliferation was
determined with CCK-8 assays of HCT 116 and SW 480 cells treated
with the indicated agents for 48 h in 0.5% FBS. The concentrations
of agents were as follows: cetuximab 0.7 µM, simvastatin 3 µM, GGPP
10 µM, squalene 10 µM, GGTI298 5 µM, FTI277 5 µM, and YM53601 5 µM.
(D, E, I and J) Cell proliferation was determined with CCK-8 assays
of HCT 116 and SW 480 cells treated with the indicated agents for
48 h in 10% FBS. The concentrations of the agents were as follows:
Gefitinib (HCT 116) 6 µM or (SW 480) 12 µM, simvastatin 1 µM, GGPP
10 µM, squalene 10 µM, GGTI298 5 µM, FTI277 5 µM, and YM53601 5 µM.
(F and K) Cells were lysed and analyzed by immunoblotting as
indicated. HCT 116 and SW 480 were treated with the indicated
agents for 48 h. The concentrations of the agents were as follows:
Simvastatin 10 µM, cetuximab 0.7 µM, gefitinib (HCT 116) 6 µM or
(SW 480) 12 µM, GGPP 10 µM. DMSO served as the control. All the
data are displayed as the means ± SEM; n=3 biological replicates.
The two-tailed Student's t-test was used. **P<0.01. Cet,
cetuximab; Gef, gefitinib; Sim, simvastatin; Squ, squalene; DMSO,
dimethyl sulfoxide. |
Discussion
Resistance to EGFR inhibitors, such as cetuximab and
gefitinib, has become an urgent issue for both basic science and
clinical investigators (15–17).
Driver genes, such as mutant KRAS, BRAF, PTEN and PIK3CA, are
closely related to this resistance. In the present study, we
observed that YAP may be useful in identifying cetuximab resistance
in CRC cells. At present, the reported YAP inhibitors include
verteporfin (18), statins and
zoledronic acid (9,10). In the present study, we primarily
used simvastatin as a YAP inhibitor. Simvastatin could not only
inhibit total YAP protein expression but it also inhibited YAP
translocation into the nucleus. However, simvastatin did not
inhibit YAP mRNA levels, indicating that simvastatin
promoted YAP protein degradation at the post-transcriptional level.
Several studies have demonstrated that statins mainly inhibit YAP
nuclear translocation to decrease its bioactivity, while the total
YAP protein expression is not affected (9,19). We
thought this discrepancy may be due to differences in the cell
types used in each study.
The in vitro and in vivo study results
demonstrated that the combination of simvastatin and EGFR
inhibitors caused synthetic inhibition of cell proliferation. These
results were consistent to the findings of Lee et al but did
not reveal the function of YAP in cetuximab resistance (20). Compared with the cetuximab group,
cetuximab + simvastatin markedly decreased the nuclear YAP levels
in vivo. Furthermore, both pEGFR and pAKT were inhibited
more markedly with the combination treatment than with single
agents in vitro and in vivo. The aforementioned
results were consistent with the cell proliferation and xenograft
tumor growth assay results (Figs. 4
and 5).
Simvastatin induced pAKT downregulation in the
present study. We hypothesized that this decrease may be caused by
EGFR and (or) PTEN. In addition, our investigation revealed that
simvastatin inhibited pEGFR but not PTEN, which indicated that
simvastatin-induced AKT signaling was downregulated through EGFR.
While EGF caused pEGFR and pAKT upregulation, this could be
reversed by simvastatin, which verified this hypothesis.
Furthermore, addition of GGPP reversed simvastatin-induced EGFR-AKT
signal downregulation, revealing that simvastatin-mediated
inhibition of EGFR-AKT signaling may occur through GGPP.
Crosstalk between the Hippo signaling and other
pathway signals is involved in tumorigenesis, cancer progression
and drug resistance (21–24). In the present study, YAP knockdown
had nearly no effect on EGFR signaling, while EGF treatment
promoted the activation of YAP and CYR61. These results could
partly explain some epidemiological studies, which reported that no
benefit was observed in simvastatin users for cancer therapy
(25,26), and the reason may be inadequate
inhibition of YAP. In addition to EGFR signaling, YAP is also
regulated by other signals, such as RHOA, LATS and G
protein-coupled receptors (10,27).
Therefore, it is difficult to fully inhibit YAP bioactivity, and
multiple inhibition approaches may be an effective and feasible
method, as demonstrated in our study (Fig. 6F, lanes 5, 6 and 8).
Recent studies have revealed that the antitumor
effects of simvastatin are controversial (25,26,28–30).
As an HMG-CoA reductase inhibitor, simvastatin can inhibit the
mevalonate pathway specifically. The mevalonate pathway is involved
in biosynthesis of squalene, GGPP and farnesyl pyrophosphate (FPP)
(31). Squalene is an upstream
product of cholesterol synthesis, and GGPP and FPP are critical for
prenylation of small G proteins, such as those in the RAS, RHO and
RAB families (Fig. 7A). In the
present study, GGPP rescued CYR61 expression inhibited by
simvastatin, but did not markedly alter YAP expression. The
immunofluorescence analysis results revealed that GGPP partly
reversed the effects of simvastatin on nuclear YAP levels. This may
be because GGPP rescued YAP bioactivity primarily through YAP
protein transportation to the nucleus and not by increasing YAP
protein expression. Consistent with this, GGPP reversed the
synthetic cell proliferation inhibition caused by simvastatin and
the EGFR inhibitors. All of the aforementioned results revealed the
effect of YAP bioactivity on the resistance to EGFR inhibitors.
This was in accordance with the study of Lee et al, in which
a significant association between the oncogene YAP1 and cetuximab
resistance was reported (8).
However, their analysis was mainly focused on clinical patients. In
addition, combined treatment with GGTI298 and EGFR inhibitors
caused proliferation inhibition similar to that of the combination
of EGFR inhibitors and simvastatin. However, combing FTI277 (or
YM53601) and EGFR inhibitors had little effect compared with EGFR
inhibitors alone. These results demonstrated that the GGPP pathway
mainly mediated the combined effect of simvastatin and EGFR
inhibitors (Fig. 8).
Notably, the concentration of simvastatin used in
vitro was much higher (>10-fold) than that typically used
for lowering cholesterol, a dosage of approximately 0.2–1.5
mg/kg/day and thus, toxic effects at such concentrations should be
evaluated clinically.
In the present study, we found that YAP knockdown
enhanced the sensitivity of CRC cells to cetuximab, and inhibition
of YAP bioactivity by simvastatin increased the cytotoxicity of
cetuximab and gefitinib. The combination of simvastatin and EGFR
inhibitors synthetically inhibited YAP and EGFR signals. Our
findings revealed a new promising therapeutic strategy to enhance
the efficacy of CRC treatment through combined YAP and EGFR
targeting (Fig. 8) and indicated
that further studies of YAP inhibitors and the effects of YAP on
cetuximab resistance should be performed.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Key
R&D Program of China (grant no. 2016YFC1303200/2016YFC1303203),
the National Natural Science Foundation of China (grant no.
81621003) and the National Natural Science Foundation for Young
Scholars of China (grant no. 81502655).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
BSL and HWX designed and performed the experiments,
analyzed the data and prepared the manuscript. SZ, QL and NXB
contributed to studies related to Hoechst 33342 staining, real-time
PCR and in vivo experiments. QLT and JTZ conducted
immunoblotting to examine the EGFR pathway and colony formation
assays. QYG and YZN analyzed the data and contributed to the
manuscript. FB supervised and designed the experiments, analyzed
the data and prepared the manuscript. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
All animal experimental procedures used in the
present study were approved by the Experimental Animal Manage
Committee of West China Hospital, Sichuan University under contract
2016003A and were performed strictly according to the guidelines of
the Animal Ethics Committee Guidelines of the Animal Facility of
West China Hospital and the Animal Care and Use Committee of
Sichuan University (Chengdu, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
NXB is a volunteer student from Chengdu
International School (Chengdu, Sichuan).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Goldberg RM, Sargent DJ, Morton RF, Fuchs
CS, Ramanathan RK, Williamson SK, Findlay BP, Pitot HC and Alberts
SR: A randomized controlled trial of fluorouracil plus leucovorin,
irinotecan, and oxaliplatin combinations in patients with
previously untreated metastatic colorectal cancer. J Clin Oncol.
22:23–30. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Heinemann V, von Weikersthal LF, Decker T,
Kiani A, Vehling-Kaiser U, Al-Batran SE, Heintges T, Lerchenmuller
C, Kahl C, Seipelt G, et al: FOLFIRI plus cetuximab versus FOLFIRI
plus bevacizumab as first-line treatment for patients with
metastatic colorectal cancer (FIRE-3): A randomised, open-label,
phase 3 trial. Lancet Oncol. 15:1065–1075. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stintzing S, Modest DP, Rossius L, Lerch
MM, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U,
Al-Batran SE, Heintges T, et al: FOLFIRI plus cetuximab versus
FOLFIRI plus bevacizumab for metastatic colorectal cancer (FIRE-3):
A post-hoc analysis of tumour dynamics in the final RAS wild-type
subgroup of this randomised open-label phase 3 trial. Lancet Oncol.
17:1426–1434. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Camargo FD, Gokhale S, Johnnidis JB, Fu D,
Bell GW, Jaenisch R and Brummelkamp TR: YAP1 increases organ size
and expands undifferentiated progenitor cells. Curr Biol.
17:2054–2060. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu FX, Meng Z, Plouffe SW and Guan KL:
Hippo pathway regulation of gastrointestinal tissues. Annu Rev
Physiol. 77:201–227. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee KW, Lee SS, Kim SB, Sohn BH, Lee HS,
Jang HJ, Park YY, Kopetz S, Kim SS, Oh SC, et al: Significant
association of oncogene YAP1 with poor prognosis and cetuximab
resistance in colorectal cancer patients. Clin Cancer Res.
21:357–364. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Z, Wu Y, Wang H, Zhang Y, Mei L, Fang
X, Zhang X, Zhang F, Chen H, Liu Y, et al: Interplay of mevalonate
and hippo pathways regulates RHAMM transcription via YAP to
modulate breast cancer cell motility. Proc Natl Acad Sci USA.
111:E89–E98. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sorrentino G, Ruggeri N, Specchia V,
Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio
R, Piazza S, et al: Metabolic control of YAP and TAZ by the
mevalonate pathway. Nat Cell Biol. 16:357–366. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma J, Xue Y, Liu W, Yue C, Bi F, Xu J,
Zhang J, Li Y, Zhong C and Chen Y: Role of activated Rac1/Cdc42 in
mediating endothelial cell proliferation and tumor angiogenesis in
breast cancer. PLoS One. 8:e662752013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Van Cutsem E, Kohne CH, Hitre E, Zaluski
J, Chien Chang CR, Makhson A, D'Haens G, Pinter T, Lim R, Bodoky G,
et al: Cetuximab and chemotherapy as initial treatment for
metastatic colorectal cancer. N Engl J Med. 360:1408–1417. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mullen PJ, Yu R, Longo J, Archer MC and
Penn LZ: The interplay between cell signalling and the mevalonate
pathway in cancer. Nat Rev Cancer. 16:718–731. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yen LC, Uen YH, Wu DC, Lu CY, Yu FJ, Wu
IC, Lin SR and Wang JY: Activating KRAS mutations and
overexpression of epidermal growth factor receptor as independent
predictors in metastatic colorectal cancer patients treated with
cetuximab. Ann Surg. 251:254–260. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
De Roock W, Claes B, Bernasconi D, De
Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V,
Papamichael D, Laurent-Puig P, et al: Effects of KRAS, BRAF,
NRAS, and PIK3CA mutations on the efficacy of cetuximab
plus chemotherapy in chemotherapy-refractory metastatic colorectal
cancer: A retrospective consortium analysis. Lancet Oncol.
11:753–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chiu CF, Chang YW, Kuo KT, Shen YS, Liu
CY, Yu YH, Cheng CC, Lee KY, Chen FC, Hsu MK, et al: NF-κB-driven
suppression of FOXO3a contributes to EGFR mutation-independent
gefitinib resistance. Proc Natl Acad Sci USA. 113:E2526–E2535.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng
Z, Zhao L, Peyman G, Ouyang H, Jiang W, et al: Mutant Gq/11 promote
uveal melanoma tumorigenesis by activating YAP. Cancer Cell.
25:822–830. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang KC, Yeh YT, Nguyen P, Limqueco E,
Lopez J, Thorossian S, Guan KL, Li YJ and Chien S: Flow-dependent
YAP/TAZ activities regulate endothelial phenotypes and
atherosclerosis. Proc Natl Acad Sci USA. 113:11525–11530. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee J, Lee I, Han B, Park JO, Jang J, Park
C and Kang WK: Effect of simvastatin on cetuximab resistance in
human colorectal cancer with KRAS mutations. J Natl Cancer Inst.
103:674–688. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hong X, Nguyen HT, Chen Q, Zhang R, Hagman
Z, Voorhoeve PM and Cohen SM: Opposing activities of the Ras and
Hippo pathways converge on regulation of YAP protein turnover. EMBO
J. 33:2447–2457. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Song S, Honjo S, Jin J, Chang SS, Scott
AW, Chen Q, Kalhor N, Correa AM, Hofstetter WL, Albarracin CT, et
al: The hippo coactivator YAP1 mediates EGFR overexpression and
confers chemoresistance in esophageal cancer. Clin Cancer Res.
21:2580–2590. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
He C, Lv X, Hua G, Lele SM, Remmenga S,
Dong J, Davis JS and Wang C: YAP forms autocrine loops with the
ERBB pathway to regulate ovarian cancer initiation and progression.
Oncogene. 34:6040–6054. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
He C, Mao D, Hua G, Lv X, Chen X,
Angeletti PC, Dong J, Remmenga SW, Rodabaugh KJ, Zhou J, et al: The
Hippo/YAP pathway interacts with EGFR signaling and HPV
oncoproteins to regulate cervical cancer progression. EMBO Mol Med.
7:1426–1449. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pocobelli G, Newcomb PA, Trentham-Dietz A,
Titus-Ernstoff L, Hampton JM and Egan KM: Statin use and risk of
breast cancer. Cancer. 112:27–33. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Freeman SR, Drake AL, Heilig LF, Graber M,
McNealy K, Schilling LM and Dellavalle RP: Statins, fibrates, and
melanoma risk: A systematic review and meta-analysis. J Natl Cancer
Inst. 98:1538–1546. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Y, Xia H, Ge X, Chen Q, Yuan D, Chen
Q, Leng W, Chen L, Tang Q and Bi F: CD44 acts through RhoA to
regulate YAP signaling. Cell Signal. 26:2504–2513. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Farwell WR, Scranton RE, Lawler EV, Lew
RA, Brophy MT, Fiore LD and Gaziano JM: The association between
statins and cancer incidence in a veterans population. J Natl
Cancer Inst. 100:134–139. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cauley JA, McTiernan A, Rodabough RJ,
LaCroix A, Bauer DC, Margolis KL, Paskett ED, Vitolins MZ, Furberg
CD and Chlebowski RT: Women's Health Initiative Research Group:
Statin use and breast cancer: Prospective results from the Women's
Health Initiative. J Natl Cancer Inst. 98:700–707. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee J, Hong YS, Hong JY, Han SW, Kim TW,
Kang HJ, Kim TY, Kim KP, Kim SH, Do IG, et al: Effect of
simvastatin plus cetuximab/irinotecan for KRAS mutant
colorectal cancer and predictive value of the RAS signature
for treatment response to cetuximab. Invest New Drugs. 32:535–541.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Goldstein JL and Brown MS: Regulation of
the mevalonate pathway. Nature. 343:425–430. 1990. View Article : Google Scholar : PubMed/NCBI
|