Introduction
Erastin is a voltage-dependent anion channel
(VDAC)-binding small molecule and exerts cytotoxic effects on
several selective cancer cells (1,2).
Erastin exposure induces the accumulation of reactive oxygen
species (ROS) in an iron-dependent manner. The direct binding of
erastin to voltage-dependent anion-selective channel protein 2
(VDAC2) and 3 (VDAC3) is necessary to induce ferroptosis, which
involves a unique constellation of morphological, biochemical and
genetic features and is distinct from apoptosis, various forms of
necrosis and autophagy (3). Erastin
also exerts cytotoxic effects on several human cancer cell lines by
inducing oxidative stress and caspase-9-dependent apoptotic death
(4), indicating that erastin
potentially induces ferroptotic and apoptotic cell death.
Being the most well-known tumour suppressor, p53
exerts multi-functional roles in controlling cell cycle
checkpoints, apoptosis and DNA repair (5). In addition to these commonly accepted
functions mediated by activated p53, accumulating evidence
indicates that other activities of p53 are also involved in tumour
suppression, including ferroptosis. It was reported that p53
post-transcriptionally inhibited the expression of SLC7A11, a key
component of the cysteine/glutamate antiporter, leading to
inhibition of cysteine uptake and sensitization of cells to
ferroptosis (6). The suppression of
SLC7A11 by p53 subsequently leads to the reduction of glutathione
production and ROS accumulation, which are important components of
ferroptosis (6). Gao et al
identified GLS2, a p53-regulated glutaminase, as essential for
ferroptosis (7). All of these
studies support the potential relevance of p53 to ferroptosis and
the tumour-suppressing activity of p53 via the regulation of
ferroptosis. Thus, we investigated the potential mechanism
underlying how p53 participates in the regulation of
erastin-induced ferroptosis.
Erastin-induced ferroptotic cell death is dependent
on the increased level of intracellular reactive oxygen species
(ROS), which are widely believed to act as a mediator of apoptosis.
Tsai et al (8) found that,
in A549 non-small cell lung cancer cells, the activation of
p53-dependent apoptotic proteins, including PUMA, cytochrome
c, Apaf-1 and caspase-3, was dependent on the presence of
ROS. Accumulated ROS were also reported to regulate the expression
and activation of p53, and the ROS/p53 pathway was found to
regulate several cellular physiological processes, including cell
senescence (9), oxidative
protection (10) and apoptosis
(11). In erastin-induced
ferroptotic death, p53 was activated as a post-transcriptional
suppressor of SLC7A11 and induced ROS generation (6). By considering that DNA damage induced
by accumulated ROS activates p53 (8), the question was raised whether
induction of ROS by erastin exposure was attributed to p53
activity.
In the present study, we investigated the regulatory
effects of ROS induced by erastin exposure on p53 expression in
A549 cells, and we studied the effects of the activation of p53 on
cell proliferation and apoptosis. In the present study, we revealed
that the markedly increased expression of p53 in A549 cells
following erastin exposure was partially dependent on the
accumulation of ROS. We concluded that the ROS/p53 pathway
activated by erastin exposure exerted cytotoxic and cytostatic
effects on A549 cells via both ferroptosis and apoptosis.
Materials and methods
Cell culture and treatment
The human adenocarcinoma A549 and lung fibroblast
WI-38 cell lines were purchased from the American Type Culture
Collection (ATCC; Manassas, VA, USA) and maintained in Gibco™
Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific,
Inc., Paisley, UK) supplemented with 10% fetal bovine serum (FBS),
100 U/ml of penicillin and 0.1 mg/ml of streptomycin (all from
Thermo Fisher Scientific, Inc.). For different analyzing purposes,
A549 cells was treated with 3.12 µM erastin (Sigma-Aldrich, St.
Louis, MO, USA) for 24 h, NAC (Sigma-Aldrich) 5 mM for 4 h or
KU-55933 (Selleck Chemicals, Shanghai, China) 15 nM for 24 h. Then
cells were harvested as described below for further analysis.
Western blotting
Cells were harvested and pelleted by centrifugation
at 1,000 × g and 4°C for 10 min and washed with ice-cold PBS twice.
Lysis buffer (100 µl) containing 50 mM Tris-HCl, 150 mM NaCl, 0.02%
NaN3, 100 µg/ml phenylmethanesulfonyl fluoride (PMSF), 1
µg/ml aprotinin, 1 µg/ml pepstatin A, and 1% Triton X-100 was each
added into 1×106 cells for obtaining the cell lysate.
After centrifugation at 12,000 × g for 10 min at 4°C, the
supernatant was collected and assessment of protein concentration
was carried out using a bicinchoninic acid (BCA) protein assay kit
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Protein (50 µg)
was resolved by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and then transferred to a nitrocellulose
membrane. After transferring, the membrane was blocked using 5%
bovine serum albumin (BSA) in PBS for 30 min at room temperature.
The primary antibodies against p53 (1:5,000 dilution; cat. no.
ab28), phosphorylated p53 (1:2,000 dilution; cat. no. ab1431), p21
(1:1,000 dilution; cat. no. ab109520), Bax (1:2,000 dilution; cat.
no. ab32503), SLC7A11 (1:1,000 dilution; cat. no. ab37185) and
β-actin (1:5,000 dilution; cat. no. ab8226) were purchased from
Abcam (Cambridge, UK) and incubated with the blocked membrane
separately for 2 h at room temperature. After being washed 3 times
for 5 min each with PBS-T, the membrane was incubated for 1 h with
peroxidase-coupled secondary antibodies (1:5,000 dilution; cat.
nos. ab6785 or ab6721), and detected with the ECL Plus Western
Blotting Detection reagents (Pierce Biotechnology, Rockford, IL,
USA) and imaged using X-ray film.
CFSE/PI double staining
A549 cells were washed in phosphate-buffered saline
(PBS) twice and 100 µl of CFSE fluorescent dye (50 µmol/l; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) was added, followed by
incubation at 37°C for 30 min. Cells were washed in PBS twice and
incubated in medium supplemented with 10% FBS for another 24-h
incubation. After two washes with PBS, PI (20 µg/ml; Sigma-Aldrich)
was added for incubation at room temperature for 10 min. After two
washes with PBS, cells were imaged under a X71 (U-RFL-T)
fluorescence microscope (Olympus, Melville, NY, USA).
EdU staining
Cells were plated in 12-well plate and allowed to
attach overnight. In addition, 5-ethyny-2′-deoxyuridine (EdU)
(Cell-Light EdU Cell Proliferation Detection kit; Guangzhou RiboBio
Co., Ltd., Guangzhou, China) was used as a marker of cell
proliferation. EdU was added at a final concentration of 50 µmol/l
into the medium and the cells were cultured for an additional 120
min. Cells were washed twice with PBS and fixed with 4%
paraformaldehyde at room temperature for 10 min, washed with
glycine (2 mg/ml) for 5 min in a shaker, treated with 0.2% Triton
X-100 for 10 min and washed with PBS twice. Click-iT®
Cell Reaction Buffer kit (Thermo Fisher Scientific, Inc.) was added
for further incubation for 30 min. Then the cells were washed with
0.5% Triton X-100 for three times, stained with DAPI for 10 min at
room temperature, washed with 0.5% Triton X-100 for three times,
immersed in 150 µl of PBS and examined under a X71 (U-RFL-T)
fluorescence microscope (Olympus).
Cell viability assay
A549 cells were suspended and adjusted to
1×106 cells/ml and 5,000 cells/well were plated into a
96-well plate and incubated overnight. Various concentrations of
erastin (0.1, 1, 2, 4, 6, 8 and 10 µM) were added into each well.
Twenty-four hours later, the Cell Counting Kit-8 (CCK-8,
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) prepared solution
was added for a 4-h co-incubation at 37°C in the dark. Absorbance
at wavelength 450 nm (A450) was detected by a microplate reader
(Synergy 2 Multi-Mode Microplate Reader; BioTek, Winooski, VT, USA)
to determine the cell viability.
Assessment of ROS
Cells were co-incubated with [5-(and
6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate]
(carboxy-H2DCFDA; Thermo Fisher Scientific, Inc.) at a
final concentration of 5 µmol/l for 30 min. Subsequently the medium
was removed and washed twice with PBS. The green fluorescence was
imaged using a X71 (U-RFL-T) fluorescence microscope (Olympus). For
quantitative measurement, stained cells were suspended using 0.25%
trypsin (Gibco; Thermo Fisher Scientific, Inc.) and the green
fluorescence was assessed using a 3-laser Navios flow cytometer
(Beckman Coulter Inc., Brea, CA, USA).
RNA interference
Invitrogen™ Oligofectamine transfection reagent
(Thermo Fisher Scientific, Inc.) was used for transfecting 50 nM
p53 siRNA (p53si), SLC7A11 siRNA (SLC7A11si) or negative control
siRNA (CTLsi) purchased from Ambion (Life Technologies; Thermo
Fischer Scientific, Inc.) into A549 cells according to the
manufacturer's instructions and then after 24 h, the cells were
used for subsequent experiments.
Immunofluorescence assay
Cells were washed with ice-cold PBS for 5 min, 3
times and fixed with ice-cold alcohol for 15 min at 4°C.
Subsequently, the cells were blocked with 1% BSA, 0.1% Triton X-100
in PBS for 1 h at room temperature. Then the cells were incubated
with anti-γ H2AX antibody (cat. no. ab26350; Abcam) at a 1:200
dilution for 2 h at room temperature. After 3 washes with ice-cold
PBS, the cells were incubated with Alexa Fluor®
647-conjugated donkey anti-mouse IgG secondary antibody (cat. no.
ab150107; Abcam) at a 1:2,000 dilution for 1 h at room temperature.
After 3 washes with ice-cold PBS, the cells were incubated with 5
µg/ml DAPI for 10 min. The slices were analyzed under a X71
(U-RFL-T) fluorescence microscope (Olympus) at a magnification of
×200.
Cell cycle analysis
Cells (1×106) were collected and fixed in
70% ice-cold ethanol at −20°C overnight. Then the cells were
collected and resuspended in PBS supplemented with 100 ng/ml RNase
A and 50 ng/ml propidium iodide (PI) for 30 min. After staining,
samples were analyzed for cell cycle distribution with a 3-laser
Navios flow cytometer (Beckman Coulter). Experiments with
duplicates were performed independently thrice.
Assessment of caspase-3 activity
A caspase-3 colorimetric assay kit (R&D Systems,
Minneapolis, MN, USA) was used to detect the enzyme activity of
caspase-3. The total cell lysate was qualified by the BCA protein
assay kit (Sigma-Aldrich; Merck KGaA). Total protein (50 µg) for
each sample was moved to a 96-well microplate and quantified using
a microplate reader (Synergy 2 Multi-Mode Microplate Reader;
BioTek).
Cell apoptosis
Cells (5×105) were collected and
co-incubated with 5 µl Annexin V-fluorescein isothiocyanate
(Annexin V-FITC) and 10 µl PI supplied by an Annexin V/PI apoptosis
kit (BioVision, San Francisco, CA, USA) for 10 min in the dark.
After staining, the ells were subjected to flow cytometric analysis
(3-laser Navios flow cytometer; Beckman Coulter).
Statistical analysis
All data are presented as the mean ± SD. Statistical
differences among different groups were analyzed by one way
analysis of variance (ANOVA) with Dunnett's post hoc test using
Prism 6 (GraphPad Software, Inc., San Diego, CA, USA). A value of
P<0.05 or P<0.01 was considered to indicate a statistically
significant difference.
Results
ROS upregulate and activate p53 in
response to erastin exposure
To ascertain whether erastin exerts a cytotoxic
effect and increases ROS in A549 cells and the non-tumour cell line
WI-38, both cell types were treated with increasing concentrations
of erastin (0.1–10 µM) and were subjected to the CCK-8 assay 24 h
later to assess cell viability. As displayed in Fig. 1A, exposure to 0–10 µM erastin for 24
h significantly decreased A549 cell viability in the A549 cells,
but not that of WI-38 cells. Erastin has been reported to play a
critical role in inducing ferroptosis via ROS accumulation
(3). We quantified fluorescence in
erastin-treated (IC30 concentration for A549 cells, 3.12
µM) A549 or WI-38 cells stained with a redox-sensitive probe.
Consistent with previous literature (6), erastin treatment increased the ROS
level in A549 cells but not that in WI-38 cells (Fig. 1B), indicating that NSCLC A549 cells
are much more sensitive than non-tumour WI-38 lung cells. Thus,
subsequent experiments were focused on the effects of erastin
treatment on A549 cells. To determine the role of ROS induced by
erastin exposure on p53, the expression levels of p53 protein and
its transcriptional targets Bax and p21 were detected. As displayed
in Fig. 1C, the upregulation and
activation of p53 were evidenced by upregulated p53, Bax and p21.
Following pretreatment with N-acetyl-1-cysteine (NAC), an ROS
scavenger, erastin exposure failed to significantly affect p53
activation, indicating that activation was dependent on the
presence of ROS induced by erastin exposure (Fig. 1C, right panel). According to the
literature, activated p53 transcriptionally suppresses SLC7A11, a
key component of the cysteine/glutamate antiporter, thus promoting
ferroptosis induced by erastin exposure (12). This prompted us to detect the
expression of SLC7A11 in A549 cells with or without p53 knockdown
after erastin exposure. In the CTLsi group, with the upregulation
of p53 induced by erastin treatment, SLC7A11 was obviously
downregulated (Fig. 1D). While
there was no obvious effect of erastin treatment on the expression
of SLC7A11 in A549 cells with p53 knockdown, progressive
suppression of the SLC7A11 protein level was observed in A549 cells
exposed to erastin (Fig. 1D). These
results indicated that ROS induced by erastin potentially
contributed to ferroptosis induction via activating p53
transcriptional activity.
Erastin-induced ROS lead to the DNA
damage response and stimulate p53 in A549 cells
By considering the effect of accumulated ROS on the
DNA damage response (DDR) (13,14),
we then tested whether erastin-induced ROS caused DDR in A549
cells. As expected, erastin exposure produced some typical γ-H2AX
positive-stained cells. Conversely, co-exposure of erastin with NAC
only produced few γ-H2AX positive-stained cells (Fig. 2A). Without disturbing p53 mRNA
levels (data not shown), the activation of p53 by erastin was
abolished by NAC co-incubation (Fig.
2B). DDR is responsible for inducing p53
post-transcriptionally, which is phosphorylated by activated ataxia
telangiectasia mutated (ATM) kinase (15), and prompted us to determine whether
ROS induction of p53 is dependent on DDR and subsequent activation
of phosphorylation. Thus, KU-55933, a specific ATM inhibitor, was
employed to inhibit the phosphorylation of p53 at S15 by
DDR-activated ATM, and then the effects of accumulated ROS on p53
were detected. As displayed in Fig. 2C
and D, NAC treatment, but not KU-55933, induced DDR, and both
NAC and KU-55933 treatment clearly inhibited p53 phosphorylation
following erastin exposure, indicating that the activating effects
of erastin-induced ROS on p53 were exerted via ATM kinetic activity
after causing DDR (Fig. 2E).
Expression of p53 increases
erastin-induced ROS generation partially dependent on decreasing
the expression of SCL7A11
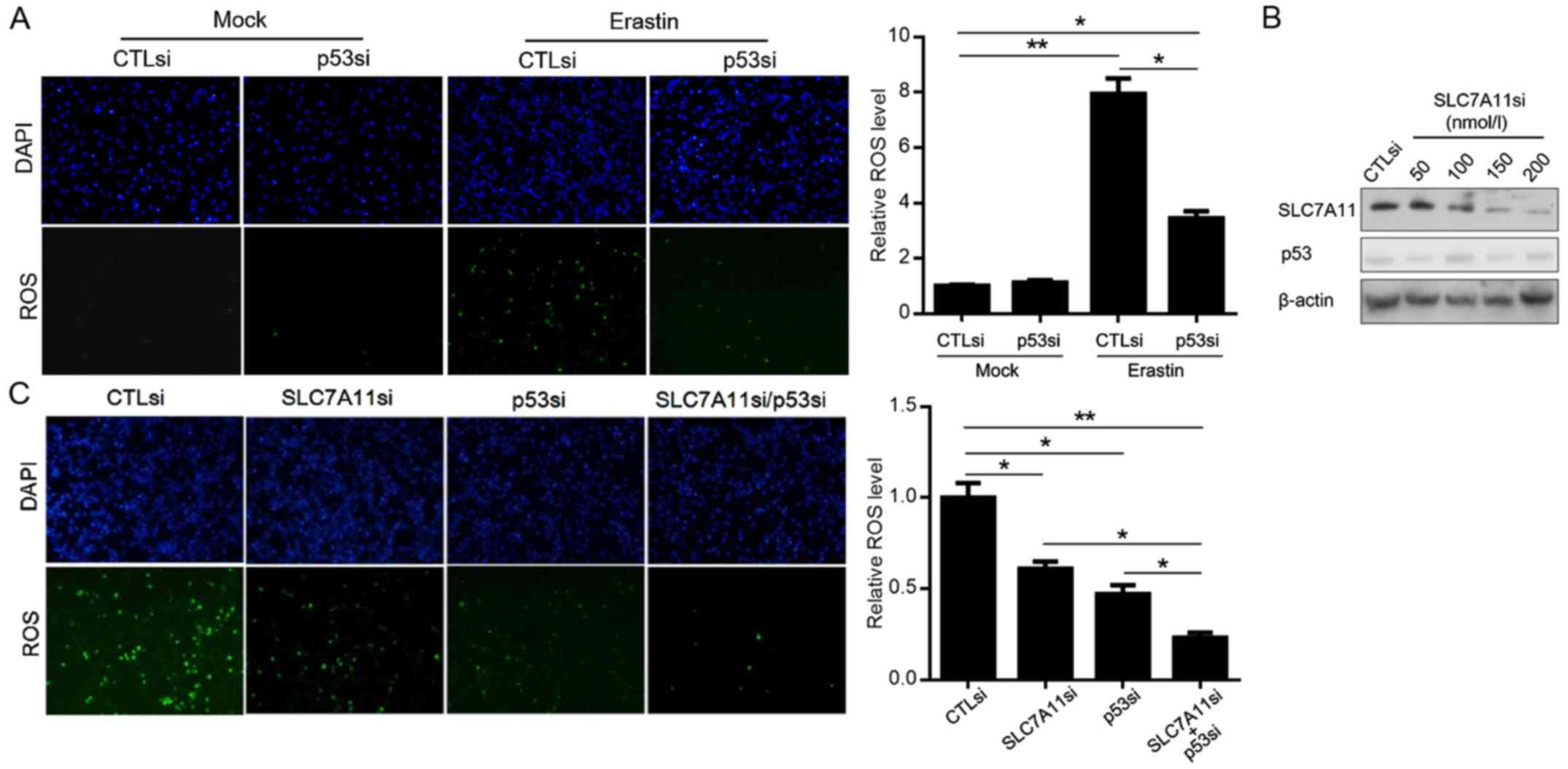
To determine the role of activated p53 on ROS
generation induced by erastin, we assessed the ROS level in A549
cells with or without p53 knockdown in A549 cells after erastin
exposure. As illustrated in Fig.
3A, while erastin exposure induced accumulating ROS in both
A549-p53si and A549-CTLsi cells, the ROS level in A549-p53si cells
was lower than that in A549-CTLsi cells. According to research,
activated p53 suppresses post-transcriptionally the expression of
SLC7A11, a key component of the cysteine/glutamate antiporter, thus
inducing ROS accumulation (12).
This prompted us to ascertain whether ROS induction by activated
p53 after erastin exposure was dependent on the suppression of
SLC7A11. We employed SCL7A11si to efficiently knock down the
expression of SCL7A11 without disturbing the expression of p53
(Fig. 3B). After erastin exposure,
both A549-SLC7A11si and A549-SLC7A11si/p53si cells presented
decreased ROS levels. By comparison with the SCL7A11si-transfected
group, co-transfection of SCL7A11si and p53si presented a
significantly lower level of ROS, indicating that, potentially, ROS
induction by p53 occurred partially by suppressing SCL7A11
(Fig. 3C).
Expression of p53 induced by erastin
exposure contributes to the cytotoxic effect on A549 cells, leading
to ferroptotic and apoptotic death
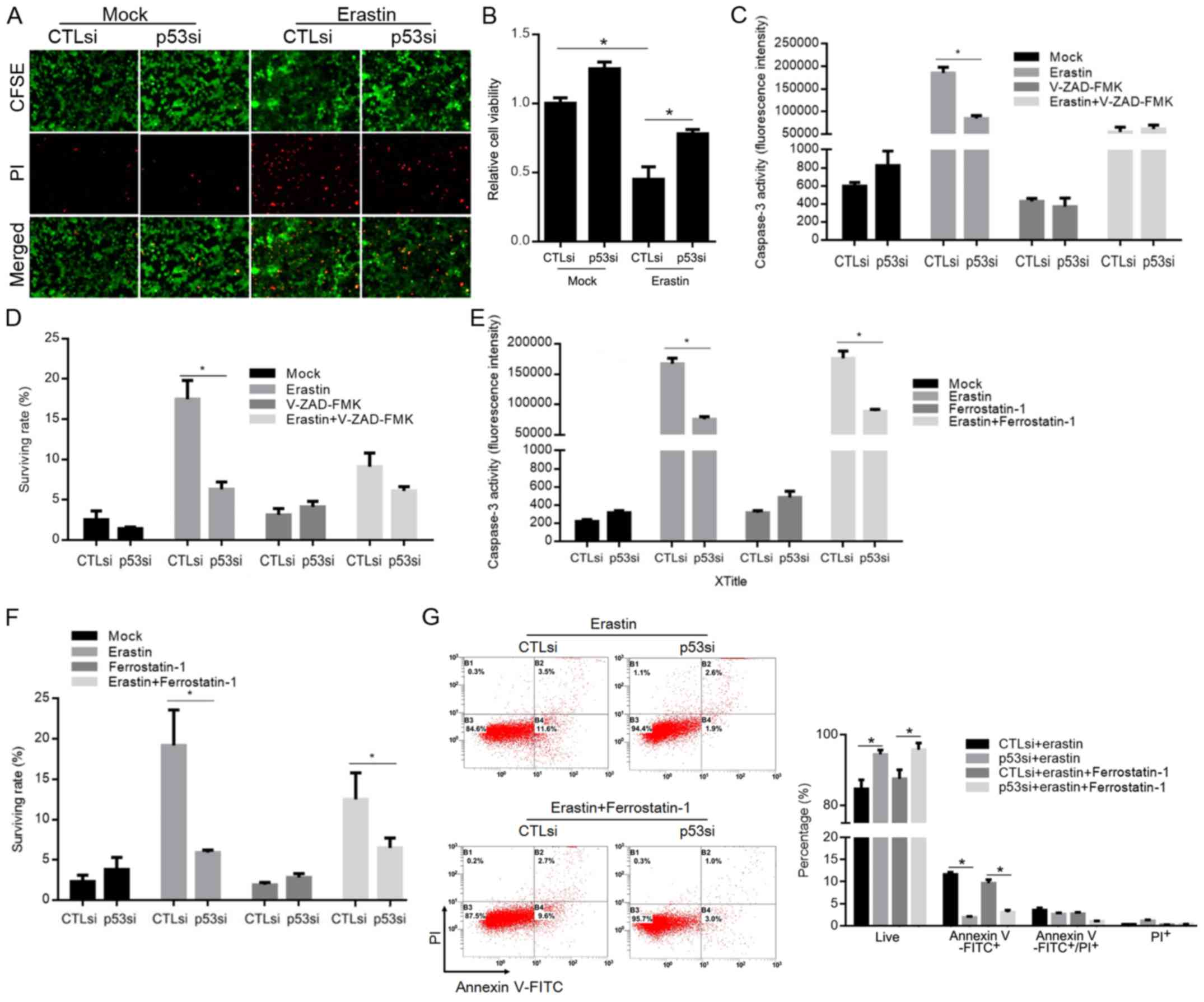
To evaluate the effects of p53 induced by erastin
exposure on A549 cells, the CFSE/PI double staining or CCK-8 assay
was performed to detect the survival rate or cell viability of A549
cells exposed to the IC50 concentration of erastin for
24 h. As illustrated in Fig. 4A and
B, erastin potently inhibited A549 cell survival, as evidenced
by CCK-8 optical density (OD) reduction. Knockdown of p53
attenuated erastin-exerted cytotoxicity. According to research,
erastin triggers ferroptosis, which is a unique iron-dependent form
of non-apoptotic death, prompting us to identify the
p53-contributed cytotoxic effect on A549 cells after erastin
exposure. Under the erastin-exposed condition, ferrostatin-1
(Fer-1) or Z-VAD-FMK was employed to inhibit ferroptotic or
apoptotic death, respectively, and the role of p53 in inducing
cytotoxicity was assessed. After confirming the significant
decrease in cell viability following erastin exposure compared with
untreated cells (data not shown), Fer-1 or Z-VAD-FMK was applied to
reveal the effects of p53 on erastin-induced ferroptotic and
apoptotic death. By inhibiting apoptotic death confirmed by
detecting caspase-3 activity (Fig.
4C), knockdown of p53 inhibited erastin-induced cell death, as
detected by CFSE/PI double staining (Fig. 4D). By inhibiting ferroptotic death
without disturbing caspase-3 activity (Fig. 4E), knockdown of p53 also exerted an
inhibitory effect of apoptotic death induced by erastin exposure
(Fig. 4F and G).
p53 induced by erastin exposure exerts
cytostatic effects on A549 cells
To test the effect of p53 induced by erastin on A549
cell proliferation, A549 cells were treated with the
IC30 concentration of erastin (3.12 µM) for 24 h. We
subjected the treated cells to EdU staining, which is a
proliferation marker, and observed that erastin treatment promoted
stalled replication, which was greatly rescued by p53 knockdown, as
shown by the EdU incorporation (Fig.
5A). This result indicated that erastin treatment inhibited
cellular proliferation via the presence of p53. Consistently,
analysis of the cell cycle distribution by flow cytometry
demonstrated that erastin treatment induced slight accumulation in
the G0/G1 phase that was reversed by p53
knockdown (Fig. 5B). Since both
accumulated ROS and activated p53 mediate the cell accumulation in
the G0/G1 phase (13), we next examined the related
kinetics. While activation of p53 or G0/G1
phase accumulation was detectable at 12, 18 or 24 h (Fig. 5C), accumulation of ROS was detected
at 3 h after erastin treatment (Fig.
5D). We also determined the effects of erastin-induced ROS on
the cell cycle arrest by employing the ROS scavenger
N-L-acetylcysteine (NAC) with erastin. In Fig. 5E, scavenging of erastin-induced ROS
was eliminated by pretreatment of the cells with NAC, leading to
entry into the cell cycle. Collectively, erastin exposure generated
ROS, thus activating p53, which demonstrated critical cytotoxic and
cytostatic effects in A549 cells.
Discussion
Erastin exposure has been found to exert a cytotoxic
effect in numerous cancer cells, including colon (16), lung cancer (17) and leukaemia cells (18), via inducing apoptotic cell death.
Recently, it was also revealed that erastin treatment exerts
cytotoxic effects via promoting ferroptotic cell death, which is
morphologically, biochemically and genetically distinct from
apoptosis, necrosis and autophagy (4). Erastin exposure rapidly causes
iron-dependent accumulation of lipid ROS, which is a direct trigger
of cell death (3). It is also
well-known that the accumulation of ROS results in the release of
cytochrome c into the cytosol, thus activating caspase
cleavage and initiating the apoptotic process (19,20).
p53, as the most well-known tumour suppressor, is tightly
associated with the regulation of ROS generation and the
ROS-induced apoptotic process (21). However, how ROS-induced activated
p53 following erastin exposure exerts cytotoxic or cytostatic
effects or how induced ROS regulates the activity of p53 remains
largely elusive.
In the present study, we demonstrated that
erastin-induced ROS regulated and activated p53, as well as
partially exerted cytotoxic and cytostatic effects on A549 cells.
Following the addition of erastin to A549 cells, we observed an
increased ROS level and increased expression of p53 and
phosphorylated p53 over time (Fig. 1A
and B). This increase was followed by an increased expression
of p53 downstream target genes Bax and p21 (Fig. 1C) and decreased expression of
SLC7A11 (Fig. 1D). Since NAC
scavenging generated ROS by erastin exposure (22), no detectable change in the p53
expression level and its downstream target genes was observed,
indicating that erastin-induced ROS may play a critical role in
inducing p53 and subsequently transcriptionally activating its
downstream target genes. By considering that a decrease in the
SLC7A11 level increased the ROS level (6), we tested whether knockdown of SLC7A11
was accompanied by changes in the ROS level. Notably, we observed
that not only SLC7A11 was responsible for the ROS level, but
knockdown of p53 also partially decreased the ROS level in an
SLC7A11-independent mechanism (Fig.
3). We hypothesised that ROS generation may collaborate with
other p53-dependent mechanisms.
Our results demonstrated that erastin exposure led
to apoptotic and ferroptotic cell death that was partially
dependent on p53 (Fig. 4). We
speculated that activated p53 may play a critical role in promoting
both apoptotic and ferroptotic cell death, possibly through the
activity of its downstream target genes. For example, Bax
translocates from the cytosol to the mitochondria upon stress,
leading to cytochrome c release and subsequent caspase
cascade (23), which is a direct
target gene of p53 and was found to be regulated via p53 after
erastin exposure (Fig. 1). As
expected, both p53 knockdown and inhibition of apoptotic cell death
by involving the caspase inhibitor Z-VAD-FMK attenuated the
cytotoxic effect of erastin treatment (Fig. 4).
A previous study has shown that activated p53
transcriptionally regulates genes such as p21, 14-3-3σ, Reprimo and
GADD45 to inhibit cell cycle entry (24). Our results indicated that, following
exposure to a comparatively low concentration of erastin, activated
p53 may activate one or several pathways that limit cell-cycle
progression. This outcome was consistent with the studies of
ferritin showing that upregulated ferritin facilitated growth
arrest via the induction of cyclin-dependent kinase inhibitor p21
(25).
In summary, exposure to erastin induced ROS
generation and subsequent p53 activation. As a feedback loop,
activated p53 partially contributed to induce ROS generation,
potentially through post-transcriptional suppression of SLC7A11. In
addition, p53 activation contributed to erastin-induced cytostatic
effects via arresting the cell cycle at the G1-phase. Collectively,
the presence of p53 sensitised lung cancer cells to erastin-induced
cytotoxic and cytostatic effects.
Acknowledgements
We would like to thank Dr Tao Hong (Department of
Anesthesiology, Chongqing People's Hospital, Chongqing, China) for
his helpful English editing work.
Funding
The present study was supported by the Science
Foundation of Sichuan Provincial Hospital (no. 30305031023).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
CH and RJ designed part of the experiments. MY was
involved in performing cell culture relative experiments. JD and PL
performed the gene expressing analysis, cell transfection and
treatment experiments. WS was involved in the molecular
experiments, data analysis and writing of the manuscript. All
authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yagoda N, von Rechenberg M, Zaganjor E,
Bauer AJ, Yang WS, Fridman D, Wolpaw AJ, Smukste I, Peltier JM,
Boniface JJ, et al: RAS-RAF-MEK-dependent oxidative cell death
involving voltage-dependent anion channels. Nature. 447:864–868.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dolma S, Lessnick SL, Hahn WC and
Stockwell BR: Identification of genotype-selective antitumor agents
using synthetic lethal chemical screening in engineered human tumor
cells. Cancer Cell. 3:285–296. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of non-apoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huo H, Zhou Z, Qin J, Liu W, Wang B and Gu
Y: Erastin Disrupts mitochondrial permeability transition pore
(mPTP) and induces apoptotic death of colorectal cancer cells. PLoS
One. 11:e01546052016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jiang L, Kon N, Li T, Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao M, Monian P, Quadri N, Ramasamy R and
Jiang X: Glutaminolysis and transferrin regulate ferroptosis. Mol
Cell. 59:298–308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsai MH, Liu JF, Chiang YC, Hu SC, Hsu LF,
Lin YC, Lin ZC, Lee HC, Chen MC, Huang CL and Lee CW: Artocarpin,
an isoprenyl flavonoid, induces p53-dependent or independent
apoptosis via ROS-mediated MAPKs and Akt activation in non-small
cell lung cancer cells. Oncotarget. 8:28342–28358. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou Z, Yin Y, Chang Q, Sun G, Lin J and
Dai Y: Downregulation of B-myb promotes senescence via the
ROS-mediated p53/p21 pathway, in vascular endothelial cells. Cell
Prolif. 50:2017. View Article : Google Scholar
|
|
10
|
Assaily W, Rubinger DA, Wheaton K, Lin Y,
Ma W, Xuan W, Brown-Endres L, Tsuchihara K, Mak TW and Benchimol S:
ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation
in response to nutritional stress. Mol Cell. 44:491–501. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seo SU, Cho HK, Min KJ, Woo SM, Kim S,
Park JW, Kim SH, Choi YH, Keum YS, Hyun JW, et al: Thioridazine
enhances sensitivity to carboplatin in human head and neck cancer
cells through downregulation of c-FLIP and Mcl-1 expression. Cell
Death Dis. 8:e25992017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao
Y and Gu W: Acetylation is crucial for p53-mediated ferroptosis and
tumor suppression. Cell Rep. 17:366–373. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shi Y, Nikulenkov F, Zawacka-Pankau J, Li
H, Gabdoulline R, Xu J, Eriksson S, Hedström E, Issaeva N, Kel A,
et al: ROS-dependent activation of JNK converts p53 into an
efficient inhibitor of oncogenes leading to robust apoptosis. Cell
Death Differ. 21:612–623. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang J, Zhao X, Tang M, Li L, Lei Y, Cheng
P, Guo W, Zheng Y, Wang W, Luo N, et al: The role of ROS and
subsequent DNA-damage response in PUMA-induced apoptosis of ovarian
cancer cells. Oncotarget. 8:23492–23506. 2017.PubMed/NCBI
|
|
15
|
An JJ, Shi KJ, Wei W, Hua FY, Ci YL, Jiang
Q, Li F, Wu P, Hui KY, Yang Y and Xu CM: The ROS/JNK/ATF2 pathway
mediates selenite-induced leukemia NB4 cell cycle arrest and
apoptosis in vitro and in vivo. Cell Death Dis. 4:e9732013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suh DH, Kim MK, Kim HS, Chung HH and Song
YS: Mitochondrial permeability transition pore as a selective
target for anti-cancer therapy. Front Oncol. 3:412013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yamaguchi H, Hsu JL, Chen CT, Wang YN, Hsu
MC, Chang SS, Du Y, Ko HW, Herbst R and Hung MC:
Caspase-independent cell death is involved in the negative effect
of EGFR inhibitors on cisplatin in non-small cell lung cancer
cells. Clin Cancer Res. 19:845–854. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Aresvik DM, Pettersen RD, Abrahamsen TG
and Wright MS: 5-Fluorouracil-induced death of Jurkat T-cells-A
role for caspases and MCL-1. Anticancer Res. 30:3879–3887.
2010.PubMed/NCBI
|
|
19
|
Siskind LJ: Mitochondrial ceramide and the
induction of apoptosis. J Bioenerg Biomembr. 37:143–153. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Colombini M: Ceramide channels and their
role in mitochondria-mediated apoptosis. Biochim Biophys Acta.
1797:1239–1244. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lago CU, Sung HJ, Ma W, Wang PY and Hwang
PM: p53, Aerobic metabolism, and cancer. Antioxid Redox Signal.
15:1739–1748. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Downs I, Liu J, Aw TY, Adegboyega PA and
Ajuebor MN: The ROS scavenger, NAC, regulates hepatic Vα14iNKT
cells signaling during Fas mAb-dependent fulminant liver failure.
PLoS One. 7:e380512012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jürgensmeier JM, Xie Z, Deveraux Q,
Ellerby L, Bredesen D and Reed JC: Bax directly induces release of
cytochrome c from isolated mitochondria. Proc Natl Acad Sci
USA. 95:4997–5002. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Horn HF and Vousden KH: Coping with
stress: Multiple ways to activate p53. Oncogene. 26:1306–1316.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang F, Wang W, Tsuji Y, Torti SV and
Torti FM: Post-transcriptional modulation of iron homeostasis
during p53-dependent growth arrest. J Biol Chem. 283:33911–33918.
2008. View Article : Google Scholar : PubMed/NCBI
|