The two emerging hallmarks of metabolic
reprogramming and evasion of immune destruction represent
significant conceptual progress in the field of tumor research
(1). Tumor cells preferentially
uptake and utilize glucose via aerobic glycolysis, even in the
presence of sufficient oxygen to support the mitochondrial
oxidative respiration, a phenomenon referred to as the ‘Warburg
effect’ (2). The role of the immune
system in tumor cell recognition and elimination is becoming
increasingly ambiguous, as tumor cells have developed several
mechanisms to avoid immune responses (3–5). The
functional impairment of effector T cells (Teffs) and the induction
of immunosuppressive T cells in the tumor micromilieu may lead to
immune escape or evasion by cancer cells (6–8). The
mechanisms responsible for T-cell dysfunction or hyporesponsiveness
are complicated (9), and the
association between tumor metabolic switch and immune tolerance has
been attracting increasing attention (1,10–13).
The immune functions of Teffs are intimately
associated with their metabolic regulation (14–18).
Clonal expansion and antitumor function acquisition of T cells upon
activation, which are energy-demanding processes, are accompanied
by a marked shift in metabolism from energy-oriented catabolic
oxidation in naive T cells to biosynthetic aerobic glycolysis in
activated T cells (14). The
metabolic and immunological functions of Teffs may be impaired by
tumor cells due to the tumor-imposed nutrient depletion and
accumulation of tumor-derived immunomodulatory metabolic
intermediates, including lactate, in the tumor microenvironment
(11–13). The manipulation of metabolic
reprogramming in T cells is currently considered as a potential
therapeutic target to regulate the antitumor function and fate of
Teffs (19,20).
Although several intracellular signaling pathway
molecules in the metabolic reprogramming of tumor cells and Teffs
have been identified, including hypoxia-inducible factor-1 (HIF-1),
c-Myc and phosphatidylinositol 3-kinase (PI3K)/protein kinase B
(Akt) (15,21–26),
how tumor cells cope with extracellular metabolic signals and
transduce extracellular signals to intracellular stimuli remain to
be fully elucidated. CD147, also referred to as HAb18G/CD147 in
humans, is a transmembrane protein that has been reported to be
overexpressed on the surface of various types of malignant tumor
cells (27,28). Upregulation of the expression of
CD147 has been found to contribute significantly to malignant
potential and poor prognosis through triggering the production and
release of extracellular matrix metalloproteinase (MMP) and
vascular endothelial growth factor (29–33).
An increased dependence on aerobic glycolysis inevitably results in
an increased production of lactic acid, and this surplus of lactic
acid has to be exported in order to prevent cellular acidosis and
maintain cellular homeostasis (34). Monocarboxylate transporters (MCTs)
catalyze the transport of monocarboxylates, including L-lactate,
across plasma membranes (35,36).
CD147 has also been described as a chaperone assisting in the
folding, stability, membrane expression and functionality of MCTs
(37), suggesting the involvement
of CD147 in metabolic regulation (34). The aim of the present review was to
highlight the immunosuppression in the tumor microenvironment
induced by underlying glycolytic mechanism reprogramming and
discuss the therapeutic potential of targeted metabolic
manipulation in tumor immunotherapy.
An emerging theme in immunology is that metabolic
adaption and lymphocyte function are intimately linked, and changes
in cellular metabolism have been shown to be associated with
altered immunological function (18). Tumor cells have been the focus of
investigations on the metabolic switch, although the metabolic
reprogramming of immune cells, particularly tumor-infiltrating
lymphocytes (TILs), has not been investigated as extensively.
Warburg was also one of the first to examine leukocyte metabolism,
and found that leukocyte stimulation led to a shift towards aerobic
glycolysis from oxidative phosphorylation (OXPHOS), which is
primarily used by resting leukocytes (38).
Resting or naive T cells predominantly oxidize
glucose-derived pyruvate, in addition to lipids and amino acids,
via OXPHOS, to maintain quiescence and immune surveillance
(39). Upon T cell activation,
lipid oxidation is sharply reduced, and the cells rely instead on
increased aerobic glycolysis to support extensive proliferation and
Teff differentiation and function (14,40).
At the end of an immune response, a small population of
antigen-specific T cells survives to become long-lived memory T
cells, which revert back to a metabolic program comparable with
that of resting T cells (14,18,41).
However, memory T cells exhibit an increased capacity for efficient
energy generation, characterized by an increase in mitochondrial
mass and, consequently, maximal mitochondrial spare respiratory
capacity, which allows for the rapid and vigorous production of ATP
upon repeat encounter with antigens (42,43).
In addition to Teffs, activated T cells can differentiate into
regulatory T cells (Tregs), which serve a critical role in
self-tolerance and immunosuppression (3,44,45).
Unlike Teffs, Tregs primarily use glucose-derived pyruvate and
fatty acids to efficiently produce ATP through the tricarboxylic
acid cycle and lipid β-oxidation (46). Distinct metabolic programs are
required for functionally different T-cell lineage differentiation
and commitment (47,48). A comprehensive study by Michalek
et al demonstrated that Teffs, including T helper (Th)1, Th2
and Th17 cells, were selectively increased in glucose transporter
(GLUT)1 transgenic mice, and were dependent on a highly glycolytic
metabolism (48). Tregs, by
contrast, expressed a low level of GLUT1 and relied on high rates
of lipid oxidation (48). Gene
array analysis on CD8+ cytotoxic T cells under
conditions of glucose deprivation or incubated in the presence or
absence of 2-deoxy-D-glucose, which inhibits glycolysis,
demonstrated that multiple key gene expression events and effector
functions were selectively inhibited, including the production of
interferon-γ (IFN-γ), cell cycle progression and cytolytic activity
(49). Consistently, impaired
T-cell metabolism directly contributed to T-cell dysfunction and
exhaustion in leukemia, whereas the genetically increased
expression of GLUT1 and hexokinase 2 (HK2) may partially restore
T-cell function (50). The
upregulation of glycolysis by the transgenic overexpression of
GLUT1 or glycolytic genes was sufficient to augment T-cell
activation, ultimately resulting in lymphadenopathy and a systemic
lupus erythematosus-like autoimmunity in aging mice (17).
The identification of transcription factors
potentially responsible for the metabolic reprogramming upon T-cell
activation revealed c-Myc and HIF-1α as two of the top-ranked
candidates, as both were found to be induced at the mRNA and
protein levels upon T-cell stimulation (21). c-Myc specifically upregulates the
expression of all glycolytic genes, including GLUT1, lactate
dehydrogenase type A (LDHA), HK2 and pyruvate kinase muscle isoform
2 (PKM2). Subsequently, the acute genetic deletion of c-Myc
markedly inhibits the upregulated glycolytic activity. In addition,
an HIF-1α-mediated glycolytic switch regulates the balance of
Th17/Treg differentiation (22,51).
Th17- but not Treg-polarizing conditions elicited a
HIF-1α-dependent acceleration of glycolysis via upregulation of
glycolytic enzyme expression. By contrast, the inhibition of
glycolytic metabolism resulted in the inhibition of Th17
differentiation and promotion of Treg development. Upon
investigation of the underlying molecular mechanism, HIF-1α was
found to be selectively induced in Th17 differentiation through the
mammalian target of rapamycin (mTOR) signaling pathway, whereas the
deficiency of HIF-1α led to decreased Th17 commitment but enhanced
generation of Treg, which protected mice from experimental
autoimmune encephalomyelitis (22).
PI3K/Akt is activated by various stimuli in T
lymphocytes, including T cell antigen receptor, costimulatory
molecules, cytokine receptors and chemokine receptors (23,24,52),
and PI3K/Akt signaling serves a fundamental role in T-cell
activity. For example, the trafficking of GLUTI to the cell surface
and prevention of internalization in T cells are promoted by Akt
(53). Of note, mTOR, as a
downstream target of Akt, is activated by Akt and serves a key role
in linking the activation of PI3K to Th-cell differentiation
(54,55). mTOR is a catalytic unit of two
distinct multi-protein assemblies, referred to as mTOR complex
(mTORC)1 and mTORC2. The activation of mTORC1 can initiate a
signaling cascade, which leads to metabolic reprogramming
characterized by increased aerobic glycolysis. Of note, mTOR
differentially regulates Teff and Treg lineage commitments through
the activation of specific signal transducer and activator of
transcription pathways and, consequently, the induction of
lineage-specific transcription factors (54). By contrast, rapamycin treatment,
which targets mTORC1, has been shown to exhibit an inhibitory
effect on glycolytic switching upon T-cell activation (56). AMP-activated protein kinase (AMPK),
as a well-known evolutionarily conserved energy sensor, is
activated by an increased AMP/ATP ratio and acts in opposition to
mTORC1 to maximize energy production via promoting mitochondrial
phosphorylation (57).
AMPKα1−/− T cells exhibit an impaired ability to transit
from an anabolic and glycolytic metabolism to a catabolic and lipid
oxidative state under metabolic stress (58).
It is well established that malignant transformation
is associated with a disrupted balance between oncogenes and tumor
suppressor genes. From a metabolic perspective, this is associated
with a reprogrammed metabolism and constitutes a molecular basis
for the accelerated aerobic glycolysis in tumors (59–62).
Accumulating evidence has confirmed that the MYC
oncogene, the PI3Ks/Akt/mTOR pathway and HIF-1 (62,63),
in addition to tumor suppressor p53, are implicated in the
metabolic reprogramming of tumor cells (64–66)
(Fig. 1). The MYC oncogene encodes
a transcription factor, c-Myc, which links altered cellular
metabolism to tumorigenesis (67).
Generally, c-Myc directly and/or indirectly regulates the
expression of genes involved in glucose, glutamine and nucleotide
metabolism. For example, glycolytic genes, including LDHA are
directly upregulated by c-Myc (68); however, c-Myc can repress the
expression of microRNA-23a/b to indirectly promote the protein
expression of glutaminase and metabolism of glutamine (69). The depletion of c-Myc has been shown
to result in the repression of several genes encoding enzymes
rate-limiting for deoxyribonucleoside triphosphates (dNTPs)
metabolism, including thymidylate synthase, inosine monophosphate
dehydrogenase 2 and phosphoribosyl pyrophosphate synthetase 2. The
depletion of c-Myc also leads to a decrease in dNTPs and inhibited
cell proliferation (70). A number
of glycolytic genes have been documented to be directly regulated
by c-Myc in screens for c-Myc target genes, including GLUT1, HK2
and muscle phosphofructokinase (71,72).
In addition, c-Myc may cooperatively serve a pivotal role in
hypoxic adaptation with HIF-1 through upregulating pyruvate
dehydrogenase kinase 1 under non-normoxic conditions, thereby
accelerating glycolytic metabolism by favoring the conversion of
pyruvate to lactate and suppressing mitochondrial oxidative
respiration (72–74).
The PI3K/Akt/mTOR signaling pathway has been found
to be activated at a high level and contribute to the metabolic
transformation of tumors (75,76)
(Fig. 1). Akt, a serine/threonine
kinase, has been shown to be constitutively activated in tumor
cells through the amplification of PI3K, which phosphorylates
membrane-associated phosphatidylinositol 4,5-bisphosphate (PIP2) to
generate phosphatidylinositol 3,4,5-trisphosphate as an upstream
activator of Akt (77). Human
glioblastoma cells with constitutive Akt activity exhibit high
rates of aerobic glycolysis through the direct effect of Akt on
glucose metabolism, including upregulating the expression and/or
localization of glucose transporters and glycolytic enzymes,
including GLUT1 and HK2 (75). Akt
also activates mTOR, which also contributes to the glucose
metabolic reprogramming of tumor cells (78,79).
mTOR is also an upstream activator of HIF-1 and c-Myc in tumor
cells, and high levels of Akt and mTOR activity lead to high HIF-1
activity and adaption to hypoxia (78,79).
In conclusion, relevant transporters and receptors
on tumor cells integrate signals from growth factors, cytokines and
nutrient availability in the tumor microenvironment to activate the
PI3K/Akt/mTOR signaling pathway, which regulates the expression of
various transcription factors, including c-Myc, HIF-1 and p53,
leading to the reprogramming of glucose metabolism in tumors. Of
note, a reciprocal interaction exists between molecular signaling
pathways regulating c-Myc, HIF-1 and p53, forming a complicated and
intricate regulatory network controlling the metabolic switch in
tumors (85).
A number of mechanisms for the immune evasion of
tumor cells have been elaborated (86), including the downregulation of
tumor-associated antigen and costimulatory molecule expression, and
the upregulation of inhibitory immunomodulatory molecules and
immunosuppressive cells (3,9,87). The
metabolic interplay between tumor cells and infiltrating
lymphocytes has been suggested to be an important metabolic
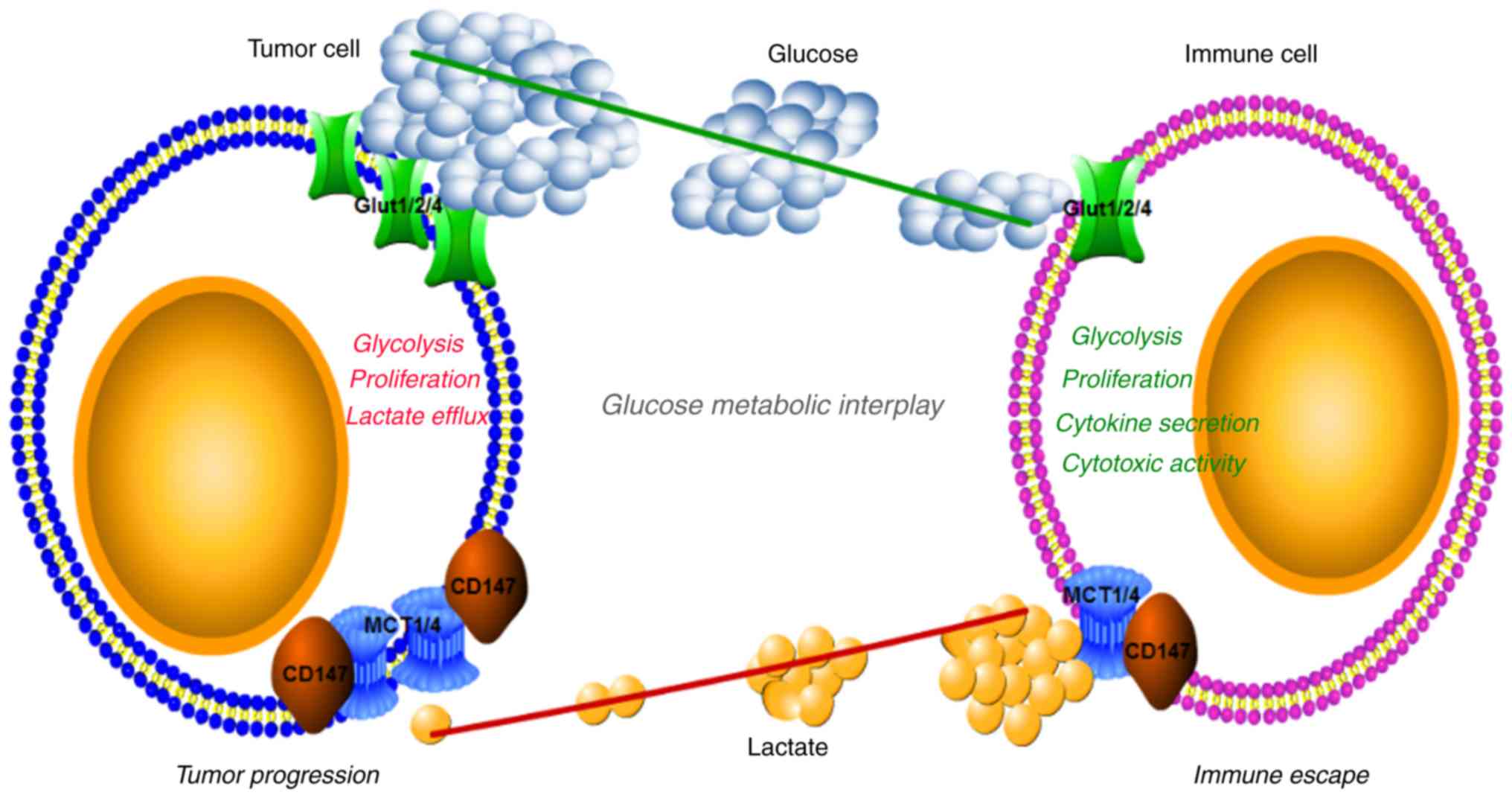
mechanism underlying immunological escape of tumor cells (88) (Fig.
2). The similarity of tumor cell and activated lymphocyte
metabolism is not coincidental, as is it essential that their
metabolism matches the functional demands of the cells. Rapid
growth and proliferation are necessary for tumor cells and
activated lymphocytes; therefore, they preferentially select the
more biosynthesis-efficient aerobic glycolysis and anabolism over
the energy-oriented mitochondrial OXPHOS.
It is likely that intense nutrient competition
exists between tumor cells and TILs in the tumor microenvironment,
as tumor cells may deplete nutrients due to their dependence on
enhanced aerobic glycolysis (10).
This tumor-imposed glucose restriction may lead to TIL dysfunction
due to reduced glucose uptake and metabolic reprogramming. In an
established mouse model of progressing and regressing tumors, the
progressing tumors exhibited higher rates of glycolytic activity
compared with regressing tumors, suggesting that progressing tumors
consume more glucose. Consistently, T cells in progressing tumors
exhibited decreased phosphorylation of eukaryotic translation
initiation factor 4E-binding protein 1 (4EBP1) and S6 kinase
compared with that in regressing tumors. However, Tregs and M2
macrophages, neither of which depend on enhanced aerobic
glycolysis, but rather on fatty acid oxidation, are unaffected by
the glucose-depleted tumor microenvironment, and may suppress the
antitumor immune response.
In addition to tumor-imposed nutrient limitation for
tumor-infiltrating immune cells, metabolites, including the
enhanced lactate production by tumors due to increased dependence
on glycolysis (59,62,89),
are suggested to be key metabolic components in the communication
between tumor cells and tumor-infiltrating immune cells (34,90).
It is important that the excess cellular lactate produced by tumors
is exported, mainly by MCTs (MCT1/MCT4), in order to prevent
acidosis in tumor cells, which leads to the accumulation of lactate
in the tumor milieu (91). Lactate
has been shown to promote cancer cell stemness (92) and metastasis (93) through the increased production of
several tumor progression-promoting factors, including transforming
growth factor (TGF)-β (94,95), hyaluronic acid and CD44 (96). In addition to its direct effect on
tumor cells, lactate act as an immunomodulator, mediating the
immune evasion of tumor cells (97). The exogenous lactate treatment of
natural killer (NK) cells inhibited their cytotoxicity directly and
indirectly by increasing the number of myeloid-derived suppressor
cells (MDSCs), which may repress NK cell function (12,98).
By contrast, LDHA-deleted pancreatic cancer cell xenografts, with a
defect in lactate production, exhibited improved cytolytic function
of NK cells in C57BL/6 mice, with higher expression of perforin and
granzyme and a decreased number of MDSCs in the spleen (12,98).
Furthermore, the immunomodulatory effects of lactic acid have been
demonstrated not only for dendritic cells, but also for T cells
(13). However, the export of
excess lactic acid by activated T cells is inhibited due to the
lactic acid gradient between the cytoplasm and extracellular
milieu, due to the accumulation of lactic acid secreted by
surrounding tumor cells with high rates of glycolysis. To conclude,
the accumulation of tumor-derived lactate in the extracellular
milieu may lead to a metabolic obstruction in cytotoxic T
lymphocytes (CTLs); subsequently, CTLs become hyporesponsive,
exhibiting decreased proliferation, cytokine secretion and
cytotoxic activity (13,99,100).
Tregs and M2 macrophages are not affected by the presence of high
levels of lactate, as their distinct metabolic program relies
mainly on fatty acid oxidation rather than aerobic glycolysis
(48).
Given the profound effect of HIF-1 on gene
regulation, T-cell differentiation is likely controlled by HIF-1
(15,22). Furthermore, TGF-β1 may stabilize
HIF-1 through the inhibition of prolyl hydroxylase 2 under hypoxic
conditions (106). A screening of
key transcription factors for T-cell differentiation during
inflammatory hypoxia of the mucosa revealed forkhead box (FOX)P3 as
a direct target of HIF-1, and it has also been demonstrated that
the hypoxic induction of FOXP3 and accumulation of Treg require
both HIF-1 and TGF-β1 (107).
HIF-1 has also been identified as a decisive factor in T-cell
differentiation to Th17 or Tregs by promoting Th17-polarization and
inhibiting Treg differentiation (22,51).
TGF-β1 may be induced in hypoxia (108), and is also key role the
differentiation of Th17 and Tregs (109). It is reasonable to hypothesize
that the Th17/Treg balance is an integral outcome of HIF-1, TGF-β1
and inflammatory cytokine interplay in the local tumor milieu
(22,51,107).
The exposure of human breast and prostate cells, and mouse melanoma
and mammary carcinoma cells to hypoxia resulted in the upregulation
of programmed cell death ligand-1 (PD-L1), an important
immunoinhibitory molecule, on the surface of tumor cells in an
HIF-1-dependent manner, which eventually contributed to tumor cell
evasion from antitumor immunity via the increased apoptosis of CTLs
due to the enhanced PD-1/PD-L1 interaction (110). Although the mechanisms underlying
increased the expression of PD-L1 on tumor cells remain to be fully
elucidated, the hypoxia-induced upregulation of PD-L1 on tumor
cells may represent a novel mechanism responsible for
hypoxia-mediated immune evasion of tumor cells. In addition to the
previously known inhibitory effects of TGF-β1 on T-cell
differentiation and function, TGF-β1 has also been found to mediate
T-cell hyporesponsiveness, in part through the enhanced expression
of PD-1 on tumor-infiltrating antigen-specific T cells induced by
mothers against decapentaplegic homolog 3 (Smad3)-dependent
signaling (111,112).
CD147 may significantly contribute to tumor growth,
invasion, metastasis and angiogenesis (29,30,113),
particularly in human hepatocellular carcinoma (HCC), mainly via
triggering the production of MMP and interacting with various
ligands involved in neoplastic cell behavior, including integrin
α3β1 (114) and α6β1 (115), and Annexin II (116). The non-metabolic molecular
mechanisms responsible for the tumor progression associated with
the upregulation of CD147 were discussed in a previous review
(38).
In addition to non-metabolic molecular mechanisms
responsible for the tumor progression associated with the
upregulation of CD147, a metabolic molecular basis has become a
focus of investigations. Blocking CD147 with a targeted monoclonal
antibody or silencing CD147 by small interfering RNA has been shown
to result in a marked decrease in glycolytic energy metabolism
(117,118). Consistently, a study by Huang
et al demonstrated that CD147 acts as an important regulator
of cell proliferation through promoting glucose metabolic
reprogramming by the post-transcriptional inhibition of p53 via the
activation of PI3K/Akt/Mdm2 signaling promoted by MCT1-induced
lactate export in HCC (119)
(Fig. 1). CD147 is increasingly
recognized as being implicated in glucose metabolism reprogramming
in tumors through gain/loss-of-function studies (92,120).
According to the regulation of CD147, it is well
established that the tumor micromilieu serves an important role in
the overexpression of CD147 on the surface of tumor cells. As is
known, TGF-β1 induces epithelial-to-mesenchymal transition (EMT)
via Smad-dependent and -independent signaling pathways (121–123). A study by Wu et al
demonstrated a positive correlation between the expression of CD147
and typical EMT markers, revealed that CD147 is a Slug target gene,
and demonstrated that the upregulation of CD147 involves activation
of the PI3K/Akt-GSK3β-Snail-Slug signaling pathway through the
stimulation of TGF-β1 (124). A
series of transcription factors have been found to be implicated in
the fundamental metabolic adaptation of tumors to hypoxia, among
which HIF-1 is critical to this process (125,126). HIF-1 is a heterodimer that
consists of a constitutively expressed HIF-1β subunit and an
oxygen-sensitive HIF-1α subunit (127). Under hypoxic conditions, HIF-1α
binds to a conserved DNA consensus, referred to as
hypoxia-responsive element (HRE), on the promoters of numerous
hypoxia-responsive genes. There are two HIF-1-binding sites and
three specificity protein 1 (SP1)-binding sites in the 3′ and 5′
flanking regions of the CD147 gene, respectively (128,129). Consistently, a genome-wide
chromatin immunoprecipitation-on-chip assay and immunohistochemical
staining identified CD147 as a novel hypoxia-responsive molecule.
The identification of key molecules engaged in epithelial solid
tumor glycolytic switch confirmed that the upregulation of CD147
was mainly mediated through the combined effect of HIF-1α and SP1
on activation of the CD147 promoter (120). A study by Kong et al also
reported that upregulation of the expression of CD147 was mediated
by promoter hypomethylation through increased SP1 binding in human
HCC and lung cancer (127,130).
In conclusion, the signaling pathways responsible
for the overexpression of CD147 on tumor cells mainly include
TGF-β1 and HIF-1α, which are pivotal in tumor glycolysis and
immunosuppression.
Successful chemotherapeutic tumor treatment
generally depends on the rapid proliferation of tumor cells.
However, undesirable side-effects on normal proliferating cells are
inevitable due to the non-specific nature of this treatment.
Therefore, therapeutic strategies based on specifically targeting
the ‘metabolic transformation’ of tumor cells may be a preferred
approach (63). Various potential
agents targeted against the altered metabolism of tumor cells are
currently in clinical trials, and several more are under
development.
Apart from tumor cells themselves, manipulating the
metabolic activity of immune cells to prevent immune cell
hyporesponsiveness in tumors is currently considered a promising
approach in cancer therapy (11,19,20).
The fundamental principle of modulating the metabolism of T cells
is to favor anabolic glycolysis rather than catabolic oxidative
respiration. Specific antibodies against nutrient transporters have
been confirmed as potential pharmacological agents targeting T-cell
metabolism. For example, blockade of GLUT1 on T cells has been
found to decrease glucose uptake, thus leading to T-cell
dysfunction (17). The blocking of
co-inhibitory receptors has been suggested as a promising
immunotherapy option for enhancing antitumor immunity to eliminate
tumor cells (131–134). Cytotoxic T-lymphocyte-associated
protein 4 (CTLA-4) and PD-1, two well-known inhibitory receptors on
T cells, are induced upon T-cell activation to control and moderate
excessive immune responses, acting as checkpoints (131). However, CTLA-4 and PD-1 signaling
have been shown to restrict T-cell activation and function by
downregulating aerobic glycolysis (10,135).
Therefore, it is hypothesized that checkpoint blocking may relieve
the suppression of antitumor immunity, in part through remodeling
T-cell metabolic programming to enhance nutrient uptake and
glycolytic metabolism, consequently restoring their capacity to
kill tumor cells (11). PD-L1 also
has a PD-L1 and PD-1 interaction-independent metabolic function
(111,136). PD-L1 on tumor cells is important
for Akt/mTOR signaling, which in turn increases the rate of
glycolysis through promoting the translation of glycolytic enzymes.
Blocking PD-L1 may directly decrease glycolysis in tumors,
increasing the nutrient availability in the extracellular tumor
milieu for infiltrating lymphocytes (10,11).
In general, therapeutic drugs targeting the
metabolic adaptation of tumor cells may be divided in two
categories, namely direct and indirect. Indirect drugs target
aberrant signaling pathways relevant to metabolic transformation in
tumor cells, including the HIF-1α (137), c-Myc (67,138),
PI3K/Akt/mTOR (139–141) and AMPK (142) pathways. For example, metformin, a
drug originally designed to treat patients with type 2 diabetes
(143), may activate the AMPK
signaling pathway to oppose mTORC1, subsequently decreasing
glycolytic metabolism and increasing OXPHOS in tumor cells to
control tumor progression (144,145). Consistently, patients with type 2
diabetes who were treated with metformin were more likely to remain
cancer-free over 8 years compared with those who received other
treatments (146,147). Metformin is currently in phase I
and II clinical trials for cancer therapy. Direct drugs comprise
antagonists against multiple metabolic enzymes and several
metabolites in glucose, amino acid, lipid and nucleotide metabolism
(63). This review focuses on
glycolytic metabolism. Almost all enzymes involved in every stage
of glycolysis may represent potential targets, particularly
tumor-specific enzyme isoforms and glycolytic metabolites,
including PKM2 (129,148) and lactic acid (90,102).
In terms of CD147, it has been reported in patients with HCC that
targeted radioimmunotherapy with 131I-labeled HAb18
F(ab′)2 metuximab monoclonal antibody injection (licartin), which
is a radiolabelled anti-CD147 monoclonal antibody, effectively
prevented the recurrence and metastasis of HCC following
hepatectomy and liver transplantation (149). Based on the evidence described
above, it is reasonable, to a certain extent at least, to attribute
the antineoplastic capacity of licartin to its ability to inhibit
the glycolytic metabolism of HCC cells. Combination therapy of
131I-labeled metuximab and other metabolic
transformation-targeting drugs may be more beneficial for antitumor
treatment compared with monotherapy.
Regardless of the modulation of cellular metabolism
in tumor cells or T cells, targeted delivery of specific drugs in
the body is crucial for preventing off-target effects.
Transporter-facilitated drug uptake (150,151), bi-specific antibodies (152,153) and nanoparticle-mediated delivery
(154,155) have been developed to optimize drug
efficacy. Optimal Teff function in the tumor microenvironment is
necessary for successful adoptive T-cell immunotherapy. Combination
treatment comprising metabolic intervention and adoptive T-cell
immunotherapy appears promising for metabolic reprogramming of T
cells to exert effective antitumor immunity (19,20).
As reported previously, CD147 serves an important role in the
reprogramming of glucose metabolism and cell proliferation in HCC
cells (119), and a targeted
radiolabeled anti-CD147 monoclonal antibody (licartin) effectively
prevented the recurrence and metastasis of HCC following
hepatectomy and liver transplantation (149). The blocking of CD147 inhibited the
enhanced glycolysis of HCC cells and contribute to improved
antitumor immunity in the tumor microenvironment, which is exactly
what current endeavors are aiming to prove. Combination therapy
comprising CD147 intervention and tumor immunotherapy is likely to
lead to more marked antitumor effects than monotherapy. However,
the timing of adoptive Teffs entering the local tumor milieu is an
important issue requiring consideration. Using inhibitors of
glycolysis prior to the adoptive transfer of T cells may assist in
remodeling metabolic function in T cells in a hospitable tumor
milieu with nutrient repletion (19,20).
CD147 exhibits high expression on the surface of a
variety of malignant tumor cells, and serves an important role in
neoplastic cell behavior via non-metabolic and metabolic molecular
mechanisms. Specifically, the involvement of CD147 in tumor glucose
metabolism reprogramming has been suggested, as CD147 can assist in
the folding, stability, membrane expression and functionality of
MCTs as a chaperone in the transport of monocarboxylates, including
L-lactate, across the plasma membrane in tumor glycolysis. The
metabolic interplay between tumor cells and infiltrating
lymphocytes has been increasingly recognized as an important
metabolic mechanism underlying the immune escape of tumor cells,
including intense competition for nutrients between tumor cells and
TILs in the tumor microenvironment, and an accumulation of
tumor-derived lactate in the extracellular milieu. HIF-1α, c-Myc,
PI3K/Akt/mTOR and AMPK signaling are considered to be important
metabolic pathways responsible for metabolic reprogramming and
antitumor immunoediting in tumors. Therefore, the manipulation of
cellular metabolism may be of value for the treatment of
immunological disorders and tumor immunotherapy.
The authors would like to thank Professor Juan Li of
Tianjin Medical University for their assistance in improving
schematic diagrams and in manuscript submission.
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81601377 and
81501984), the National Science and Technology Major Project (grant
no. 2013ZX09303001), the Tianjin Natural Science Fund (grant nos.
16JCZDJC35200 and 17JCYBJC25100), the Incubation Project of
National Clinical Research Center for Cancer (grant no. N14B09) and
the Tianjin Medical University Cancer Institute and Hospital Fund
(grant no. Y1601).
Not applicable.
XL and WX conceived, designed the study and wrote
the manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Kareva I and Hahnfeldt P: The emerging
‘hallmarks’ of metabolic reprogramming and immune evasion: Distinct
or linked? Cancer Res. 73:2737–2742. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Warburg O: On respiratory impairment in
cancer cells. Science. 124:269–270. 1956.PubMed/NCBI
|
|
3
|
Li X, Peng J, Pang Y, Yu S, Yu X, Cheng
PC, Wang WZ, Han WL, Zhang J, Yin YH and Zhang Y: Identification of
a FOXP3+CD3+CD56+ population with
immunosuppressive function in cancer tissues of human
hepatocellular carcinoma. Sci Rep. 5:147572015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kalathil SG and Thanavala Y: High
immunosuppressive burden in cancer patients: A major hurdle for
cancer immunotherapy. Cancer Immunol Immunother. 650:813–819. 2016.
View Article : Google Scholar
|
|
5
|
Wargo JA, Reddy SM, Reuben A and Sharma P:
Monitoring immune responses in the tumor microenvironment. Curr
Opin Immunol. 41:23–31. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Josefowicz SZ, Lu LF and Rudensky AY:
Regulatory T cells: Mechanisms of differentiation and function.
Annu Rev Immunol. 30:531–564. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hansen M and Andersen MH: The role of
dendritic cells in cancer. Semin Immunopathol. 9:307–316. 2017.
View Article : Google Scholar
|
|
8
|
Ni L and Dong C: New checkpoints in cancer
immunotherapy. Immunol Rev. 276:52–65. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Speiser DE, Ho PC and Verdeil G:
Regulatory circuits of T cell function in cancer. Nat Rev Immunol.
16:599–611. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chang CH, Qiu J, O'Sullivan D, Buck MD,
Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ,
et al: Metabolic competition in the tumor microenvironment is a
driver of cancer progression. Cell. 162:1229–1241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Siska PJ and Rathmell JC: T cell metabolic
fitness in antitumor immunity. Trends Immunol. 36:257–264. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gottfried E, Kunz-Schughart LA, Ebner S,
Mueller-Klieser W, Hoves S, Andreesen R, Mackensen A and Kreutz M:
Tumor-derived lactic acid modulates dendritic cell activation and
antigen expression. Blood. 107:2013–2021. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fischer K, Hoffmann P, Voelkl S,
Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G,
Hoves S, et al: Inhibitory effect of tumor cell-derived lactic acid
on human T cells. Blood. 109:3812–3819. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
MacIver NJ, Michalek RD and Rathmell JC:
Metabolic regu-lation of T lymphocytes. Annu Rev Immunol.
31:259–283. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gerriets VA and Rathmell JC: Metabolic
pathways in T cell fate and function. Trends Immunol. 33:168–173.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chang CH, Curtis JD, Maggi LB Jr, Faubert
B, Villarino AV, O'Sullivan D, Huang SC, van der Windt GJ, Blagih
J, Qiu J, et al: Posttranscriptional control of T cell effector
function by aerobic glycolysis. Cell. 153:1239–1251. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Macintyre AN, Gerriets VA, Nichols AG,
Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen
BJ, Hale LP, et al: The glucose transporter Glut1 is selectively
essential for CD4 T cell activation and effector function. Cell
Metab. 20:61–72. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pearce EL, Poffenberger MC, Chang CH and
Jones RG: Fueling immunity: Insights into metabolism and lymphocyte
function. Science. 42:12424542013. View Article : Google Scholar
|
|
19
|
Chang CH and Pearce EL: Emerging concepts
in immunotherapy-T cell metabolism as a therapeutic target. Nat
Immunol. 17:364–368. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
O'Sullivan D and Pearce EL: Targeting T
cell metabolism for therapy. Trends Immunol. 36:71–80. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang R, Dillon CP, Shi LZ, Milasta S,
Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger
J, et al: The transcription factor Myc controls metabolic
reprogramming upon T lymphocyte activation. Immunity. 35:871–882.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shi LZ, Wang R, Huang G, Vogel P, Neale G,
Green DR and Chi H: HIF1alpha-dependent glycolytic pathway
orchestrates a metabolic checkpoint for the differentiation of TH17
and Treg cells. J Exp Med. 208:1367–1376. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
So L and Fruman DA: PI3K signalling in B-
and T-lymphocytes: New developments and therapeutic advances.
Biochem J. 442:465–841. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Han JM, Patterson SJ and Levings MK: The
role of the PI3K signaling pathway in CD4+ T cell
differentiation and function. Front Immunol. 3:2452012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Powell JD, Pollizzi KN, Heikamp EB and
Horton MR: Regulation of immune responses by mTOR. Annu Rev
Immunol. 30:39–68. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chi HB: Regulation and function of mTOR
signaling in T cell fate decisions. Nat Rev Immunol. 12:325–338.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Weidle UH, Scheuer W, Eggle D, Klostermann
S and Stockinger H: Cancer-related issues of CD147. Cancer Genomics
Proteomics. 7:157–169. 2010.PubMed/NCBI
|
|
28
|
Riethdorf S, Reimers N, Assmann V,
Kornfeld JW, Terracciano L, Sauter G and Pantel K: High incidence
of EMMPRIN expression in human tumors. Int J Cancer. 119:1800–1810.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xu J, Xu HY, Zhang Q, Song F, Jiang JL,
Yang XM, Mi L, Wen N, Tian R, Wang L, et al: HAb18G/CD147 functions
in invasion and metastasis of hepatocellular carcinoma. Mol Cancer
Res. 5:605–614. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Q, Zhou J, Ku XM, Chen XG, Zhang L,
Xu J, Chen GS, Li Q, Qian F, Tian R, et al: Expression of CD147 as
a significantly unfavorable prognostic factor in hepatocellular
carcinoma. Eur J Cancer Prev. 16:196–202. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zheng HC, Takahashi H, Murai Y, Cui ZG,
Nomoto K, Miwa S, Tsuneyama K and Takano Y: Upregulated
EMMPRIN/CD147 might contribute to growth and angiogenesis of
gastric carcinoma: A good marker for local invasion and prognosis.
Br J Cancer. 95:1371–1378. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tang Y, Nakada MT, Kesavan P, McCabe F,
Millar H, Rafferty P, Bugelski P and Yan L: Extracellular matrix
metalloproteinase inducer stimulates tumor angiogenesis by
elevating vascular endothelial cell growth factor and matrix
metalloproteinases. Cancer Res. 65:3193–3199. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tang Y, Nakada MT, Rafferty P, Laraio J,
McCabe FL, Millar H, Cunningham M, Snyder LA, Bugelski P and Yan L:
Regulation of vascular endothelial growth factor expression by
EMMPRIN via the PI3K-Akt signaling pathway. Mol Cancer Res.
4:371–377. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kennedy KM and Dewhirst MW: Tumor
metabolism of lactate: The influence and therapeutic potential for
MCT and CD147 regulation. Future Oncol. 6:127–148. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Halestrap AP and Price NT: The
proton-linked monocarboxylate transporter (MCT) family: Structure,
function and regulation. Biochem J. 343:281–299. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Halestrap AP and Wilson MC: The
monocarboxylate transporter family-role and regulation. IUBMB Life.
64:109–119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kirk P, Wilson MC, Heddle C, Brown MH,
Barclay AN and Halestrap AP: CD147 is tightly associated with
lactate transporters MCT1 and MCT4 and facilitates their cell
surface expression. EMBO J. 19:3896–3904. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li XF, Yu XZ, Song XY, Dai D and Xu WG:
The altered glucose metabolism in tumor and a tumor acidic
microenvironment associated with extracellular matrix
metalloproteinase inducer and monocarboxylate transporters.
Oncotarget. 7:23141–23155. 2016.PubMed/NCBI
|
|
39
|
Kroemer G and Pouyssegur J: Tumor Cell
Metabolism: Cancer's Achilles' Heel. Cancer Cell. 13:472–482. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hsu PP and Sabatini DM: Cancer cell
metabolism: Warburg and beyond. Cell. 134:703–707. 2014. View Article : Google Scholar
|
|
41
|
Levine AJ and Puzio-Kuter AM: The control
of the metabolic switch in cancers by oncogenes and tumor
suppressors genes. Science. 330:1340–1344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tennant DA, Duran RV and Gottlieb E:
Targeting metabolic transformation for cancer therapy. Nat Rev
Cancer. 10:267–277. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Madan E, Gogna R, Bhatt M, Pati U,
Kuppusamy P and Mahdi AA: Regulation of glucose metabolism by p53:
Emerging new roles for the tumor suppressor. Oncotarget. 2:948–957.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang C, Liu J, Wu R, Liang Y, Lin M, Liu
J, Chan CS, Hu W and Feng Z: Tumor suppressor p53 negatively
regulates glycolysis stimulated by hypoxia through its target RRAD.
Oncotarget. 5:5535–5546. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schwartzenberg-Bar-Yoseph F, Armoni M and
Karnieli E: The tumor suppressor p53 down-regulates glucose
transporters GLUT1 and GLUT4 gene expression. Cancer Res.
64:2627–2633. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dang CV, Le A and Gao P: MYC-induced
cancer cell energy metabolism and therapeutic opportunities. Clin
Cancer Res. 15:6479–6483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shim H, Dolde C, Lewis BC, Wu CS, Dang G,
Jungmann RA, Dalla-Favera R and Dang CV: c-Myc transactivation of
LDH-A: Implications for tumor metabolism and growth. Proc Natl Acad
Sci USA. 94:6658–6663. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Michalek RD, Gerriets VA, Jacobs SR,
Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG and
Rathmell JC: Cutting Edge: Distinct glycolytic and lipid oxidative
metabolic programs are essential for effector and regulatory
CD4+ T cell subsets. J Immunol. 186:3299–3303. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Osthus RC, Shim H, Kim S, Li Q, Reddy R,
Mukherjee M, Xu Y, Wonsey D, Lee LA and Dang CV: Deregulation of
glucose transporter 1 and glycolytic gene expression by c-Myc. J
Biol Chem. 275:21797–21800. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kim JW, Gao P, Liu YC, Semenza GL and Dang
CV: Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively
induce vascular endothelial growth factor and metabolic switches
hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol.
27:7381–7393. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dang CV, Kim JW, Gao P and Yustein J: The
interplay between MYC and HIF in cancer. Nat Rev Cancer. 8:51–56.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kim JW, Tchernyshyov I, Semenza GL and
Dang CV: HIF-1-mediated expression of pyruvate dehydrogenase
kinase: A metabolic switch required for cellular adaptation to
hypoxia. Cell Metab. 3:177–185. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Elstrom RL, Bauer DE, Buzzai M, Karnauskas
R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM and
Thompson CB: Akt stimulates aerobic glycolysis in cancer cells.
Cancer Res. 64:3892–3899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Makinoshima H, Takita M, Saruwatari K,
Umemura S, Obata Y, Ishii G, Matsumoto S, Sugiyama E, Ochiai A, Abe
R, et al: Signaling through the phosphatidylinositol 3-kinase
(PI3K)/mammalian target of rapamycin (mTOR) axis is responsible for
aerobic glycolysis mediated by glucose transporter in epidermal
growth factor receptor (EGFR)-mutated lung adenocarcinoma. J Biol
Chem. 290:17495–17504. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Laplante M and Sabatini DM: mTOR signaling
at a glance. J Cell Sci. 122:3589–3594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Woo YM, Shin Y, Lee EJ, Lee S, Jeong SH,
Kong HK, Park EY, Kim HK, Han J, Chang M, et al: Inhibition of
aerobic glycolysis represses Akt/mTOR/HIF-1α axis and restores
tamoxifen sensitivity in antiestrogen-resistant breast cancer
cells. PLoS One. 10:e01322852015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cheng SC, Quintin J, Cramer RA, Shepardson
KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JH, Rao
NA, Aghajanirefah A, et al: mTOR- and HIF-1α-mediated aerobic
glycolysis as metabolic basis for trained immunity. Science.
345:12506842014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Denko NC: Hypoxia, HIF1 and glucose
metabolism in the solid tumour. Nat Rev Cancer. 8:705–713. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lu H, Forbes RA and Verma A:
Hypoxia-inducible factor 1 activation by aerobic glycolysis
implicates the Warburg effect in carcinogenesis. J Biol Chem.
277:23111–23115. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu J, Zhang C, Hu WW and Feng ZH: Tumor
suppressor p53 and its mutants in cancer metabolism. Cancer Lett.
356:197–203. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Vousden KH and Ryan KM: p53 and
metabolism. Nat Rev Cancer. 9:691–700. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yeung SJ, Pan J and Lee MH: Roles of p53,
MYC and HIF-1 in regulating glycolysis-the seventh hallmark of
cancer. Cell Mol Life Sci. 65:3981–3999. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ogawara Y, Kishishita S, Obata T, Isazawa
Y, Suzuki T, Tanaka K, Masuyama N and Gotoh Y: Akt enhances
Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem.
277:21843–21850. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Warburg O, Gawehn K and Geissler AW:
Metabolism of leukocytes. Z Naturforsch B. 13B:515–516. 1958.(In
German). View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Jacobs SR, Michalek RD and Rathmell JC:
IL-7 is essential for homeostatic control of T cell metabolism in
vivo. J Immunol. 184:3461–3469. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang R and Green DR: Metabolic checkpoints
in activated T cells. Nat Immunol. 13:907–915. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Vander-Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Van der Windt GJ, Everts B, Chang CH,
Curtis JD, Freitas TC, Amiel E, Pearce EJ and Pearce EL:
Mitichondrial respiratory capacity is a critical regulator of
CD8+ T cell memory development. Immunity. 36:68–78.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
van der Windt GJ, O'Sullivan D, Everts B,
Huang SC, Buck MD, Curtis JD, Chang CH, Smith AM, Ai T, Faubert B,
et al: CD8 memory T cells have a bioenergetic advantage that
underlies their rapid recall ability. Proc Natl Acad Sci USA.
110:14336–14341. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Mannava S, Grachtchouk V, Wheeler LJ, Im
M, Zhuang D, Slavina EG, Mathews CK, Shewach DS and Nikiforov MA:
Direct role of nucleotide metabolism in C-MYC-dependent
proliferation of melanoma cells. Cell Cycle. 7:2392–2400. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Shen Y, Wei Y, Wang Z, Jing Y, He H, Yuan
J, Li R, Zhao Q, Wei L, Yang T, et al: TGF-β regulates
hepatocellular carcinoma progression by inducing Treg cell
polarization. Cell Physiol Biochem. 35:1623–1632. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kalathil S, Lugade AA, Miller A, Iyer R
and Thanavala Y: Higher frequencies of
GARP+CTLA-4+Foxp3+ T regulatory
cells and myeloid-derived suppressor cells in hepatocellular
carcinoma patients are associated with impaired T-cell
functionality. Cancer Res. 73:2435–2444. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Galgani M, De Rosa V, La Cava A and
Matarese G: Role of metabolism in the immunobiology of regulatory T
cells. J Immunol. 197:2567–2575. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Procaccini C, Carbone F, Di Silvestre D,
Brambilla F, De Rosa V, Galgani M, Faicchia D, Marone G, Tramontano
D, Corona M, et al: The protemic landscape of human ex vivo
regulatory and conventional T cells reveals specific metabolic
requirements. Immunity. 44:406–421. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gao P, Tchernyshyov I, Chang TC, Lee YS,
Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et
al: c-Myc suppression of miR-23a/b enhances mitochondrial
glutaminase expression and glutamine metabolism. Nature.
458:762–765. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Cham CM, Driessens G, O'Keefe JP and
Gajewski TF: Glucose deprivation inhibits multiple key gene
expression events and effector functions in CD8+ T
cells. J Immunol. 38:2438–2450. 2008.
|
|
77
|
Siska PJ, van der Windt GJ, Kishton RJ,
Cohen S, Eisner W, MacIver NJ, Kater AP, Weinberg JB and Rathmell
JC: Suppression of Glut1 and glucose metabolism by decreased
Akt/mTORC1 signaling drives T cell impairment in B cell leukemia. J
Immunol. 197:2532–2540. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Dang EV, Barbi J, Yang HY, Jinasena D, Yu
H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, et al: Control of
TH17/Treg balance by hypoxia-inducible factor
1. Cell. 146:772–784. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Chen L and Flies DB: Molecular mechanisms
of T cell co-stimulation and co-inhibition. Nat Rev Immunol.
3:227–242. 2013. View Article : Google Scholar
|
|
80
|
Wieman HL, Wofford JA and Rathmell JC:
Cytokine stimulation promotes glucose uptake via
phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and
trafficking. Mol Biol Cell. 18:1437–1446. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Delgoffe GM, Kole TP, Zheng Y, Zarek PE,
Matthews KL, Xiao B, Worley PF, Kozma SC and Powell JD: The mTOR
kinase differentially regulates effector and regulatory T cell
lineage commitment. Immunity. 30:832–844. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Liu C, Chapman NM, Karmaus PW, Zeng H and
Chi H: mTOR and metabolic regulation of conventional and regulatory
T cells. J Leukoc Biol. 7:837–847. 2015. View Article : Google Scholar
|
|
83
|
Procaccini C, De Rosa V, Galgani M, Abanni
L, Calì G, Porcellini A, Carbone F, Fontana S, Horvath TL, La Cava
A, et al: An oscillatory switch in mTOR kinase activity sets
regulatory T cell responsiveness. Immunity. 33:929–941. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Mihaylova MM and Shaw RJ: The AMPK
signalling pathway coordinates cell growth, autophagy and
metabolism. Nat Cell Biol. 13:1016–1023. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Blagih J, Coulombe F, Vincent EE, Dupuy F,
Galicia-Vázquez G, Yurchenko E, Raissi TC, van der Windt GJ,
Viollet B, Pearce EL, et al: The energy sensor AMPK regulates T
cell metabolic adaptation and effector responses in vivo. Immunity.
42:41–54. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Dunn GP, Bruce AT, Ikeda H, Old LJ and
Schreiber RD: Cancer immunoediting: From immunosurveilance to tumor
escape. Nat Immunol. 3:991–998. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Liu Y and Cao XT: Immunosuppressive cells
in tumor immune escape and metastasis. J Mol Med. 94:509–522. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wang TT, Liu GW and Wang RN: The
intercellular metabolic interplay between tumor and immune cells.
Front Immunol. 5:3582014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Slomiany MG, Grass GD, Robertson AD, Yang
XY, Maria BL, Beeson C and Toole BP: Hyaluronan, CD44, and emmprin
regulate lactate efflux and membrane localization of
monocarboxylate transporters in human breast carcinoma cells.
Cancer Res. 69:1293–1301. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Hirschhaeuser F, Sattler UG and
Mueller-Klieser W: Lactate: A metabolic key player in cancer.
Cancer Res. 71:6921–6925. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Le Floch R, Chiche J, Marchiq I, Naiken T,
Ilc K, Murray CM, Critchlow SE, Roux D, Simon MP and Pouysségur J:
CD147 subunit of lactate/H+ symporters MCT1 and
hypoxia-inducible MCT4 is critical for energetics and growth of
glycolytic tumors. Proc Natl Acad Sci USA. 108:16663–16668. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Martinez-Outschoorn UE, Prisco M, Ertel A,
Tsirigos A, Lin Z, Pavlides S, Wang C, Flomenberg N, Knudsen ES,
Howell A, et al: Ketones and lactate increase cancer cell
‘stemness,’ driving recurrence, metastasis and poor clinical
outcome in breast cancer: Achieving personalized medicine via
Metabolo-Genomics. Cell Cycle. 10:1271–1286. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Goetze K, Walenta S, Ksiazkiewicz M,
Kunz-Schughart LA and Mueller-Klieser W: Lactate enhances motility
of tumor cells and inhibits monocyte migration and cytokine
release. Int J Oncol. 39:453–463. 2011.PubMed/NCBI
|
|
94
|
Wong TY, Phillips AO, Witowski J and
Topley N: Glucose-mediated induction of TGF-β1 and MCP-1 in
mesothelial cells in vitro is osmolality and polyol pathway
dependent. Kidney Int. 63:1404–1416. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Kottmann RM, Kulkarni AA, Smolnycki KA,
Lyda E, Dahanayake T, Salibi R, Honnons S, Jones C, Isern NG, Hu
JZ, et al: Lactic acid is elevated in idiopathic pulmonary fibrosis
and induces myofibroblast differentiation via pH-dependent
activation of transforming growth factor-β. Am J Respir Crit Care
Med. 186:740–751. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Rudrabhatla SR, Mahaffey CL and Mummert
ME: Tumor microenvironment modulates hyaluronan expression: The
lactate effect. J Invest Dermatol. 126:1378–1387. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Gottfried E, Kreutz M and Mackensen A:
tumor metabolism as modulator of immune response and tumor
progression. Semin Cancer Biol. 22:335–341. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Husain Z, Huang Y, Seth P and Sukhatme VP:
Tumor-derived lactate modifies antitumor immune response: Effect on
myeloid-derived suppressor cells and NK cells. J Immunol.
191:1486–1495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Feder-Mengus C, Ghosh S, Weber WP, Wyler
S, Zajac P, Terracciano L, Oertli D, Heberer M, Martin I, Spagnoli
GC, et al: Multiple mechanisms underlie defective recognition of
melanoma cells cultured in three-dimensional architectures by
antigen-specific cytotoxic T lymphocytes. Br J Cancer.
96:1072–1082. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Mendler AN, Hu B, Prinz PU, Kreutz M,
Gottfried E and Noessner E: Tumor lactic acidosis suppresses CTL
function by inhibition of p38 and JNK/c-Jun activation. Int J
Cancer. 131:633–640. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Gatenby RA, Gawlinski ET, Gmitro AF,
Kaylor B and Gillies RJ: Acid-mediated tumor invasion: A
multidisciplinary study. Cancer Res. 66:5216–5223. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
McCarty MF and Whitaker J: Manipulating
tumor acidification as a cancer treatment strategy. Altern Med Rev.
15:264–272. 2010.PubMed/NCBI
|
|
103
|
Hu XY and Ivashkiv LB: Cross-regulation of
signaling pathways by interferon-gamma: Implications for immune
responses and autoimmune diseases. Immunity. 31:539–550. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Nathan I, Groopman JE, Quan SG, Bersch N
and Golde DW: Immune (gamma) interferon produced by a human
T-lymphoblast cell line. Nature. 292:842–844. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Mekhail K, Gunaratnam L, Bonicalzi ME and
Lee S: HIF activation by pH-dependent nucleolar sequestration of
VHL. Nat Cell Biol. 6:642–647. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
McMahon S, Charbonneau M, Grandmont S,
Richard DE and Dubois CM: Transforming growth factor beta1 induces
hypoxia-inducible factor-1 stabilization through selective
inhibition of PHD2 expression. J Biol Chem. 281:24171–24181. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Clambey ET, McNamee EN, Westrich JA,
Glover LE, Campbell EL, Jedlicka P, de Zoeten EF, Cambier JC,
Stenmark KR, Colgan SP, et al: Hypoxia-inducible factor-1
alpha-dependent induction of FoxP3 drives regulatory T-cell
abundance and function during inflammatory hypoxia of the mucosa.
Proc Natl Acad Sci USA. 109:E2784–E2793. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Hung SP, Yang MH, Tseng KF and Lee OK:
Hypoxia-induced secretion of TGF-β1 in mesenchymal stem cell
promotes breast cancer cell progression. Cell Transplant.
22:1869–1882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Sanjabi S, Oh SA and Li MO: Regulation of
the immune response by TGF-β: From conception to autoimmunity and
infection. Cold Spring Harb Perspect Biol. 9(pii): a0222362017.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Barsoum IB, Smallwood CA, Siemens DR and
Graham CH: A mechanism of hypoxia-mediated escape from adaptive
immunity in cancer cells. Cancer Res. 74:665–674. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Park BV, Freeman ZT, Ghasemzadeh A,
Chattergoon MA, Rutebemberwa A, Steigner J, Winter ME, Huynh TV,
Sebald SM, Lee SJ, et al: TGF-β1-mediated SMAD3 enhances PD-1
expression on antigen-specific T Cells in cancer. Cancer Discov.
6:1366–1381. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Wei S, Shreiner AB, Takeshita N, Chen L,
Zou W and Chang AE: Tumor-induced immune suppression of in vivo
effector T-cell priming is mediated by the B7-H1/PD-1 axis and
transforming growth factor beta. Cancer Res. 68:5432–5438. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Mamori S, Nagatsuma K, Matsuura T, Ohkawa
K, Hano H, Fukunaga M, Matsushima M, Masui Y, Fushiya N, Onoda H,
et al: Useful detection of CD147 (EMMPRIN) for pathological
diagnosis of early hepatocellular carcinoma in needle biopsy
samples. World J Gastroenterol. 13:2913–2917. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Tang J, Wu YM, Zhao P, Yang XM, Jiang JL
and Chen ZN: Overexpression of HAb18G/CD147 promotes invasion an
metastasis via alpha3beta1 integrin mediated FAK-paxilli and
FAK-PI3K-Ca2+ pathways. Cell Mol Life Sci. 65:2933–2942.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Dai JY, Dou KF, Wang CH, Zhao P, Lau WB,
Tao L, Wu YM, Tang J, Jiang JL and Chen ZN: The interaction of
HAb18G/CD147 with integrin α6β1 and its implications for the
invasion potential of human hepatoma cells. BMC Cancer. 9:337–346.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zhao P, Zhang W, Tang J, Ma XK, Dai JY, Li
Y, Jiang JL, Zhang SH and Chen ZN: Annexin II promotes invasion and
migration of human hepatocellular carcinoma cells in vitro via its
interaction with HAb18G/CD147. Cancer Sci. 101:387–395. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Baba M, Inoue M, Itoh K and Nishizawa Y:
Blocking CD147 induces cell death in cancer cells through
impairment of glycolytic energy metabolism. Biochem Biophys Res
Commun. 374:111–116. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Su J, Chen X and Kanekura TA:
CD147-targeting siRNA inhibits the proliferation, invasiveness, and
VEGF production of human malignant melanoma cells by
down-regulating glycolysis. Cancer Lett. 273:140–147. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Huang QC, Li JB, Xing JL, Li WW, Li HW, Ke
X, Zhang J, Ren TT, Shang YK, Yang HS, et al: CD147 promotes
reprogramming of glucose metabolism and cell proliferation in HCC
cells by inhibiting the p53-dependent signaling pathway. J Hepatol.
61:859–866. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Ke X, Fei F, Chen YK, Xu L, Zhang Z, Huang
QC, Zhang HX, Yang HS, Chen ZN and Xing JL: Hypoxia upregulates
CD147 through a combined effect of HIF-1alpha and Sp1 to promote
glycolysis and tumor progression in epithelial solid tumors.
Carcinogenesis. 33:1598–1607. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Murata M, Matsuzaki K, Yoshida K, Sekimoto
G, Tahashi Y, Mori S, Uemura Y, Sakaida N, Fujisawa J, Seki T, et
al: Hepatitis B virus X protein shifts human hepatic transforming
factor (TGF)-beta signaling from tumor suppression to oncogenesis
in early chronic hetatitis B. Hepatology. 49:1203–1217. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Wu J, Ru NY, Zhang Y, Li Y, Wei D, Ren Z,
Huang XF, Chen ZN and Bian H: HAb18G/CD147 promotes
epithelial-mesenchymal transition through TGF-β signaling and is
transcriptionally regulated by Slug. Oncogene. 30:4410–4427. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Wang GL, Jiang BH, Rue EA and Semenza GL:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS
heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci
USA. 92:5510–5514. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Christofk HR, Vander Heiden MG, Harris MH,
Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and
Cantley LC: The M2 splice isoform of pyruvate kinase is important
for cancer metabolism and tumour growth. Nature. 52:230–233. 2008.
View Article : Google Scholar
|
|
127
|
Kong LM, Liao CG, Chen L, Yang HS, Zhang
SH, Zhang Z, Bian HJ, Xing JL and Chen ZN: Promoter hypomethylation
up-regulates CD147 expression through increasing Sp1 binding and
associates with poor prognosis in human hepatocellular carcinoma. J
Cell Mol Med. 15:1415–1428. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Guo H, Majmudar G, Jensen TC, Biswas C,
Toole BP and Gordon MK: Characterization of the gene for human
EMMPRIN, a tumor cell surface inducer of matrix metalloproteinases.
Gene. 220:99–108. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Yang H, Zou W and Chen BL: Overexpression
of CD147 in ovarian cancer is initiated by the hypoxic
microenvironment. Cell Biol Int. 37:1139–1142. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Kong LM, Liao CG, Fei F, Guo X, Xing JL
and Chen ZN: Transcription factor Sp1 regulates expression of
cancer-associated molecule CD147 in human lung cancer. Cancer Sci.
101:1463–1470. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Kono K: Current status of cancer
immunotherapy. J Stem Cells Regen Med. 10:8–13. 2014.PubMed/NCBI
|
|
132
|
Sangro B, Gomez-Martin C, de la Mata M,
Iñarrairaegui M, Garralda E, Barrera P, Riezu-Boj JI, Larrea E,
Alfaro C, Sarobe P, et al: A clinical trial of CTLA-4 blockade with
tremelimumab in patients with hepatocellular carcinoma and chronic
hepatitis C. J Hepatol. 59:81–88. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Prat A, Navarro A, Paré L, Reguart N,
Galván P, Pascual T, Martínez A, Nuciforo P, Comerma L, Alos L, et
al: Immune-related gene expression profiling after PD-1 blockade in
non-small cell lung carcinoma, head and neck squamous cell
carcinoma and melanoma. Cancer Res. 77:3540–3550. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Cabel L, Riva F, Servois V, Livartowski A,
Daniel C, Rampanou A, Lantz O, Romano E, Milder M, Buecher B, et
al: Circulating tumor DNA changes for early monitoring of anti-PD1
immunotherapy: A proof-of-concept study. Ann Oncol. 28:1996–2001.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Patsoukis N, Bardhan K, Chatterjee P, Sari
D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, et al:
PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis
and promoting lipolysis and fatty acid oxidation. Nat Commun.
6:66922015. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Noman MZ, Desantis G, Janji B, Hasmim M,
Karray S, Dessen P, Bronte V and Chouaib S: PD-L1 is a novel direct
target of HIF-1α, and its blockade under hypoxia enhanced
MDSC-mediated T cell activation. J Exp Med. 211:781–790. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Kizaka-Kondoh S, Tanaka S, Harada H and
Hiraoka M: The HIF-1-active microenvironment: An environmental
target for cancer therapy. Adv Drug Deliv Rev. 61:623–632. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Li B and Simon MC: Molecular pathways:
Targeting MYC-induced metabolic reprogramming and oncogenic stress
in cancer. Clin Cancer Res. 19:5835–5841. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Okkenhaug K, Graupera M and Vanhaesebroeck
B: Targeting PI3K in cancer: Impact on tumor cells, their
protective stroma, angiogenesis, and immunotherapy. Cancer Discov.
6:1090–1105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Thorpe LM, Yuzugullu H and Zhao JJ: PI3K
in cancer: Divergent roles of isoforms, modes of activation and
therapeutic targeting. Nat Rev Cancer. 15:7–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Troncone M, Cargnelli SM, Villani LA,
Isfahanian N, Broadfield LA, Zychla L, Wright J, Pond G, Steinberg
GR and Tsakiridis T: Targeting metabolism and AMP-activated kinase
with metformin to sensitize non-small cell lung cancer (NSCLC) to
cytotoxic therapy; translational biology and rationale for current
clinical trials. Oncotarget. 8:57733–57754. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Luchsinger JA, Ma Y, Christophi CA, Florez
H, Golden SH, Hazuda H, Crandall J, Venditti E, Watson K, Jeffries
S, et al: Diabetes Prevention Program Research Group: Metformin,
lifestyle intervention, and cognition in the diabetes prevention
program outcomes study. Diabetes Care. 40:958–965. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Anisimov VN, Berstein LM, Egormin PA,
Piskunova TS, Popovich IG, Zabezhinski MA, Kovalenko IG, Poroshina
TE, Semenchenko AV, Provinciali M, et al: Effect of metformin on
life span and on the development of spontaneous mammary tumors in
HER-2/neu transgenic mice. Exp Gerontol. 40:685–693. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Dowling RJ, Zakikhani M, Fantus IG, Pollak
M and Sonenberg N: Metformin inhibits mammalian target of
rapamycin-dependent translation initiation in breast cancer cells.
Cancer Res. 67:10804–10812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Libby G, Donnelly LA, Donnan PT, Alessi
DR, Morris AD and Evans JM: New users of metformin are at low risk
of incident cancer: A cohort study among people with type 2
diabetes. Diabetes Care. 32:1620–1625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Jiralerspong S, Palla SL, Giordano SH,
Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi
GN and Gonzalez-Angulo AM: Metformin and pathologic complete
responses to neoadjuvant chemotherapy in diabetic patients with
breast cancer. J Clin Oncol. 27:3297–3302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Christofk HR, Vander Heiden MG, Wu N,
Asara JM and Cantley LC: Pyruvate kinase M2 is a
phosphotyrosine-binding protein. Nature. 452:181–186. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Bian H, Zheng JS, Nan G, Li R, Chen C, Hu
CX, Zhang Y, Sun B, Wang XL, Cui SC, et al: Randomized trial of
[131I] metuximab in treatment of hepatocellular
carcinoma after percutaneous radiofrequency ablation. J Natl Cancer
Inst. 106(pii): dju2392014.PubMed/NCBI
|
|
150
|
Calvaresi EC and Hergenrother PJ: Glucose
conjugation for the specific targeting and treatment of cancer.
Chem Sci. 4:2319–2333. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Ciuleanu TE, Pavlovsky AV, Bodoky G, Garin
AM, Langmuir VK, Kroll S and Tidmarsh GT: A randomised Phase III
trial of glufosfamide compared with best supportive care in
metastatic pancreatic adenocarcinoma previously treated with
gemcitabine. Eur J Cancer. 45:1589–1596. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Baeuerle PA and Reinhardt C: Bispecific
T-cell engaging antibodies for cancer therapy. Cancer Res.
69:4941–4944. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Lameris R, de Bruin RC, Schneiders FL, van
Bergen en Henegouwen PM, Verheul HM, de Gruijl TD and van der Vliet
HJ: Bispecific antibody platforms for cancer immunotherapy. Crit
Rev Oncol Hematol. 92:153–165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Smith DM, Simon JK and Baker JR Jr:
Applications of nanotechnology for immunology. Nat Rev Immunol.
13:592–605. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Metcalfe SM and Fahmy TM: Targeted
nanotherapy for induction of therapeutic immune responses. Trends
Mol Med. 18:72–80. 2012. View Article : Google Scholar : PubMed/NCBI
|