Introduction
Lung cancer is the second most common cancer and the
leading cause of cancer-associated mortality worldwide. The vast
majority of lung cancer cases are non-small cell lung cancer
(NSCLC) (1,2). Epidermal growth factor receptor (EGFR)
is a receptor tyrosine kinase that belongs to the ErbB family and
mutations in this gene are a major driver of lung cancer (3–9).
Mutations in the tyrosine kinase region of EGFR involving the L858R
substitution or small internal deletions of exon 19, lead to
constitutive activation of EGFR (10). EGFR tyrosine kinase inhibitors
(TKIs) such as gefitinib (Gef) and erlotinib (Erl) inhibit the
phosphorylation of EGFR, preventing the subsequent activation of
the downstream signaling networks, which would lead to cancer cell
proliferation and survival (11).
EGFR TKIs are standard first-line therapies for patients with
advanced NSCLC with activating EGFR mutations, whereas they
show low efficacy in patients with wild-type EGFR (12–16).
Therefore, additional therapeutic strategies are urgently necessary
for NSCLC patients with wild-type EGFR.
Since the first TKI drug imatinib (Ima) was approved
for the treatment of chronic myeloid leukemia in 2001, more than 20
TKIs have been approved by the FDA. Lapatinib (Lap), which is used
to treat advanced or metastatic breast cancer, is a reversible dual
inhibitor of EGFR and ERBB2. The response to Lap is strongly
associated with ERBB2 overexpression, which inhibits the
phosphorylation of ERBB2 and its subsequent signaling molecules
(17). Sorafenib (Sor) is the first
anti-tumor drug targeting RAF kinase and VEGFR, that is approved
for treating renal cell and hepatocellular carcinomas. This agent
can directly inhibit tumor cell proliferation by blocking the
RAF/MEK/ERK signaling pathway and by inhibiting VEGFR-mediated
angiogenesis (18).
Fingolimod (FTY720), a sphingosine analog, was
recently shown to be highly effective for treating
relapsing-remitting multiple sclerosis (19). In addition to its immunomodulatory
effects, FTY720 showed preclinical antitumor efficacy in several
cancer models (20–23). In most cases, the anticancer
mechanism of FTY720 involves inhibition of the proto-oncogene
sphingosine kinase 1; however, the anticancer properties of FTY720
may be attributable to its effects on several other molecular
targets (24,25). Furthermore, FTY720 was reported to
modulate autophagy; a number of studies have reported the induction
of autophagy by FTY720 (26–29),
while others have reported autophagy suppression (30–33).
Autophagy exhibits complex, context-dependent properties in cancer,
and interventions that stimulate or inhibit autophagy have been
proposed as potential anti-cancer therapies (34).
Drug repositioning, which is the discovery of new
medical indications for existing drugs that are different from
their original indications, is an increasingly attractive mode of
therapeutic discovery (35–37). We previously reported that macrolide
antibiotics, such as clarithromycin and azithromycin, block
autophagy flux and that the combination of Gef with macrolide
antibiotics enhances the cytotoxic effect in NSCLC cells via
endoplasmic reticulum (ER)-stress loading (38). Under remediable levels of ER stress,
the homeostatic unfolded protein response (UPR) outputs activate
transcriptional and translational changes that promote cellular
adaptation (39–41). Contrary to its pro-survival roles,
prolonged UPR activation caused by severe or unresolved ER stress
leads to cell death (39–41). In the present study, in order to
discover more effective combinations of the existing drugs, the
cytotoxic effects of various TKIs against EGFR wild-type
NSCLC cells were investigated, using TKIs in single or combination
treatments with FTY720 as the repurposed candidate drug.
Materials and methods
Reagents
Gef, Erl, Lap, Sor and FTY720 were purchased from
Cayman Chemical Company (Ann Arbor, MI, USA). Ima was obtained from
Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). These drugs were
dissolved in dimethyl sulfoxide (DMSO) at a concentration of 20 mM
as a stock solution. FTY720 (S)-phosphate (FTY720-P) was also
purchased from Cayman Chemical Company and dissolved in DMSO at a
concentration of 0.5 mM as a stock solution. Thapsigargin (Tg) and
bafilomycin A1 (BafA1) were purchased from Wako Pure Chemical
Industries, Ltd. (Osaka, Japan). Tunicamycin (Tm) was purchased
from Nacalai Tesque, Inc. (Kyoto, Japan). Z-VAD-FMK (Z-VAD), a
pan-caspase inhibitor, was purchased from Peptide Institute, Inc.
(Osaka, Japan). Necrostatin-1 (Nec-1), a specific inhibitor of
receptor-interacting serine/threonine-protein kinase 1 (RIPK1), was
purchased from Enzo Life Sciences, Inc. (Farmingdale, NY, USA).
Azithromycin was purchased from Tokyo Chemical Industry Co., Ltd.
These reagents: Tg, BafA1, Tm, Z-VAD, Nec-1 and azithromycin were
also dissolved in DMSO as a stock solution.
Cell lines
Human NSCLC cell lines A549, H596, and H226, as well
as the ERBB2-positive breast cancer cell line BT474 were purchased
from American Type Culture Collection (Manassas, VA, USA). These
cell lines were cultured in RPMI-1640 medium (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany), supplemented with 10% heat inactivated
fetal bovine serum (Biosera, Nuaillé, France) at 37°C in a 5%
CO2 atmosphere. Stable A549/GFP-LC3-RFP-LC3ΔG cell lines
were generated as follows: A549 cells (2×106 cells/100
mm dish) were transfected with the plasmid DNA (GFP-LC3-RFP-LC3ΔG
plasmid, 10 µg) using Lipofectamine® 3000 (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) according to the manufacturer's
instructions. The GFP-LC3-RFP-LC3ΔG plasmid used in the present
study was a kind gift from Dr N. Mizushima (University of Tokyo,
Tokyo, Japan). After selecting transfected cells using puromycin (2
µg/ml), single clones of the cells were isolated, and GFP-LC3 and
RFP-LC3ΔG expression was confirmed by immunoblotting using specific
antibodies against green fluorescent protein (GFP), red fluorescent
protein (RFP), and microtubule associated proteins 1A/1B light
chain 3B (MAP1LC3B).
Cell viability assay
The number of viable cells was assessed by the
CellTiter Blue cell viability assay kit (Promega Corporation,
Madison, WI, USA) according to the manufacturer's instructions.
Briefly, cells were incubated with the CellTiter Blue reagent for 2
h at 37°C in 5% CO2 atmosphere. Fluorescence (560 nm for
excitation and 590 nm for emission) was measured using the
POWERSCAN HT 96-well plate reader (BioTek Instruments, Inc.,
Winooski, VT, USA). In order to determine IC50
(half-maximal inhibitory concentration) values, A549 cells treated
with different concentrations of TKIs or FTY720 were tested for
cell viability at 48 h after the treatments. Dose response curves
were fitted and IC50 values were analyzed using the
four-parameter logistic regression analysis by means of ImageJ
software (version 1.52a).
Immunoblotting
Total cellular proteins were extracted using
radioimmunoprecipitation assay lysis buffer containing 50 mM
Tris-HCl (pH 8.0), 150 mM NaCl, 1.0% Nonidet P-40, 0.5% sodium
deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and a protease
inhibitor cocktail (Nacalai Tesque, Inc.). Each sample was
sonicated for 20 pulses (0.5 sec on/off) to disrupt the aggregated
proteins, using a Branson 450D Sonifier (Emerson, Danbury, CT,
USA). Protein concentrations were measured using a Bicinchoninic
Acid Protein Assay kit (Pierce; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Equal amounts of
proteins were resolved by SDS-PAGE (5–20% gradient gel) and
transferred onto Immobilon-P membranes (EMD Millipore, Billerica,
MA, USA). The membranes were then blocked with 5% non-fat milk for
1 h at room temperature and probed with the following primary
antibodies at 4°C overnight: Anti-MAP1LC3B (cat. no. NB600-1384;
1:4,000 dilution; Novus Biologicals, Inc. Littleton, CO, USA).
Anti-sequestosome (SQSTM1; cat. no. sc-28359; 1:1,000 dilution),
anti-GAPDH (cat. no. sc-32233; 1:2,000 dilution), anti-heat shock
protein family A member 5 (HSPA5; cat. no. sc-13968; 1:1,000
dilution), anti-β-actin (cat. no. sc-47778; 1:2,000 dilution),
anti-EGFR (cat. no. sc-03; 1:1,000 dilution), and
anti-phosphorylated (p)-EGFR (Tyr1173; cat. no. sc-101668, 1:1,000
dilution) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA); anti-cyclin D (CCND)1 (cat. no. 2978, 1:1,000 dilution),
anti-CCND3 (cat. no. 2936, 1:1,000 dilution), anti-cyclin-dependent
kinase (CDK)4 (cat. no. 12790, 1:1,000 dilution), anti-CDK6 (cat.
no. 3136, 1:1,000 dilution), anti-DNA damage-inducible transcript 3
protein (DDIT3; cat. no. 2895, 1:1,000 dilution), anti-eukaryotic
translation initiation factor 2-α kinase 3 (EIF2AK3; cat. no. 3192,
1:1,000 dilution), anti-p-CDK substrate motif [(K/H)pSP] MultiMab™
(cat. no. 9477, 1:1,000 dilution), anti-CCNA2 (cat. no. 4656,
1:1,000 dilution), anti-CCNB1 (cat. no. 4138, 1:1,000 dilution),
anti-autophagy protein 5 (ATG5; cat. no. 12994, 1:1,000 dilution),
anti-RIPK1 (cat. no. 3493, 1:1,000 dilution), anti-ERBB2 (cat. no.
4290, 1:1,000 dilution), and anti-p-ERBB2 (Tyr1221/1222) (cat. no.
2243, 1:1,000 dilution) from Cell Signaling Technology (Danvers,
MA, USA). Anti-CDK1 (A17) antibody was a kind gift from Dr J Gannon
and Dr T Hunt (Francis Crick Institute, London, UK). The membranes
were followed by incubation with horseradish peroxidase-conjugated
secondary antibodies [anti-mouse immunoglobulin G (IgG); cat. no.
115-035-003; 1:5,000 dilution; or anti-rabbit IgG; cat. no.
711-035-152; 1:5,000 dilution; Jackson ImmunoResearch Laboratories,
Inc., West Grove, PA, USA] for 1 h at room temperature.
Immunoreactive proteins were detected with enhanced
chemiluminescence reagent (Immobilon Western Chemiluminescent HRP
Substrate; EMD Millipore). Densitometry analysis was performed
using the WSE-6300 Luminograph III molecular imager (ATTO
Corporation, Tokyo, Japan) and ATTO CS Analyzer 4 densitograph
software (version 2.3.1, ATTO Corporation).
Gene expression analysis
Total RNA was extracted from A549 cells using a
NucleoSpin RNA kit (Takara Bio, Inc., Otsu, Japan) and reverse
transcribed to cDNA using PrimeScript RT Master mix (Takara Bio,
Inc.) according to the manufacturer's instructions. The expression
of ER stress-associated genes was determined by quantitative
polymerase chain reaction (qPCR) using SYBR Premix Ex Taq II Tli
RNase H Plus (Takara Bio, Inc.). The sequences of validated primers
and reaction conditions were as follows (42,43):
5′-CCTAGCTGTGTCAGAATCTCCATCC-3′ and 5′-GTTTCAATGTCACCATCCAAGATCC-3′
for HSPA5; 5′-AAATCAGAGCTGGAACCTGAGGA-3′ and
5′-CCATCTCTGCAGTTGGATCAGTC-3′ for DDIT3;
5′-AACCAGCAGTTCCCTTCCTG-3′ and 5′-TTGCCTCTCGCTCACCATAC-3′ for
protein phosphatase 1 regulatory subunit 15A (PPP1R15A);
5′-AAGTGCCGCACAGGGTGTCC-3′ and 5′-GCTGGGACTTCCCCACTGTGC-3′ for
tumor necrosis factor receptor superfamily member 10B
(TNFRSF10B); 5′-CCCGATCGTGAAGCAGTTAGA-3′ and
5′-CAGAACCACCTTTATAGGTCCTGAA-3′ for ER to nucleus signaling 1
(ERN1); 5′-AAGCCCTGATGGTGCTAACTGAA-3′ and
5′-CATGTCTATGAACCCATCCTCGAA-3′ for activating transcription factor
6 (ATF6); and 5′-GCACCGTCAAGGCTGAGAAC-3′ and
5′-TGGTGAAGACGCCAGTGGA-3′ for GAPDH. qPCR was performed in a
Thermal Cycler Dice Real-Time System TP800 (Takara Bio, Inc.) under
the following conditions: Initial denaturation at 95°C for 30 sec,
followed by 45 cycles of the sequence of denaturation at 95°C for 5
sec and simultaneous annealing and extension at 60°C for 30 sec.
Data were analyzed using Thermal Cycler Dice Real-Time System
Software (Takara Bio, Inc.) and the comparative Cq method
(2−ΔΔCq) was used for relative quantification of gene
expression (44). The data were
standardized to GAPDH as an internal control.
Assessment of autophagy flux using the
GFP-LC3-RFP- LC3ΔG system
A549/GFP-LC3-RFP-LC3ΔG cells (8×103
cells/well) were plated on a 96-well plate 24 h before treatment
with FTY720. Fluorescence intensities derived from GFP-LC3B and
RFP-LC3ΔG were monitored during the 24 h exposure to the drug,
using an Incu-Cyte ZOOM cell imaging system (Essen BioScience,
Ltd., Ann Arbor, MI, USA). Autophagy flux was measured as
alterations in the relative intensities of GFP/RFP, using
DMSO-treated groups as the control (45).
Lysotracker staining
A549 cells (8×104 cells/well) were seeded
onto glass coverslips in a 24-well culture plate for 24 h. Next,
A549 cells were treated with FTY720 or BafA1 for 4 h and then
incubated for 30 min in 50 nM LysoTracker Red DND-99 (Molecular
Probes; Thermo Fisher Scientific, Inc.). Coverslips were washed
twice with PBS and cells were fixed in 2% paraformaldehyde for 10
min at room temperature. Following washing, the coverslips were
mounted in ProLong Diamond Antifade Mountant (Thermo Fisher
Scientific, Inc.). The cells were visualized using an LSM 700
confocal laser scanning fluorescence microscope (Zeiss GmbH, Jena,
Germany) equipped with Plan-Apochromat 40×/1.4 oil DIC (Zeiss
GmbH). All images were acquired and processed equally using ZEN
2012 software (version 8.1.0.484, Zeiss GmbH).
Cell cycle analysis
A549 cells (1×106 cells/100 mm dish) were
plated on 100 mm dishes. After seeding for 24 h, A549 cells were
treated with the indicated drugs for 24 h. The cells were harvested
and fixed in 75% ethanol for 1 h at 4°C. Next, the cells were
treated with RNaseA (0.1 mg/ml) at 37°C for 30 min and stained with
propidium iodide (PI) (25 µg/ml) for 15 min at room temperature.
DNA content was determined using Attune Acoustic Focusing Flow
Cytometer (Applied Biosystems; Thermo Fisher Scientific, Inc.) and
the cell cycle distribution was analyzed using ModFit LT version
5.0 (Verity Software House, Inc., Topsham, ME, USA).
Immunofluorescence analysis
A549 cells (8×104 cells/well) were seeded
onto glass coverslips in a 24-well culture plate for 24 h. A549
cells were subsequently treated with FTY720 or FTY720-P for 1 or 4
h. Coverslips were washed twice with PBS and cells were fixed in 2%
paraformaldehyde for 10 min at room temperature. Following washing,
cells were permeabilized in 0.1% Triton X-100 for 5 min, followed
by washing twice with PBS. The coverslips were blocked with 10%
newborn calf serum (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) for 1 h at room temperature, then incubated with a primary
anti-sphingosine 1-phosphate receptor 1 (S1PR1) antibody (cat. no.
ab11424; 1:200 dilution; Abcam, Cambridge, MA, USA) at 4°C
overnight. Following three washes, coverslips were incubated with
Alexa Fluor 488-conjugated anti-Rabbit IgG (cat. no. A-11034;
1:1,000 dilution; Molecular Probes; Thermo Fisher Scientific, Inc.)
and DAPI (1 µM; Sigma-Aldrich; Merck KGaA) at 37°C for 1 h.
Following washing, the coverslips were mounted in ProLong Diamond
Antifade Mountant (Thermo Fisher Scientific, Inc.). Cells were
visualized using an LSM 700 confocal laser scanning fluorescence
microscope (Zeiss GmbH) equipped with Plan-Apochromat 40×/1.4 oil
DIC (Zeiss GmbH) and acquired images were analyzed using ZEN 2012
software (version 8.1.0.484; Zeiss GmbH).
May-Grünwald-Giemsa staining
A549 (4×104 cells/well) cells were seeded
onto glass coverslips in a 24-well culture plate. After 24 h, the
cells were treated with the indicated reagents for 24 or 48 h.
Coverslips were washed twice with PBS and fixed in methanol for 10
min. These cells were stained with May-Grünwald's stain solution
(Muto Pure Chemicals Co., Ltd., Tokyo Japan) for 3 min at room
temperature. After adding equal volume of PBS, coverslips were left
to stand for another 3 min. Once the May-Grünwald's stain solution
was removed, the cells were stained with diluted (1:20 with water)
Giemsa's stain solution (Muto Pure Chemicals Co., Ltd.) for 20 min
at room temperature. After washing and drying, the cells were
examined under a BZ-8100 digital microscope (Keyence Co., Osaka,
Japan) equipped with PlanApo 20× NA0.75 (Nikon Corporation, Tokyo,
Japan).
Knockout of ATG5 gene by
CRISPR/Cas9-mediated genome editing
Target sequences for CRISPR interference for human
ATG5, AACTTGTTTCACGCTATATC (exon 2) or AAGAGTAAGTTATTTGACGT
(exon 3), were derived from a previous report (46). In total, two complementary
oligonucleotides with BpiI restriction sites for guide RNAs
(gRNAs) were synthesized at FASMAC (Kanagawa, Japan), and cloned
into the pX459 CRISPR/Cas9-Puro vector (Addgene, Inc., Cambridge,
MA, USA) deposited by the Feng Zhang Laboratory. A549 cells
(2×106 cells/100 mm dish) were transfected with
pX459-gRNA using Lipofectamine® 3000 according to the
manufacturer's instructions. The day after transfection, cells were
treated with 2 µg/ml puromycin for 2 days. Surviving cells were
diluted in growth medium (RPMI-1640 medium supplemented with 10%
heat inactivated fetal bovine serum) to prepare cell suspension (5
cells/ml) and distributed in 100 µl of the cell suspension per well
in a 96-well plate. The expression of ATG5 in the expanded colonies
was detected by immunoblotting using anti-ATG5 antibody (cat. no.
12994; 1:1,000 dilution) at 4°C overnight to select the
ATG5-depleted colonies. The genome sequences of the edited locus in
selected colonies were also confirmed by Sanger DNA sequencing
performed at FASMAC, which demonstrated that the expected deletion
and frameshifting were present in each exon of ATG5.
Gene silencing of RIPK1
A549 cells (2×106 cells/100 mm dish) were
transfected with either a control small interfering RNA (siRNA; 15
nM; cat. no. 46-2001; Thermo Fisher Scientific, Inc.) or Stealth
siRNA (15 nM) targeting human RIPK1 (HSS112847; cat. no.
10620318; Thermo Fisher Scientific, Inc.) using Lipofectamine
RNAiMAX (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. A total of 24 h post-transfection,
these cells were re-plated on a 96-well plate for cell viability
assays.
Statistical analysis
All data are shown as the mean ± standard deviation.
Statistical analysis was performed using a one-way analysis of
variance (ANOVA), followed by Dunnett's post hoc test for
comparisons with a control group, or with the Student-Newman-Keuls
post hoc test for multiple comparisons between all pairs of groups.
These statistical analyses were performed using Excel Statistical
Program File ystat2008 (Igakutosho-shuppan, Ltd., Tokyo, Japan).
P<0.01 was considered to indicate a statistically significant
difference.
Results and Discussion
Cell growth inhibition in NSCLC cells
after single or combined treatment with TKIs and FTY720
A549 cells, expressing wild-type EGFR, were treated
with different concentrations (0–40 µM) of the TKIs Sor, Gef, Erl,
Lap, and Ima (Fig. 1A). The
half-maximal inhibition concentration (IC50) values of
each tested compound were shown in Table I. Sor markedly decreased A549 cell
viability in a dose- and time-dependent manner. The effectiveness
of Sor may be attributable to the activating KRAS proto-oncogene,
GTPase (KRAS) mutations in A549 cells (47). A target molecule of Sor is Raf
kinase, a downstream effector molecule of KRAS (48). In contrast, Gef, Erl, and Lap only
slightly decreased A549 cell viability, while Ima showed nearly no
effects on A549 cell viability.
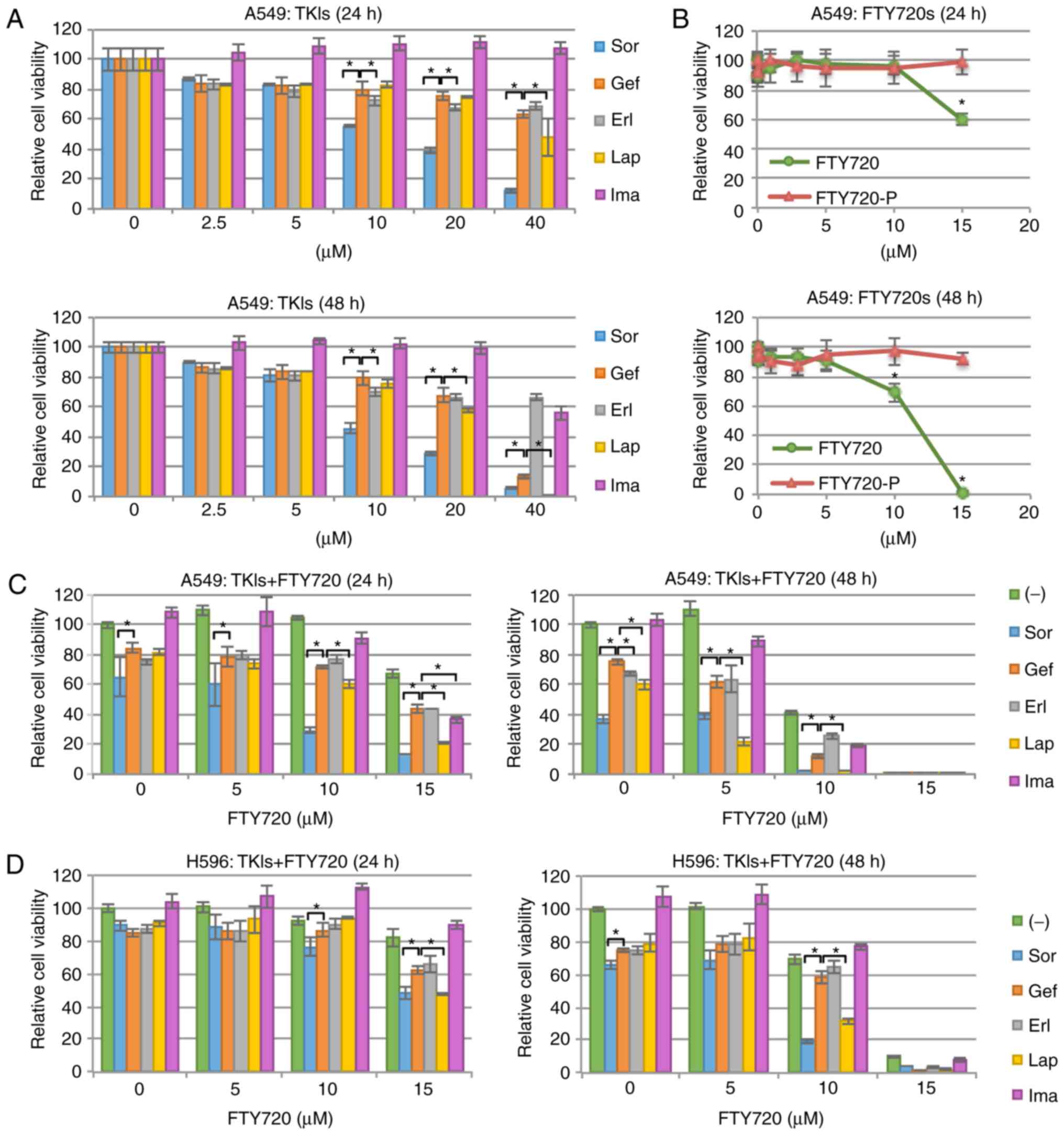 | Figure 1.Cell growth inhibition in NSCLC
cells. Cell viability was assessed at 24 and 48 h following
treatment with the indicated drug. (A) A549 cells were treated with
one of the following tyrosine kinase inhibitors: Sor, Gef, Erl, Lap
or Ima, at the indicated concentrations. *P<0.01 vs. treatment
with Gef. Gef-treated group were used for comparisons because Gef
is a well-known tyrosine kinase inhibitors for the treatment of
non-small cell lung cancer. (B) A549 cells were treated with FTY720
or FTY720-P at the indicated concentrations. *P<0.01 vs. 0 µM.
(C) A549 cells were treated with FTY720 at the indicated
concentrations, combined with Sor, Gef, Erl, Lap or Ima (at 10 µM).
*P<0.01 vs. combined treatment with Gef and FTY720. (D) H596
cells were also treated with reagents and cell viability was
assessed. *P<0.01 vs. combined treatment with Gef and FTY720.
Sor, sorafenib; Gef, gefitinib; Erl, erlotinib; Lap, lapatinib;
Ima, imatinib; FTY720-P, FTY720 (S)-phosphate. |
 | Table I.IC50 values of the drugs
used in this study in A549 cells. |
Table I.
IC50 values of the drugs
used in this study in A549 cells.
| Drugs | IC50
(µM) |
|---|
| Sorafenib | 11.1 |
| Gefitinib | 25.6 |
| Erlotinib | >40 |
| Lapatinib | 21.7 |
| Imatinib | >40 |
| FTY720 | 10.5 |
| FTY720-P | ND |
Treatment with 15 µM FTY720 resulted in apparent
cytotoxicity, although FTY720 at a concentration of ≤5 µM showed no
cytotoxicity (Fig. 1B). It is well
known that one of the biological effects of FTY720 is attributed to
its phosphorylated form, FTY720-P: It strongly binds to S1PR1,
inducing the internalization and degradation of S1PR1 at
submicromolar concentrations (19,25).
However, in the current study, the cytocidal activity of FTY720 in
A549 cells was observed only at micromolar concentrations, and
FTY720-P was not found to be cytocidal (Fig. 1B). In addition, most S1PR1 molecules
were localized in the nucleus in A549 cells and the localization of
S1PR1 did not change following treatment with FTY720 or FTY720-P
(Fig. S1). Therefore,
FTY720-mediated cytotoxicity in A549 cells was not attributed to
its phosphorylated form, FTY720-P.
Next, the effects of combined treatment with TKIs
and FTY720 was evaluated. It was found that the combined treatment
with Lap or Sor and FTY720 effectively enhanced A549 cell
cytotoxicity (Fig. 1C). To
determine what type of combinatory effect was exerted, the
combination index (CI) values were calculated. Specifically, CI
<1, CI=1, and CI >1 indicate synergistic, additive and
antagonistic effects, respectively (49). The CI value of combined treatment
with Sor (10 µM) and FTY720 (10 µM) was 0.74, and that of Lap (10
µM) and FTY720 (10 µM) was 0.77 (data not shown); therefore, both
combinations showed synergistic effects on A549 cells. In contrast,
FTY720 only marginally enhanced Gef- or Erl-induced cytotoxicity
(Fig. 1C). This synergistic effect
of Lap and FTY720 or Sor and FTY720 was also observed in another
NSCLC cell line H596, which also expresses wild-type EGFR (Fig 1D). These data demonstrated that the
cytotoxicity of NSCLC cells with wild-type EGFR was
increased following combined treatment with Lap or Sor and
FTY720.
Changes of autophagy flux in NSCLC
cells following treatment with FTY720
As described above, it has been previously reported
that macrolide antibiotics suppress autophagy flux and promote the
cytotoxic effects of TKIs in various cancer cell lines, including
A549 (38,50). Therefore, whether the modulation of
autophagy flux was involved in the enhanced cytotoxicity exerted by
FTY720 was examined. Treating A549 cells with each TKI resulted in
an increased expression of MAP1LC3B-II, a lipidated form of
MAP1LC3B, which is a hallmark of autophagosome formation (51) (Fig.
2A). Additionally, SQSTM1, a substrate of autophagy (51), decreased in response to Sor
treatment and accumulated in response to Gef, Lap, or Ima treatment
(Fig. 2A). These results suggested
that Sor induced autophagy flux and Gef, Lap or Ima suppressed
autophagy flux in A549 cells. Treating A549 cells with FTY720
resulted in an increased expression of MAP1LC3B-II and an
accumulation of SQSTM1 (Fig. 2B).
In addition, treatment with BafA1, a well-known lysosomal inhibitor
(52), also increased the
expression of MAP1LC3B-II, by blocking the catabolic flux of
autophagy. However, combined treatment with FTY720 and BafA1 failed
to further increase MAP1LC3B-II expression, compared with the
treatment with either FTY720 or BafA1 alone (Fig. 2B). This result indicated that FTY720
suppressed autophagy flux, but does not induce autophagy.
 | Figure 2.Alterations in autophagy flux. (A)
A549 cells were treated with Sor, Gef, Erl, Lap or Ima at the
indicated concentrations for 24 h, and with (B) FTY720 and BafA1
for 24 h at the indicated concentrations. The immunoblots show
MAP1LC3B and SQSTM1 expression. GAPDH was used as an internal
control. Band intensities were determined by densitometry and the
ratios of MAP1LC3B-II/GAPDH and SQSTM1/GAPDH are presented.
Representative data from multiple experiments are shown. (C)
A549/GFP-LC3-RFP-LC3ΔG cells were treated with Sor, Lap or FTY720
at the indicated concentrations. Fluorescence intensities derived
from GFP-LC3B and RFP-LC3ΔG were monitored over 24 h. Autophagy
flux was assessed as alterations in the relative intensities of
GFP/RFP, using DMSO-treated groups as a control. (D) A549 cells
were treated with FTY720 (10 µM) or BafA1 (10 nM) for 4 h and
stained with 50 nM LysoTracker Red DND-99. All images were acquired
and processed equally. Sor, sorafenib; Gef, gefitinib; Erl,
erlotinib; Lap, lapatinib; Ima, imatinib; DMSO, dimethyl sulfoxide;
GFP, green fluorescent protein; RFP, red fluorescent protein;
MAP1LC3B, microtubule-associated proteins 1A/1B light chain 3B;
SQSTM1, sequestosome; BafA1, bafilomycin A1. |
To analyze the autophagy flux, a GFP-LC3-RFP-
LC3ΔG-based system was used (45).
GFP-LC3-RFP-LC3ΔG is cleaved into equimolar amounts of GFP-LC3 and
RFP-LC3ΔG. GFP-LC3 is degraded by autophagy, while RFP-LC3ΔG
remains in the cytosol, functioning as an internal control. Thus,
autophagy flux can be estimated by calculating the GFP/RFP signal
ratio. In the present study, FTY720 or Lap evidently suppressed
autophagy flux in a dose-dependent manner, as indicated by an
increased GFP/RFP ratio, whereas Sor induced autophagy flux in a
dose-dependent manner, as shown by a reduced GFP/RFP ratio
(Fig. 2C). These results, namely
that Lap-mediated autophagy suppression and Sor-mediated autophagy
induction, obtained in A549 cells were consistent with a previous
study in HeLa cells (45).
Alterations in autophagy flux after treatment with Gef, Erl, or Ima
were small and were not dose-dependent (data not shown).
Furthermore, the effects of FTY720 on lysosomes
using LysoTracker dye were investigated, which stains lysosomes and
other acidic organelles. After treatment with FTY720 or BafA1 for 4
h, the intensity of LysoTracker dye was clearly decreased (Fig. 2D). This suggested that FTY720
inhibited the acidification of lysosomes and autolysosomes,
limiting protein degradation in these organelles and subsequently
inhibiting autophagy flux.
ER stress loading in NSCLC cells after
combined treatment with TKIs and FTY720
During the inhibition of intracellular proteolytic
processes, ER stress loading is pronounced, which reduces cell
proliferation and viability (43,53,54).
Because FTY720 suppressed autophagy flux (Fig. 2), whether ER stress loading was
involved in the enhanced cytotoxicity resulting from combination
treatment with FTY720 was investigated (Fig. 3A). Expression of ER
stress-associated proteins such as HSPA5 and DDIT3 was upregulated
in response to single treatment with Sor, but not with Gef or Lap.
However, combined treatment with Lap and 10 µM FTY720 markedly
upregulated the expression of these proteins, although single
treatment of FTY720 had no effects on protein expression.
Additionally, combined treatment of Lap with 5 µM FTY720 minimally
upregulated the expression of these proteins, indicating that the
expression of ER stress-associated proteins was dependent on the
concentration of FTY720, similar to cell viability. Furthermore,
following combined treatment with Lap and FTY720, a mobility shift
of EIF2AK3, an ER stress sensor protein, was detected (Fig. 3A), likely as a result of its
phosphorylation and activation (55).
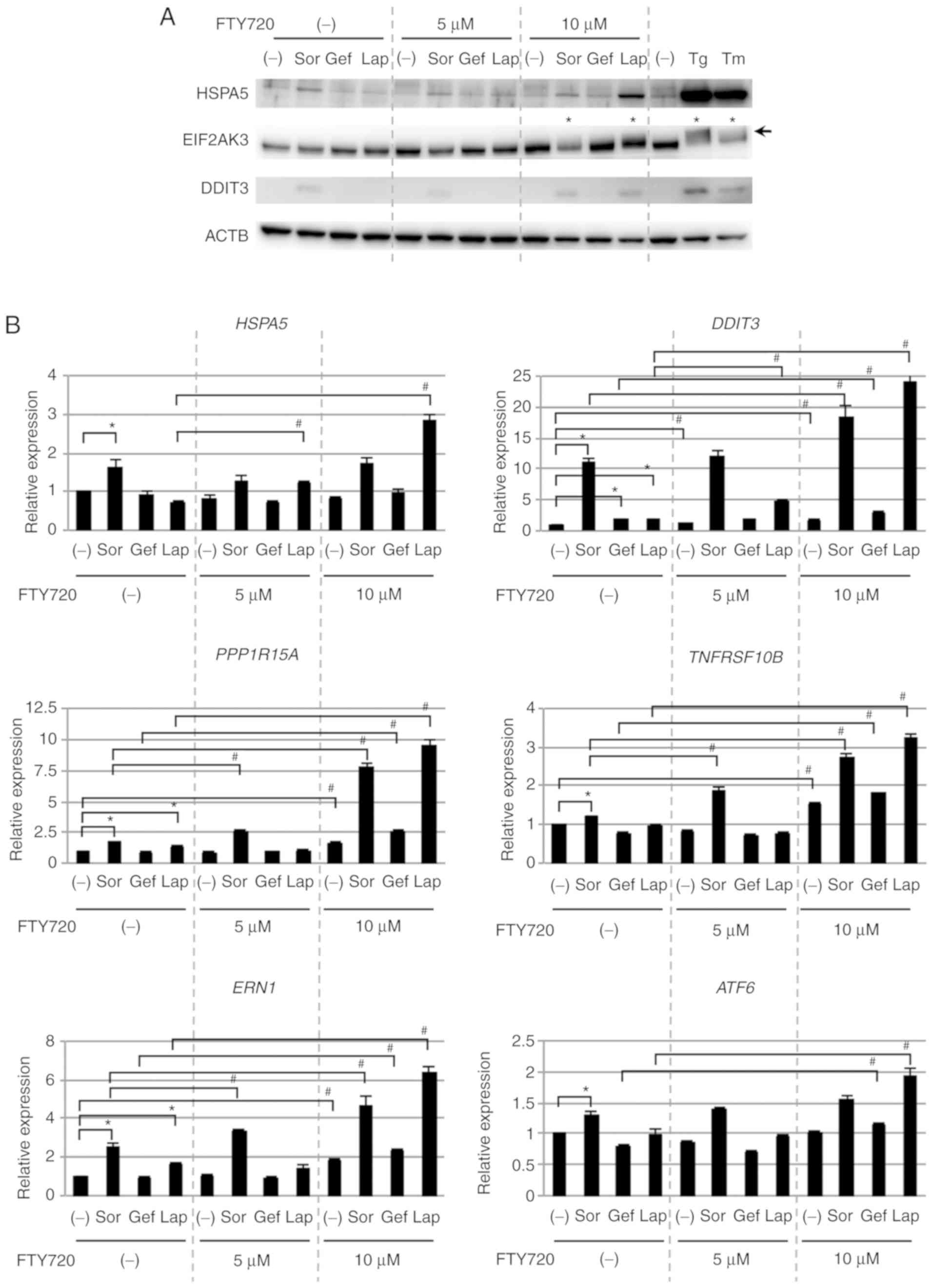 | Figure 3.ER stress loading in NSCLC cells. (A)
A549 cells were treated with one of the following tyrosine kinase
inhibitors (10 µM): Sor, Gef, or Lap in the absence or presence of
FTY720 (5 or 10 µM) for 24 h. Immunoblots showing expression of
HSPA5, EIF2AK3, and DDIT3. ACTB was used as an internal control. Tg
and Tm were used to induce ER stress as positive control reagents.
The arrow indicates the position of shifted EIF2AK3 bands.
Asterisks show lanes in which shifted EIF2AK3 bands were detected.
(B) A549 cells were treated as in Fig.
3A, and ER stress-associated gene expression was determined by
quantitative polymerase chain reaction analysis and normalized to
GAPDH. The expression level of each gene in dimethyl
sulfoxide-treated cells was designated as 1.0. *P<0.01 vs.
non-treated only in the absence of FTY720; #P<0.01
vs. each TKI-treated in the absence of FTY720. Sor, sorafenib; Gef,
gefitinib; Erl, erlotinib; Lap, lapatinib; Ima, imatinib; Tg,
thapsigargin; Tm, tunicamycin; ER, endoplasmic reticulum; HSPA5,
heat shock protein family A member 5; EIF2AK3, eukaryotic
translation initiation factor 2-α kinase 3; DDIT3, DNA
damage-inducible transcript 3 protein; ACTB, β-actin. |
qPCR data for HSPA5 and DDIT3
expression supported the immunoblotting data (Fig. 3B). Additionally, DDIT3-regulated
genes such as PPP1R15A and TNFRSF10B showed similar
regulation in response to Sor, Lap and FTY720 treatment.
Furthermore, the expression of the ER stress sensor genes,
ERN1 and ATF6 was upregulated after treatment with
Sor alone and after the combined treatment with Lap or Sor and
FTY720. Taken together, these data demonstrated that ER stress
loading was enhanced following treatment with Sor alone or in
combination with FTY720, as well as in response to Lap and FTY720
combined treatment, suggesting that ER stress loading was involved
in enhancing the cytotoxicity exerted by the combination treatment
with FTY720.
Cytostatic effects on NSCLC cells
after combined treatment with TKIs and FTY720
It has been reported that the exposure of cells to
ER stress loading leads to the activation of EIF2AK3,
phosphorylation of eIF2α, and subsequent repression of cyclin D
translation, resulting in cell cycle arrest (40,56–59).
Therefore, whether combination treatment with FTY720 induced
cytostatic effects in NSCLC cells was examined. The protein
expression of CCND1/D3 and CDK4/6 was suppressed after the
treatment with Sor alone and following combined treatment with Lap
and FTY720 (Fig. 4A). Additionally,
the protein expression of CCNA2, CCNB1, and CDK1, which are
regulators for G2/M phase of the cell cycle, was also
suppressed, particularly following combined treatment with Sor and
FTY720, or Lap and FTY720 (Fig.
4B). Furthermore, the expression of phosphorylated CDK
substrate motif was also reduced in response to these drugs
(Fig. 4C). Whether cell cycle
progression was affected by the combined treatments was further
investigated. Fig. 4D showed that
combined treatment with Lap and FTY720 or Sor and FTY720 caused
significant accumulation of A549 cells in
G0/G1 and G2/M phases, with a
concomitant decrease in the number of cells in S phase
(non-treated, 34.8%; Sor and FTY720, 14.7%; Lap and FTY720, 5.7%).
These data indicated that combined treatment with Lap and FTY720 or
Sor and FTY720 exerted cytostatic effects on NSCLC cells; these
effects were associated with the expression of ER stress-associated
proteins and A549 cell viability.
 | Figure 4.Cytostatic effects on NSCLC cells.
A549 cells were treated with one of the following tyrosine kinase
inhibitors (10 µM): Sor, Gef, or Lap in the absence or presence of
FTY720 (5 or 10 µM) for 24 h. Immunoblots showing the expression of
(A) CCND1, CCND3, CDK4, CDK6, (B) CCNA2, CCNB1 and CDK1, as well as
(C) proteins containing the phosphorylated CDK substrate motif.
GAPDH was used as an internal control. (D) A549 cells were treated
with Sor (10 µM) or Lap (10 µM) in the presence or absence of
FTY720 (10 µM) for 24 h. Cells were stained with PI prior to
analysis using a flow cytometer. Cell cycle distribution was
analyzed using ModFit LT version 5.0. *P<0.01 vs. non-treated
cells in each phase. PI, propidium iodide; CCN, cyclin; CDK,
cyclin-dependent kinase; Sor, sorafenib; Gef, gefitinib; Lap,
lapatinib; NSCLC, non-small cell lung cancer. |
EGFR TKIs are standard first-line therapies for
patients with advanced NSCLC with activating EGFR mutations.
However, it is essential to identify additional therapeutic
strategies to block the growth of NSCLC in patients with wild-type
EGFR. In our previous study, it was demonstrated that
macrolide antibiotics enhance the cytotoxic effect of Gef in NSCLC
cells; however, a relatively high dose of Gef (25 µM) was required
to achieve these synergistic effects (38). In order to discover novel effective
combination treatments, the effects of various TKIs combined with
FTY720 were investigated in EGFR wild-type NSCLC cells. The
current study showed that FTY720 enhanced the cytotoxicity of TKIs
(10 µM), particularly Lap and Sor, whereas the macrolide antibiotic
azithromycin only minimally enhanced the TKI-mediated cytotoxicity
(Fig. S2).
It was shown that FTY720 or Lap alone suppressed
autophagy flux, whereas Sor induced autophagy flux. To investigate
whether the suppression of autophagy flux by FTY720 was required to
exert the synergistic cytotoxicity of FTY720 and TKIs,
ATG5-knockout A549 cells were generated. It was confirmed
that conversion of MAP1LC3B-I to MAP1LC3B-II-a critical step in the
formation of autophagosomes-was completely abolished in
ATG5-depleted colonies, even in the presence of BafA1, by
immunoblotting using anti-MAP1LC3B antibody (Fig. S3). The results showed that FTY720
and/or TKIs exert a similar effect in ATG5-knockout and
parental A549 cells (Fig. S3),
suggesting that the suppression of autophagy flux by FTY720 was
dispensable for synergistic cytotoxicity of FTY720 and TKIs.
However, since an ATG5-independent autophagy pathway has also been
reported (60,61), further experiments are needed to
clarify the necessity of the autophagy flux modulation.
Notably, although FTY720 enhanced the cytotoxicity
of Lap or Sor, no evident morphological apoptotic features, such as
nuclear fragmentation and apoptotic body formation, were detected
(Fig. S4). Additionally, the
decreased cell viability observed following combined treatment with
TKIs and FTY720 was not suppressed by co-treatment with the
apoptosis inhibitor Z-VAD-FMK or necroptosis inhibitor
necrostatin-1 (Fig. S5).
Furthermore, the transient siRNA-mediated knockdown of
RIPK1, an indispensable regulator for necroptosis, did not
affect cell sensitivity to TKIs combined with FTY720 in A549 cells
(Fig. S6). Several lines of
research using A549 cells have reported Sor-induced apoptosis,
Lap-induced apoptosis or FTY-induced necroptosis (62–64);
however, in the present study, typical features of apoptosis or
necroptosis were not found in A549 cells treated with a combination
of TKIs and FTY720. All these data suggest that further experiments
are required in order to clarify the mechanisms of cell death
induced by combined treatment with TKIs and FTY720.
It is also necessary to consider that, although
FTY720 markedly enhanced Lap-mediated cytotoxicity (Fig. 1C and D, and Fig. S7), the enhanced effects of FTY720
on Gef or Erl were less pronounced (Fig. 1C and D). Gef and Erl inhibit the
phosphorylation of EGFR. In contrast, Lap inhibits the
phosphorylation of ERBB2 and EGFR (65). Therefore, the differences in the
enhanced effects of FTY720 may be due to its effects on ERBB2.
However, the expression of ERBB2 and phosphorylated ERBB2 were not
detected in A549, H596 and H226 cells (Fig. S8). Additionally, it has been
previously reported that FTY720 decreases the sensitivity to Lap in
SKBR3 cells, an ERBB2-overexpressing breast cancer cell line
(66). Therefore, further studies
are needed to determine the underlying molecular mechanism of the
combined effects of Lap and FTY720.
In conclusion, it was shown that the cytotoxic
effects of Lap and Sor against EGFR wild-type NSCLC cells
were markedly enhanced by the combined treatment with FTY720,
making FTY720 a strong candidate for improving the efficacy of Lap
treatment in NSCLC patients with wild-type EGFR. Sor is
another good candidate for improving the therapeutic options,
either as a single drug or in combination with FTY720. There are
many existing drugs with new therapeutic indications (e.g. the
anti-osteoporotic drug raloxifene for breast cancer and the
anti-inflammatory/analgesic drug aspirin for colorectal cancer),
which have been already evaluated in terms of safety and toxicity
(37); therefore, the present study
has the possibility to rapidly improve the treatment of NSCLC
patients with wild-type EGFR. Additionally, the benefits in
NSCLC patients with activating EGFR mutations resulting from
treatment with EGFR TKIs are often limited by the eventual
development of resistance to these agents (67,68).
The present study could be useful to increase the treatment
benefits in such patients.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Noboru Mizushima
(The University of Tokyo, Graduate School and Faculty of Medicine,
Tokyo, Japan) for gifting the GFP-LC3-RFP-LC3ΔG plasmid, and Dr J
Gannon and Dr T Hunt (The Francis Crick Institute, London, UK) for
gifting of the anti-CDK1 (A17) antibody. The authors would also
like to thank Dr Akihisa Abe, Ms. Ayako Hirota, Ms. Yumiko Yamada,
and Ms. Mayumi Tokuhisa for their technical assistance and Editage
(www.editage.jp) for English language editing.
Funding
This study was supported by funds provided through
the MEXT-Supported program of the Strategic Research Foundation at
Private Universities (grant no. S1411011; 2014-2018) from the
Ministry of Education, Culture, Sports, Science and Technology of
Japan to KM, and Research Funding granted by the Tokyo Medical
University President to MH.
Availability of data and materials
All data generated or analyzed during this study are
available from the corresponding author on reasonable request.
Authors' contributions
MH and KM designed the present study. KO, TO, ADL,
AY, HH, HK, SM, NT, and MH performed the experiments and analyzed
the data. MH, KO, NT, and KM were major contributors in writing the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patent consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
EGFR
|
epidermal growth factor receptor
|
|
TKIs
|
tyrosine kinase inhibitors
|
|
NSCLC
|
non-small cell lung cancer
|
|
ER
|
endoplasmic reticulum
|
|
UPR
|
unfolded protein response
|
|
Sor
|
sorafenib
|
|
Gef
|
gefitinib
|
|
Erl
|
erlotinib
|
|
Lap
|
lapatinib
|
|
Ima
|
imatinib
|
|
Tg
|
thapsigargin
|
|
Tm
|
tunicamycin
|
|
BafA1
|
bafilomycin A1
|
|
FTY720-P
|
FTY720 (S)-phosphate
|
|
Z-VAD
|
Z-VAD-FMK
|
|
Nec-1
|
Necrostatin-1
|
|
DMSO
|
dimethyl sulfoxide
|
|
SDS
|
sodium dodecyl sulfate
|
|
siRNA
|
small interfering RNA
|
|
ANOVA
|
analysis of variance
|
References
|
1
|
Torre LA, Siegel RL and Jemal A: Lung
cancer statistics. Adv Exp Med Biol. 893:1–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pao W and Girard N: New driver mutations
in non-small-cell lung cancer. Lancet Oncol. 12:175–180. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Govindan R, Ding L, Griffith M,
Subramanian J, Dees ND, Kanchi KL, Maher CA, Fulton R, Fulton L,
Wallis J, et al: Genomic landscape of non-small cell lung cancer in
smokers and never-smokers. Cell. 150:1121–1134. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Imielinski M, Berger AH, Hammerman PS,
Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M,
Sivachenko A, et al: Mapping the hallmarks of lung adenocarcinoma
with massively parallel sequencing. Cell. 150:1107–1120. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Devarakonda S, Morgensztern D and Govindan
R: Genomic alterations in lung adenocarcinoma. Lancet Oncol.
16:e342–e351. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chapman AM, Sun KY, Ruestow P, Cowan DM
and Madl AK: Lung cancer mutation profile of EGFR, ALK, and KRAS:
Meta-analysis and comparison of never and ever smokers. Lung
Cancer. 102:122–134. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gibelin C and Couraud S: Somatic
alterations in lung cancer: Do environmental factors matter? Lung
Cancer. 100:45–52. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hirsch FR, Scagliotti GV, Mulshine JL,
Kwon R, Curran WJ Jr, Wu YL and Paz-Ares L: Lung cancer: Current
therapies and new targeted treatments. Lancet. 389:299–311. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Okabe T, Okamoto I, Tamura K, Terashima M,
Yoshida T, Satoh T, Takada M, Fukuoka M and Nakagawa K:
Differential constitutive activation of the epidermal growth factor
receptor in non-small cell lung cancer cells bearing EGFR gene
mutation and amplification. Cancer Res. 67:2046–2053. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ono M and Kuwano M: Molecular mechanisms
of epidermal growth factor receptor (EGFR) activation and response
to gefitinib and other EGFR-targeting drugs. Clin Cancer Res.
12:7242–7251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Taron M, Ichinose Y, Rosell R, Mok T,
Massuti B, Zamora L, Mate JL, Manegold C, Ono M, Queralt C, et al:
Activating mutations in the tyrosine kinase domain of the epidermal
growth factor receptor are associated with improved survival in
gefitinib-treated chemorefractory lung adenocarcinomas. Clin Cancer
Res. 11:5878–5885. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee JK, Hahn S, Kim DW, Suh KJ, Keam B,
Kim TM, Lee SH and Heo DS: Epidermal growth factor receptor
tyrosine kinase inhibitors vs conventional chemotherapy in
non-small cell lung cancer harboring wild-type epidermal growth
factor receptor: A meta-analysis. JAMA. 311:1430–1437. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nan X, Xie C, Yu X and Liu J: EGFR TKI as
first-line treatment for patients with advanced EGFR
mutation-positive non-small-cell lung cancer. Oncotarget.
8:75712–75726. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fogli S, Polini B, Del Re M, Petrini I,
Passaro A, Crucitta S, Rofi E and Danesi R: EGFR-TKIs in
non-small-cell lung cancer: Focus on clinical pharmacology and
mechanisms of resistance. Pharmacogenomics. 19:727–740. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou Y, Li S, Hu YP, Wang J, Hauser J,
Conway AN, Vinci MA, Humphrey L, Zborowska E, Willson JK, et al:
Blockade of EGFR and ErbB2 by the novel dual EGFR and ErbB2
tyrosine kinase inhibitor GW572016 sensitizes human colon carcinoma
GEO cells to apoptosis. Cancer Res. 66:404–411. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu L, Cao Y, Chen C, Zhang X, McNabola A,
Wilkie D, Wilhelm S, Lynch M and Carter C: Sorafenib blocks the
RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor
cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer
Res. 66:11851–11858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chiba K, Kataoka H, Seki N, Shimano K,
Koyama M, Fukunari A, Sugahara K and Sugita T: Fingolimod (FTY720),
sphingosine 1-phosphate receptor modulator, shows superior efficacy
as compared with interferon-beta in mouse experimental autoimmune
encephalomyelitis. Int Immunopharmacol. 11:366–372. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sonoda Y, Yamamoto D, Sakurai S, Hasegawa
M, Aizu-Yokota E, Momoi T and Kasahara T: FTY720, a novel
immunosuppressive agent, induces apoptosis in human glioma cells.
Biochem Biophys Res Commun. 281:282–288. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Azuma H, Takahara S, Ichimaru N, Wang JD,
Itoh Y, Otsuki Y, Morimoto J, Fukui R, Hoshiga M and Ishihara T:
Marked prevention of tumor growth and metastasis by a novel
immunosuppressive agent, FTY720, in mouse breast cancer models.
Cancer Res. 62:1410–1419. 2002.PubMed/NCBI
|
|
22
|
Lee TK, Man K, Ho JW, Sun CK, Ng KT, Wang
XH, Wong YC, Ng IO, Xu R and Fan ST: FTY720 induces apoptosis of
human hepatoma cell lines through PI3-K-mediated Akt
dephosphorylation. Carcinogenesis. 25:2397–2405. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schmid G, Guba M, Papyan A, Ischenko I,
Brückel M, Bruns CJ, Jauch KW and Graeb C: FTY720 inhibits tumor
growth and angiogenesis. Transplant Proc. 37:110–111. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang L, Wang HD, Ji XJ, Cong ZX, Zhu JH
and Zhou Y: FTY720 for cancer therapy (Review). Oncol Rep.
30:2571–2578. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
White C, Alshaker H, Cooper C, Winkler M
and Pchejetski D: The emerging role of FTY720 (Fingolimod) in
cancer treatment. Oncotarget. 7:23106–23127. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang N, Qi Y, Wadham C, Wang L, Warren A,
Di W and Xia P: FTY720 induces necrotic cell death and autophagy in
ovarian cancer cells: A protective role of autophagy. Autophagy.
6:1157–1167. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liao A, Hu R, Zhao Q, Li J, Li Y, Yao K,
Zhang R, Wang H, Yang W and Liu Z: Autophagy induced by FTY720
promotes apoptosis in U266 cells. Eur J Pharm Sci. 45:600–605.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang L, Wang H, Ding K and Xu J: FTY720
induces autophagy-related apoptosis and necroptosis in human
glioblastoma cells. Toxicol Lett. 236:43–59. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li J, Wang SW, Zhang DS, Sun Y, Zhu CY,
Fei Q, Hu J, Zhang C and Sun YM: FTY720-induced enhancement of
autophagy protects cells from FTY720 cytotoxicity in colorectal
cancer. Oncol Rep. 35:2833–2842. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Alinari L, Mahoney E, Patton J, Zhang X,
Huynh L, Earl CT, Mani R, Mao Y, Yu B, Quinion C, et al: FTY720
increases CD74 expression and sensitizes mantle cell lymphoma cells
to milatuzumab-mediated cell death. Blood. 118:6893–6903. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ahmed D, de Verdier PJ, Ryk C, Lunqe O,
Stal P and Flygare J: FTY720 (Fingolimod) sensitizes hepatocellular
carcinoma cells to sorafenib-mediated cytotoxicity. Pharmacol Res
Perspect. 3:e001712015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tay KH, Liu X, Chi M, Jin L, Jiang CC, Guo
ST, Verrills NM, Tseng HY and Zhang XD: Involvement of vacuolar
H(+)-ATPase in killing of human melanoma cells by the sphingosine
kinase analogue FTY720. Pigment Cell Melanoma Res. 28:171–183.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li X, Wang MH, Qin C, Fan WH, Tian DS and
Liu JL: Fingolimod suppresses neuronal autophagy through the
mTOR/p70S6K pathway and alleviates ischemic brain damage in mice.
PLoS One. 12:e01887482017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Levy JMM, Towers CG and Thorburn A:
Targeting autophagy in cancer. Nat Rev Cancer. 17:528–542. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ashburn TT and Thor KB: Drug
repositioning: Identifying and developing new uses for existing
drugs. Nat Rev Drug Discov. 3:673–683. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jahchan NS, Dudley JT, Mazur PK, Flores N,
Yang D, Palmerton A, Zmoos AF, Vaka D, Tran KQ, Zhou M, et al: A
drug repositioning approach identifies tricyclic antidepressants as
inhibitors of small cell lung cancer and other neuroendocrine
tumors. Cancer Discov. 3:1364–1377. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pushpakom S, Iorio F, Eyers PA, Escott KJ,
Hopper S, Wells A, Doig A, Guilliams T, Latimer J, McNamee C, et
al: Drug repurposing: Progress challenges and recommendations. Nat
Rev Drug Discov. 2018.PubMed/NCBI
|
|
38
|
Sugita S, Ito K, Yamashiro Y, Moriya S,
Che XF, Yokoyama T, Hiramoto M and Miyazawa K: EGFR-independent
autophagy induction with gefitinib and enhancement of its cytotoxic
effect by targeting autophagy with clarithromycin in non-small cell
lung cancer cells. Biochem Biophys Res Commun. 461:28–34. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang M and Kaufman RJ: The impact of the
endoplasmic reticulum protein-folding environment on cancer
development. Nat Rev Cancer. 14:581–597. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
McConkey DJ: The integrated stress
response and proteotoxicity in cancer therapy. Biochem Biophys Res
Commun. 482:450–453. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang M, Law ME, Castellano RK and Law BK:
The unfolded protein response as a target for anticancer
therapeutics. Crit Rev Oncol Hematol. 127:66–79. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kawaguchi T, Miyazawa K, Moriya S, Ohtomo
T, Che XF, Naito M, Itoh M and Tomoda A: Combined treatment with
bortezomib plus bafilomycin A1 enhances the cytocidal effect and
induces endoplasmic reticulum stress in U266 myeloma cells:
Crosstalk among proteasome, autophagy-lysosome and ER stress. Int J
Oncol. 38:643–654. 2011.PubMed/NCBI
|
|
43
|
Moriya S, Che XF, Komatsu S, Abe A,
Kawaguchi T, Gotoh A, Inazu M, Tomoda A and Miyazawa K: Macrolide
antibiotics block autophagy flux and sensitize to bortezomib via
endoplasmic reticulum stress-mediated CHOP induction in myeloma
cells. Int J Oncol. 42:1541–1550. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kaizuka T, Morishita H, Hama Y, Tsukamoto
S, Matsui T, Toyota Y, Kodama A, Ishihara T, Mizushima T and
Mizushima N: An autophagic flux probe that releases an internal
control. Mol Cell. 64:835–849. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
O'Prey J, Sakamaki J, Baudot AD, New M,
Van Acker T, Tooze SA, Long JS and Ryan KM: Application of
CRISPR/Cas9 to autophagy research. Methods Enzymol. 588:79–108.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Krypuy M, Newnham GM, Thomas DM, Conron M
and Dobrovic A: High resolution melting analysis for the rapid and
sensitive detection of mutations in clinical samples: KRAS codon 12
and 13 mutations in non-small cell lung cancer. BMC Cancer.
6:2952006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wilhelm SM, Carter C, Tang L, Wilkie D,
McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al:
BAY 43–9006 exhibits broad spectrum oral antitumor activity and
targets the RAF/MEK/ERK pathway and receptor tyrosine kinases
involved in tumor progression and angiogenesis. Cancer Res.
64:7099–7109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mukai S, Moriya S, Hiramoto M, Kazama H,
Kokuba H, Che XF, Yokoyama T, Sakamoto S, Sugawara A, Sunazuka T,
et al: Macrolides sensitize EGFR-TKI-induced non-apoptotic cell
death via blocking autophagy flux in pancreatic cancer cell lines.
Int J Oncol. 48:45–54. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yoshii SR and Mizushima N: Monitoring and
measuring autophagy. Int J Mol Sci. 18:2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mauvezin C and Neufeld TP: Bafilomycin A1
disrupts autophagic flux by inhibiting both V-ATPase-dependent
acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome
fusion. Autophagy. 11:1437–1438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Moriya S, Komatsu S, Yamasaki K, Kawai Y,
Kokuba H, Hirota A, Che XF, Inazu M, Gotoh A, Hiramoto M and
Miyazawa K: Targeting the integrated networks of aggresome
formation, proteasome, and autophagy potentiates ER stressmediated
cell death in multiple myeloma cells. Int J Oncol. 46:474–486.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Miyahara K, Kazama H, Kokuba H, Komatsu S,
Hirota A, Takemura J, Hirasawa K, Moriya S, Abe A, Hiramoto M, et
al: Targeting bortezomib-induced aggresome formation using
vinorelbine enhances the cytotoxic effect along with ER stress
loading in breast cancer cell lines. Int J Oncol. 49:1848–1858.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kazama H, Hiramoto M, Miyahara K, Takano N
and Miyazawa K: Designing an effective drug combination for ER
stress loading in cancer therapy using a real-time monitoring
system. Biochem Biophys Res Commun. 501:286–292. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Brewer JW and Diehl JA: PERK mediates
cell-cycle exit during the mammalian unfolded protein response.
Proc Natl Acad Sci USA. 97:12625–12630. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hamanaka RB, Bennett BS, Cullinan SB and
Diehl JA: PERK and GCN2 contribute to eIF2alpha phosphorylation and
cell cycle arrest after activation of the unfolded protein response
pathway. Mol Biol Cell. 16:5493–5501. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Vincenz L, Jager R, O'Dwyer M and Samali
A: Endoplasmic reticulum stress and the unfolded protein response:
Targeting the Achilles heel of multiple myeloma. Mol Cancer Ther.
12:831–843. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Pluquet O, Pourtier A and Abbadie C: The
unfolded protein response and cellular senescence. A review in the
theme: Cellular mechanisms of endoplasmic reticulum stress
signaling in health and disease. Am J Physiol Cell Physiol.
308:C415–C425. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nishida Y, Arakawa S, Fujitani K,
Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y
and Shimizu S: Discovery of Atg5/Atg7-independent alternative
macroautophagy. Nature. 461:654–658. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yamaguchi H, Arakawa S, Kanaseki T,
Miyatsuka T, Fujitani Y, Watada H, Tsujimoto Y and Shimizu S: Golgi
membrane-associated degradation pathway in yeast and mammals. EMBO
J. 35:1991–2007. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chen C, Ju R, Shi J, Chen W, Sun F, Zhu L,
Li J, Zhang D, Ye C and Guo L: Carboxyamidotriazole synergizes with
sorafenib to combat non-small cell lung cancer through inhibition
of NANOG and aggravation of apoptosis. J Pharmacol Exp Ther.
362:219–229. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Diaz R, Nguewa PA, Parrondo R,
Perez-Stable C, Manrique I, Redrado M, Catena R, Collantes M,
Peñuelas I, Díaz-González JA and Calvo A: Antitumor and
antiangiogenic effect of the dual EGFR and HER-2 tyrosine kinase
inhibitor lapatinib in a lung cancer model. BMC Cancer. 10:1882010.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Saddoughi SA, Gencer S, Peterson YK, Ward
KE, Mukhopadhyay A, Oaks J, Bielawski J, Szulc ZM, Thomas RJ,
Selvam SP, et al: Sphingosine analogue drug FTY720 targets
I2PP2A/SET and mediates lung tumour suppression via activation of
PP2A-RIPK1-dependent necroptosis. EMBO Mol Med. 5:105–121. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Heymach JV, Nilsson M, Blumenschein G,
Papadimitrakopoulou V and Herbst R: Epidermal growth factor
receptor inhibitors in development for the treatment of non-small
cell lung cancer. Clin Cancer Res. 12:4441s–4445s. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
McDermott MS, Browne BC, Conlon NT,
O'Brien NA, Slamon DJ, Henry M, Meleady P, Clynes M, Dowling P,
Crown J and O'Donovan N: PP2A inhibition overcomes acquired
resistance to HER2 targeted therapy. Mol Cancer. 13:1572014.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Janne PA, Engelman JA and Johnson BE:
Epidermal growth factor receptor mutations in non-small-cell lung
cancer: Implications for treatment and tumor biology. J Clin Oncol.
23:3227–3234. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Gainor JF and Shaw AT: Emerging paradigms
in the development of resistance to tyrosine kinase inhibitors in
lung cancer. J Clin Oncol. 31:3987–3996. 2013. View Article : Google Scholar : PubMed/NCBI
|














