Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most
common type of non-Hodgkin lymphoma (NHL) in adults, accounting for
30–40 % of NHL cases. DLBCL is a heterogeneous disease
characterized by morphologic, phenotypic and molecular diversity
(1,2). Gene expression profiling studies
classify DLBCL into three subgroups, namely germinal center
B-cell-like (GCB), activated B-cell-like (ABC), and type 3 DLBCL
and these types differ in the cell of origin and survival
parameters (3–5). Approximately 50% of DLBCL patients
treated with the combination of retuximab plus cyclophosphamide,
doxorubicin, vincristine and prednisone (R-CHOP) combination
regimen exhibited long-term remission (6); however, patients who are resistant to
R-CHOP have a poor prognosis, underscoring the need to develop
novel targeted therapies. In addition, the etiology of DLBCL still
remains unknown. Therefore, a better understanding of the molecular
mechanisms underlying the pathogenesis of DLBCL is critical to
improve survival.
MicroRNAs (miRNAs) are small non-coding RNAs that
play roles in cell differentiation, proliferation, apoptosis,
metabolism and development (7–9).
miRNAs bind partially to complementary sequences in the target
mRNA, modulating gene expression at the post-transcriptional level
and silencing the mRNA target in at least two ways (7). miRNA microarray assays performed on
106 DLBCL and 30 reactive hyperplasia lymph node tissue samples,
revealed that miR-320d was significantly downregulated in DLBCL
tissues. White and Giffard revealed that miR-320 promoted neuronal
regeneration by inhibiting the expression of its target gene
cAMP-regulated phosphoprotein-19 (10). Hsieh et al revealed that
miR-320 prevents the stem cell-like characteristics of prostate
cancer cells by inhibiting the Wnt/β-catenin signaling pathway
(11). In stromal fibroblasts,
miR-320 is important for the phosphatase and tensin homolog (PTEN)
tumor suppressor axis (12). Zhi
et al demonstrated that miRNA-320d was frequently expressed
at high levels in acute myeloid leukemia (AML) (13), whereas Yan et al pointed out
that overexpression of miR-320d inhibited HSV-1-induced Kaposi's
sarcoma-associated herpesvirus replication by targeting
transcription activator (14).
These findings suggest that miR-320d exerts anticancer effects in
DLBCL. Although the anticancer functions of miR-320d have been
confirmed, the underlying anticancer mechanisms of action are not
fully understood. It is worth noting that Zhu et al revealed
that miR-320 negatively regulated the expression of NRP-1 by
binding to the 3′ untranslated region (3′UTR) of the NRP-1
promoter, and NRP-1 depletion inhibited cell proliferation by
upregulating p27, and downregulating cyclin E and cyclin-dependent
kinase (CDK)-2 (15). Therefore, we
speculated that miR-320d may affect the DLBCL process by regulating
CDK family members.
CDK6 is a member of the CDK family that functions
along with cyclins to modulate the phosphorylation of key cell
cycle proteins, thereby regulating cell cycle progression. Recent
studies have revealed that CDK6 is expressed at high levels and is
systematically correlated with poor prognosis in many types of
tumors (16,17). For example, CDK6 upregulation was
revealed to be positively correlated with the stage and invasive
behavior of bladder cancer (18).
CDK6 overexpression, which is driven by gene amplification on
chromosome 7q21.2, was associated with an adverse prognosis in
medulloblastoma and myxofibrosarcoma (19,20).
These data indicated that CDK6 may act as an oncogene and play a
critical role in tumor development and progression. The molecular
mechanisms underlying the response to R-CHOP treatment in DLBCL
remain unclear.
In a previous study from our group, it was revealed
that miR-320d was downregulated in DLBCL patients with a poor
outcome, and overexpression of miR-320d was able to inhibit DLBCL
cell proliferation (21). In the
present study, we investigated the role of miR-320d in DLBCL and
the underlying molecular mechanism and determined whether CDK6 acts
as a target gene of miR-320d.
Materials and methods
Patients
A total of 106 samples were obtained from patients
from the Department of Pathology of Shanxi Cancer Hospital
(Taiyuan, Shanxi, China) who were diagnosed with primary DLBCL and
accepted an RCHOP or CHOP-like initial treatment program +/-
second-line treatment between January 2010 and December 2015. All
patients agreed to be followed up for sampling. Finally, 85 cases
with complete follow-up data who met the requirements were
included. The controls consisted of 19 lymph node reactive
hyperplasia samples obtained from the Department of Pathology of
Shanxi Cancer Hospital. The present study was approved by the
Medical Ethics Committee of Shanxi Medical University.
Immunohistochemistry
Formalin-fixed and paraffin-embedded (FFPE) tissue
samples from DLBCL and the controls were collected and
immunohistochemical staining was performed using the EnVision
method for CDK6 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA). Positive signals of CDK6 were located at the
membrane/cytoplasm. The CDK6 staining results based on the staining
intensity were independently evaluated by two experienced
pathologists as follows: No staining (score=0), weak (score=1),
moderate (score=2) and strong (score=3). The percentage of CDK6
positive cells was 0% (score=0), 1–30 % (score=1), 31–60 %
(score=2), and >60% (score=3). The product of these values
provided the final CDK6 score.
Cell lines and cell culture
Human DLBCL cell lines, including OCI-LY1 (GCB
subtype) and NU-DUL-1 (ABC subtype) were provided by professor
Xiaoyan Zhou (Cancer Hosipital, Shanghai Fudan University,
Shanghai, China), maintained at our laboratory, were cultured as
previously described (21).
Lentivirus production and stable
transfection
Lentiviral vector production for pri-miR-320d was
performed as previously described (21). A CDK6-shRNA lentiviral vector and
its respective negative control vector (universal vector) were
generated (Shanghai GeneChem Co., Ltd., Shanghai, China). OCI-LY1
and NU-DUL-1 cells at 60–80 % confluency were injected with
lentiviral vectors at a multiplicity of infection (MOI) of 50 in
24-well plates (initial seeding at 105 cells/well, 1.5
ml/well). Three days after transfection, the cell lines stably
expressing miR-320d and CDK6-shRNA were generated and the infected
cells were visualized under a fluorescence microscope. Quantitative
reverse transcriptase polymerase chain reaction (qRT-PCR) was
performed to determine miR-320d and sh-CDK6 efficiency.
qRT-PCR
Total RNA was extracted from cell lines transfected
by lentiviral vectors using TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) according to the
manufacturer's protocol. CDK6 mRNA expression was quantified using
the SYBR PrimeScript RT-PCR Kit II (Takara Bio, Inc., Shiga, Japan)
and normalized to GAPDH expression using the 2−ΔΔCq
method (22). The PCR was performed
as follows: 95°C for 30 sec, 40 cycles at 95°C for 5 sec, 60°C for
30 sec; and the dissociation stage at 72°C for 30 sec, 60°C for 30
sec and 95°C for 15 sec. The PCR primers used were as follows:
GAPDH forward, 5′-CCATCAATGACCCCTTCATTG-3′ and reverse,
5′-CATGGGTGGAATCATATTGGAAC-3′; CDK6 forward,
5′-CTGAATGCTCTTGCTCCTTT-3′ and reverse, 5′-AAAGTTTTGGTGGTCCTTGA-3′.
All qRT-PCR amplifications were performed in triplicate.
Protein extraction and western
blotting
DLBCL cells transduced with lentivirus-miR-320d or
lentivirus-control were lysed in a RIPA lysis buffer (Beyotime
Institute of Biotechnology, Beijing, China) and protein
concentration was measured using the BCA Protein Assay kit (Thermo
Fisher Scientific, Inc., Pittsburgh, PA, USA). Lysates were
denatured with SDS sample buffer at 100°C for 10 min. Protein
samples (30 µg) were separated on 10% SDS-PAGE and
electrophoretically transferred onto polyvinylidene difluoride
(PVDF) membranes (Immobilon®-P). The membranes were
blocked and then probed with antibodies against CDK6 (mouse
polyclonal to CDK6; dilution 1:500; cat. no. sc-7961; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) overnight at 4°C, followed by
incubation with secondary antibodies (goat anti-mouse IgG
horseradish peroxidase conjugate; dilution 1:1,000; cat. no.
AP124F; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Loading was
normalized with GAPDH (dilution 1:3,000; Sigma-Aldrich; Merck
KGaA). Band signals were visualized using ECL (Pierce; Thermo
Fisher Scientific, Inc.). The band density was evaluated by Bio-Rad
Quantity One software (Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Cell proliferation assay
DLBCL cells transfected with lentivirus carrying
pri-miR-320d, CDK6-shRNA, and the respective control cells
transfected with control vectors were seeded in 96-well plates
(1×104 cells/well). After transfection for 24 h, cell
proliferation was assessed using a Cell Counting Kit-8 (CCK-8)
assay kit (Dojindo Laboratories, Kumamoto, Japan) according to the
manufacturer's protocol. CCK-8 reagent (20 µl) was added to the
cells in each well at 24, 48, 72 and 96 h after transfection
followed by incubation for 2 h at 37°C, in a 5% CO2
humidified incubator and measurement of the absorbance at 450 nm
was performed with a microplate reader (Thermo Fisher Scientific,
Inc.).
Dual-luciferase reporter assay
The following online software programs were used to
identify miRNA target genes: TargetScan (http://www.targetscan.org/), miRanda (http://www.microrna.org/microrna/home.do), and miRDB
(http://www.mirdb.org/miRDB/). CDK6 was
identified as a possible target of miR-320d. The wild-type sequence
was NM_001145306-3′UTR: ATTGCAGCTTTATGTT, and the mutant-type
sequence was NM_001145306-3′UTR: ATTGGTTAAGACTGTT.
DLBCL cells were seeded into 24-well plates at a
density of 3×104/well and cultured for 24 h before the
addition of 100 ng pMIR-REPORT luciferase vector (Shanghai GeneChem
Co., Ltd.) containing the CDK6 3′UTR or mutated forms. miR-320d
mimics or mimic negative control (NC) (50 nmol/l) were
co-transfected into the cells using Lipofectamine™ 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). After incubation for
48 h, luciferase activity was assessed using the Dual-Luciferase
Reporter Assay system (Promega Corporation, Madison, WI, USA). The
experiments were repeated three times.
Statistical analysis
The SPSS statistical software package (version 17.0)
(IBM Corp., Armonk, NY, USA) was used for statistical analyses, and
data were expressed as the mean ± SD from at least three
independent experiments. Kaplan-Meier was used to estimate the
survival curves. The Pearson χ2 test was used to analyze
the expression of CDK6 between the test group (DLBCL) and negative
control group (lymph node reactive hyperplasia). The comparison of
the CDK6 expression between the blank control, mimic NC and
miR-320d mimics was analyzed using one-way ANOVA, followed by least
significant difference (LSD) test for differences between groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
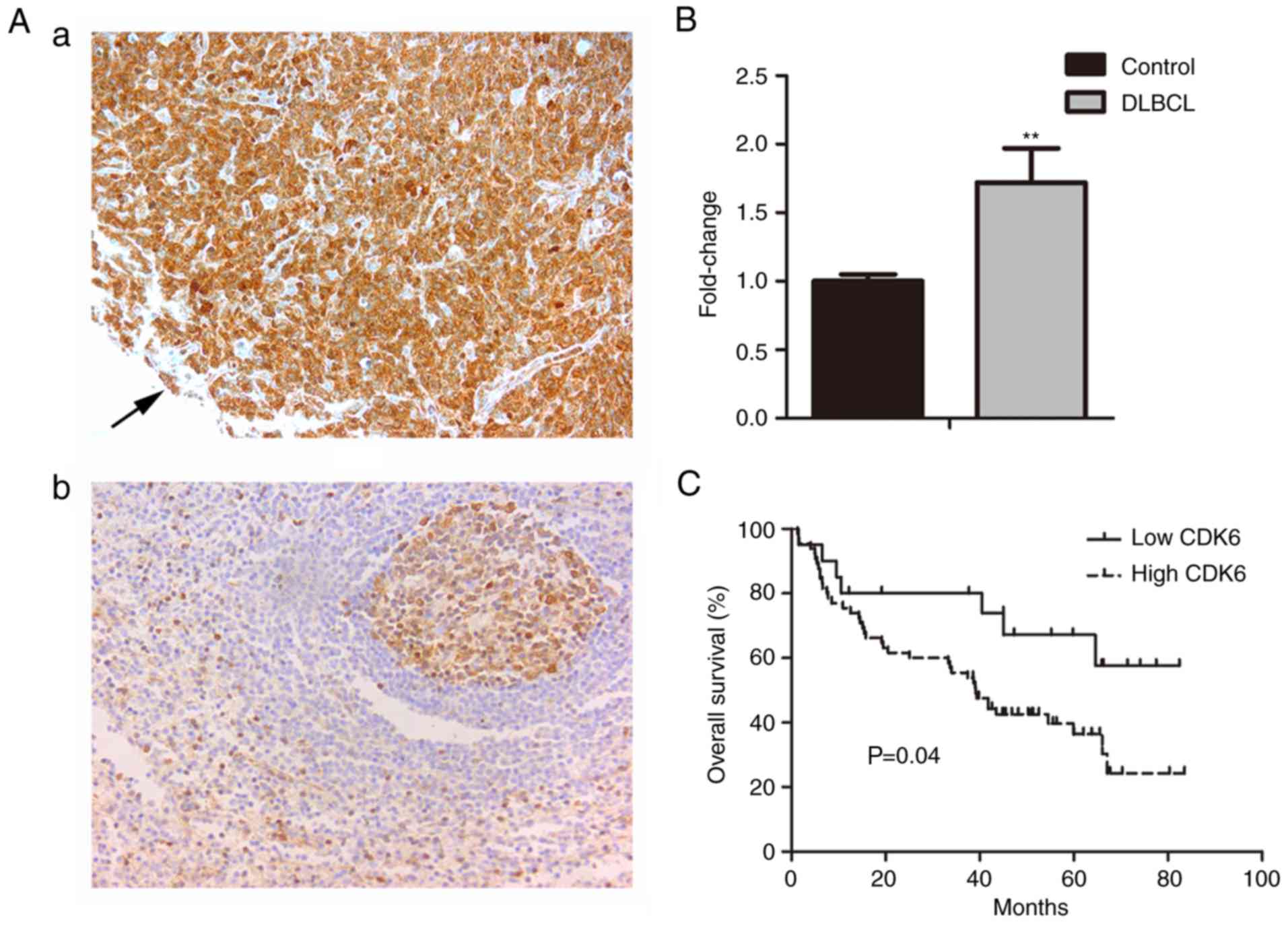
CDK6 is upregulated in DLBCL patients
with poor prognosis
A total of 85 patients with primary DLBCL, including
51 men and 34 women (sex ratio, 1.5:1) with a median age of 57
years (range, 9–81 years) were included in the analysis. The median
overall survival (OS) of patients was 39.8 months, and the OS rate
was 43.5%. The positive rate of CDK6 expression in DLBCL was
76.47%, whereas it was negatively expressed in lymph node reactive
hyperplasia (Fig. 1A). CDK6
expression was significantly higher in DLBCL than that in lymph
node reactive hyperplasia tissues (P<0.01; Fig. 1B and Table I). Patients of DLBCL with high
levels of CDK6 expression had a shorter median overall survival (39
months) than those with low levels of CDK6 expression (46.2 months)
(Fig. 1C).
 | Table I.CDK6 expression in DLBCL and lymph
node reactive hyperplasia tissues. |
Table I.
CDK6 expression in DLBCL and lymph
node reactive hyperplasia tissues.
|
|
| CDK6 expression |
|
|
|---|
|
|
|
|
|
|
|---|
| Samples | Total | Positive (%) | Negative (%) | OS rate (%) | P-value |
|---|
| DLBCL | 85 | 65 (76.47) | 20 (23.53) | 43.5 | <0.01 |
| Lymph node reactive
hyperplasia | 19 | 0 | 19 |
|
|
Overexpression of miR-320d or
knockdown of CDK6 inhibits proliferation in GCB type of DLBCL
cells
To investigate the potential role of miR-320d and
CDK6 in DLBCL proliferation, exogenous miR-320d or CDK6-shRNA
lentiviral vector was transfected into OCI-LY1 and NU-DUL-1 cells,
and miR-320d or CDK6-shRNA stably-expressing cells were
established. Both OCI-LY1 and NU-DUL-1 cells were infected with the
lentivirus containing the miR-320d and CDK6-shRNA expression
sequence (Fig. 2A). The levels of
miR-320d and CDK6-shRNA in DLBCL cells transduced respectively with
the miR-320d-expressing and CDK6-shRNA constructs were markedly
higher than those transduced with the control constructs (Fig. 2B). The CCK-8 assay revealed that the
cell proliferation of OCI-LY1 infected with lenti-miR-320d or
lenti-CDK6-shRNA was decreased compared with those infected with
lenti-NC (Fig. 2C). Although the
CCK-8 assay was not detected in the NU-DUL-1 cells, the results
indicated that miR-320d and CDK6 shRNA may participate in
inhibiting at least GCB-DLBCL cell proliferation.
Overexpression of miR-320d decreases
CDK6 expression, and CDK6 is a direct downstream target of
miR-320d
To determine the direct downstream target of
miR-320d, potential targets were predicted using TargetScan,
miRanda and miRDB. By integrating the three strategies, CDK6 was
found to be the potential target of miR-320d. Then, the 3′UTR of
CDK6 with wild-type or mutant seed-sequence-recognizing sites was
cloned into a dual-Luciferase reporter (Fig. 3A). miR-320d mimic or NC mimic with
CDK6-UTR-WT or CDK6-UTR-MUT plasmid were co-transfected into
OCI-LY1 cells, and then the luciferase activity was analyzed. The
results indicated that compared with the plasmid carrying
CDK6-UTR-MUT, the luciferase activity of the plasmid carrying
CDK6-UTR-WT was suppressed in the miR-320d mimic cells (Fig. 3B). To further analyze the expression
of CDK6 in DLBCL cells, qRT-PCR and western blotting were used to
detect CDK6 mRNA and protein expression in OCI-LY1 cells
overexpressing miR-320d. The results revealed that overexpression
of miR-320d could decrease CDK6 expression both at the protein
level and at the mRNA level (Fig. 3C
and D). Based on the results, it was indicated that miR-320d
specifically bound to the CDK6 3′UTR and may directly target
CDK6.
Discussion
The development and proliferation of tumor cells are
closely associated with the cell cycle, which is regulated by CDKs
and cell cyclins. CDK6, an important member of the CDK family, is
involved in oncogenesis and tumor development in many malignancies.
In the present study, CDK6 was highly expressed in DLBCL patients
and associated with a shorter median overall survival, suggesting
that CDK6 acts as an oncogene in DLBCL.
Increasing evidence supports the role of miRNAs in
many neoplasms, and miRNAs are differentially expressed in
different diseases. Notably, miRNAs not only contribute to
tumorigenesis but can also inhibit cancer progression. Recent
studies suggest that miR-155 (23),
miR-21 (24) and miR-17–92
(25), may function as oncogenes
and these miRNAs are overexpressed in DLBCL. Notably, miR-155 was
revealed to be highly expressed in the ABC and primary mediastinal
B cell lymphoma subtypes, but not in the GCB subtype (26). miR-1234 was upregulated in Egyptian
patients with DLBCL compared with Swedish patients, and may play a
role in the oncogenesis of ABC-DLBCL by targeting STAT3 (27). Overexpression of miR-520c-3p
resulted in the downregulation of the oncogene eIF4GII, which could
account for its antitumor activity in DLBCL (28). miR-146b-5p upregulation in papillary
thyroid carcinoma was significantly correlated with poor prognosis
(29). Among miR-320 subtypes,
miR-320d has attracted increased attention. Several studies have
revealed that miR-320d was upregulated in the serum of AML patients
(13). However, a previous study
from our group indicated that low expression of miR-320d was
related to poor prognosis in DLBCL patients treated with CHOP
(21). These studies indicated that
miR-320d plays diverse roles and this could be attributed to
differences in target genes among different cells.
We previously revealed that miR-320d expression was
downregulated in DLBCL cell lines, and the CCK-8 assay indicated
that DLBCL cells transfected with a miR-320d overexpression vector
(pri-miR-320d) exhibited decreased growth compared with control
cells (21). This led us to
hypothesize that miR-320d functions as a tumor suppressor. In the
present study, we integrated three bioinformatics prediction
software programs and identified the CDK6 gene as a target of
miR-320d. We used qRT-PCR and western blot assays to test our
hypothesis that miR-320d regulates its target gene, CDK6. The
results indicated that miR-320d overexpression downregulated CDK6
at the mRNA and protein levels, suggesting that miR-320d negatively
regulated the expression of CDK6. However, these results did not
demonstrate the direct regulation of the gene. Considering that
miRNAs bind to the 3′UTR of the target mRNA to regulate gene
expression, we constructed luciferase reporter plasmids bearing the
wild-type or a mutant CDK6 3′UTR downstream of the stop codon of
the luciferase gene. Luciferase assays revealed that overexpression
of miR-320d reduced luciferase activity in the wild-type CDK6
3′UTR, whereas it had no effect on cells transfected with the
mutant construct. These results suggested that miR-320d can
directly and negatively regulate CDK6 gene expression by binding to
the 3′UTR of CDK6.
To further validate the effect of the relation
between miRNAs and their target genes on cell proliferation, CDK6
was silenced by transfection with CDK6-shRNA, and the results
indicated that knockdown of CDK6 reduced DLBCL cell growth. This
result was consistent with that reported by van der Linden et
al (30), who revealed that
that CDK6 downregulation inhibits acute lymphoblastic leukemia
(ALL) cell proliferation. By contrast, overexpression of CDK6
suppressed the proliferation of human breast tumor cell lines
through a mechanism involving p130 and E2F4 (31). miR-29c can promote apoptosis by
downregulating CDK6 in glioma cells (32). Similarly, let-7a promoted apoptosis
by inhibiting CDK6 expression in Ewing's sarcoma (Ewing) cells
(33). These differences among
studies could be attributed to the different cell types and
different diseases, and indicate the diverse effects of CDK6 on
cell proliferation. Our previous results indicated that miR-320d
had no significant effect on DLBCL cell apoptosis; however, Classon
and Harlow revealed that CDK6 was not only associated with cell
apoptosis but also involved in cell cycle progression. They
indicated that knockdown of CDK6 can promote apoptosis and block
the cell cycle at the G0/G1 stage. CDK6 cooperation with CDK4
promote the growth of cells through the early G1 phase of cell
cycle by binding to cyclin Ds. Then, the CDK4/6-cyclin D complexes
phosphorylate members of the retinoblastoma (RB) protein family
releasing E2F transcription factors from RB-mediated inhibition
(34). Bustos et al
indicated that overexpression of miR-200a in metastatic melanoma
cells blocked the cell cycle by targeting CDK6, and reduced the
levels of phosphorylated-Rb1 and E2F-downstream targets, decreasing
cell proliferation (35).
Therefore, we speculated that knockdown of CDK6 may regulate Rb and
E2F, and then inhibit DLBCL cell proliferation.
In conclusion, we revealed that the CDK6 protein was
highly expressed in DLBCL tissues and could serve as a predictor of
poor outcome in DLBCL patients. Overexpression of miR-320d
suppressed DLBCL cell proliferation by targeting CDK6, suggesting
that miR-320d is a potential therapeutic target for the treatment
of DLBCL with high CDK6 expression.
Acknowledgements
We are grateful to Ken. H. Young and Xiangyun Guo
for their technical assistance.
Funding
The present study was supported by the Research
Project Supported by the Natural Science Foundation of Shanxi
Province of China (grant no. 2014011039-1), and the Experimental
Animals Cooperation Projects Foundation of Shanxi Province of China
(grant no. 2014k16).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
HS, MX, JC and JW were responsible for the study
design, original article drafting and editing, data acquisition,
data analysis and the editing of the images. RS was responsible for
the immunohistochemical evaluations and the critically manuscript
revision. All authors read and approved the manuscript and agree to
be accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The use of human tissues was approved by the Shanxi
Provincial Cancer Hospital Ethics Committee (no. 201733), and
patient consent was obtained.
Patient consent for publication
Publication of the clinical datasets in this study
does not compromise anonymity, or confidentiality, or breach local
data protection laws.
Competing interests
The authors declare that they have no competing
interest.
References
|
1
|
Lossos IS: Molecular pathogenesis of
diffuse large B-cell lymphoma. J Clin Oncol. 23:6351–6357. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Abramson JS and Shipp MA: Advances in the
biology and therapy of diffuse large B-cell lymphoma: Moving toward
a molecularly targeted approach. Blood. 106:1164–1174. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alizadeh AA, Eisen MB, Davis RE, Ma C,
Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al:
Distinct types of diffuse large B-cell lymphoma identified by gene
expression profiling. Nature. 403:503–511. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rosenwald A, Wright G, Chan WC, Connors
JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland
EB, Giltnane JM, et al: The use of molecular profiling to predict
survival after chemotherapy for diffuse large-B-cell lymphoma. N
Engl J Med. 346:1937–1947. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hans CP, Weisenburger DD, Greiner TC,
Gascoyne RD, Delabie J, Ott G, Müller-Hermelink HK, Campo E,
Braziel RM, Jaffe ES, et al: Confirmation of the molecular
classification of diffuse large B-cell lymphoma by
immunohistochemistry using a tissue microarray. Blood. 103:275–282.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Coiffier B, Thieblemont C, Van Den Neste
E, Lepeu G, Plantier I, Castaigne S, Lefort S, Marit G, Macro M,
Sebban C, et al: Long-term outcome of patients in the LNH-98.5
trial, the first randomized study comparing rituximab-CHOP to
standard CHOP chemotherapy in DLBCL patients: A study by the Groupe
d'Etudes des Lymphomes de l'Adulte. Blood. 116:2040–2045. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Behm-Ansmant I, Rehwinkel J and Izaurralde
E: MicroRNAs silence gene expression by repressing protein
expression and/or by promoting mRNA decay. Cold Spring Harb Symp
Quant Biol. 71:523–530. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Filipowicz W: RNAi: The nuts and bolts of
the RISC machine. Cell. 122:17–20. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
White RE and Giffard RG: MicroRNA-320
induces neurite outgrowth by targeting ARPP-19. Neuroreport.
23:590–595. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hsieh IS, Chang KC, Tsai YT, Ke JY, Lu PJ,
Lee KH, Yeh SD, Hong TM and Chen YL: MicroRNA-320 suppresses the
stem cell-like characteristics of prostate cancer cells by
downregulating the Wnt/beta-catenin signaling pathway.
Carcinogenesis. 34:530–538. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bronisz A, Godlewski J, Wallace JA,
Merchant AS, Nowicki MO, Mathsyaraja H, Srinivasan R, Trimboli AJ,
Martin CK, Li F, et al: Reprogramming of the tumour
microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol.
14:159–167. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhi F, Cao X, Xie X, Wang B, Dong W, Gu W,
Ling Y, Wang R, Yang Y and Liu Y: Identification of circulating
microRNAs as potential biomarkers for detecting acute myeloid
leukemia. PLoS One. 8:e567182013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yan Q, Li W, Tang Q, Yao S, Lv Z, Feng N,
Ma X, Bai Z, Zeng Y, Qin D, et al: Cellular microRNAs 498 and 320d
regulate herpes simplex virus 1 induction of Kaposi's
sarcoma-associated herpesvirus lytic replication by targeting RTA.
PLoS One. 8:e558322013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu H, Jiang X, Zhou X, Dong X, Xie K,
Yang C, Jiang H, Sun X and Lu J: Neuropilin-1 regulated by miR-320
contributes to the growth and metastasis of cholangiocarcinoma
cells. Liver Int. 38:125–135. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baba Y, Watanabe M, Murata A, Shigaki H,
Miyake K, Ishimoto T, Iwatsuki M, Iwagami S, Yoshida N, Oki E, et
al: LINE-1 hypomethylation, DNA copy number alterations, and CDK6
amplification in esophageal squamous cell carcinoma. Clin Cancer
Res. 20:1114–1124. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Dekken H, van Marion R, Vissers KJ,
Hop WC, Dinjens WN, Tilsanus HW, Wink JC and van Duin M: Molecular
dissection of the chromosome band 7q21 amplicon in gastroesophageal
junction adenocarcinomas identifies cyclin-dependent kinase 6 at
both genomic and protein expression levels. Genes Chromosomes
Cancer. 47:649–656. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang G, Zheng L, Yu Z, Liao G, Lu L, Xu R,
Zhao Z and Chen G: Increased cyclin-dependent kinase 6 expression
in bladder cancer. Oncol Lett. 4:43–46. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tsai JW, Li CF, Kao YC, Wang JW, Fang FM,
Wang YH, Wu WR, Wu LC, Hsing CH, Li SH, et al: Recurrent
amplification at 7q21.2 targets CDK6 gene in primary
myxofibrosarcomas and identifies CDK6 overexpression as an
independent adverse prognosticator. Ann Surg Oncol. 19:2716–2725.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Whiteway SL, Harris PS, Venkataraman S,
Alimova I, Birks DK, Donson AM, Foreman NK and Vibhakar R:
Inhibition of cyclin-dependent kinase 6 suppresses cell
proliferation and enhances radiation sensitivity in medulloblastoma
cells. J Neurooncol. 111:113–121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu PY, Zhang XD, Zhu J, Guo XY and Wang
JF: Low expression of microRNA-146b-5p and microRNA-320d predicts
poor outcome of large B-cell lymphoma treated with
cyclophosphamide, doxorubicin, vincristine, and prednisone. Hum
Pathol. 45:1664–1673. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kluiver J, Poppema S, de Jong D, Blokzijl
T, Harms G, Jacobs S, Kroesen BJ and van den Berg A: BIC and
miR-155 are highly expressed in Hodgkin, primary mediastinal and
diffuse large B cell lymphomas. J Pathol. 207:243–249. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lawrie CH, Soneji S, Marafioti T, Cooper
CD, Palazzo S, Paterson JC, Cattan H, Enver T, Mager R, Boultwood
J, et al: MicroRNA expression distinguishes between germinal center
B cell-like and activated B cell-like subtypes of diffuse large B
cell lymphoma. Int J Cancer. 121:1156–1161. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He L, Thomson JM, Hemann MT,
Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe
SW, Hannon GJ, et al: A microRNA polycistron as a potential human
oncogene. Nature. 435:828–833. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Roehle A, Hoefig KP, Repsilber D, Thorns
C, Ziepert M, Wesche KO, Thiere M, Loeffler M, Klapper W,
Pfreundschuh M, et al: MicroRNA signatures characterize diffuse
large B-cell lymphomas and follicular lymphomas. Br J Haematol.
142:732–744. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Högfeldt T, Johnsson P, Grandér D,
Bahnassy AA, Porwit A, Eid S, Österborg A, Zekri AR, Lundahl J,
Khaled MH, et al: Expression of microRNA-1234 related signal
transducer and activator of transcription 3 in patients with
diffuse large B-cell lymphoma of activated B-cell like type from
high and low infectious disease areas. Leuk Lymphoma. 55:1158–1165.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mazan-Mamczarz K, Zhao XF, Dai B,
Steinhardt JJ, Peroutka RJ, Berk KL, Landon AL, Sadowska M, Zhang
Y, Lehrmann E, et al: Down-regulation of eIF4GII by miR-520c-3p
represses diffuse large B cell lymphoma development. PLoS Genet.
10:e10041052014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Geraldo MV, Yamashita AS and Kimura ET:
MicroRNA miR-146b-5p regulates signal transduction of
TGF-β by repressing SMAD4 in thyroid cancer. Oncogene.
31:1910–1922. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van der Linden MH, Willekes M, van Roon E,
Seslija L, Schneider P, Pieters R and Stam RW: MLL fusion-driven
activation of CDK6 potentiates proliferation in MLL-rearranged
infant ALL. Cell Cycle. 13:834–844. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lucas JJ, Domenico J and Gelfand EW:
Cyclin-dependent kinase 6 inhibits proliferation of human mammary
epithelial cells. Mol Cancer Res. 2:105–114. 2004.PubMed/NCBI
|
|
32
|
Wang Y, Li Y, Sun J, Wang Q, Sun C, Yan Y,
Yu L, Cheng D, An T, Shi C, et al: Tumor-suppressive effects of
miR-29c on gliomas. Neuroreport. 24:637–645. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang Z, Huang L, Yu Z, Chen X, Yang D,
Zhan P, Dai M, Huang S, Han Z and Cao K: Let-7a functions as a
tumor suppressor in Ewing's sarcoma cell lines partly by targeting
cyclin-dependent kinase 6. DNA Cell Biol. 33:136–147. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Classon M and Harlow E: The retinoblastoma
tumour suppressor in development and cancer. Nat Rev Cancer.
2:910–917. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bustos MA, Ono S, Marzese DM, Oyama T,
Iida Y, Cheung G, Nelson N, Hsu SC, Yu Q and Hoon DSB: MiR-200a
regulates CDK4/6 inhibitor effect by targeting CDK6 in metastatic
melanoma. J Invest Dermatol. 137:1955–1964. 2017. View Article : Google Scholar : PubMed/NCBI
|