Introduction
Colorectal cancer (CRC) ranks as the third most
commonly diagnosed cancer and fourth leading cause of
cancer-related death worldwide (1,2). Surgery
and chemotherapy are the most common therapeutic strategies for
CRC, but they are frequently associated with adverse side effects
(3). However, the low efficacy of
these treatments in advanced stage CRC patients is a serious public
concern due to the high mortality rate. The limited efficacy of
these therapies is mainly due to lymph node metastasis (4). At the time of diagnosis, approximately
25% of CRC patients present with metastatic disease, and the 5-year
survival rate is approximately 10% (5). Therefore, to reduce CRC-associated
mortality through the development of therapeutic agents, it is
critical to elucidate the mechanisms underlying tumor
metastasis.
Among many solid malignancies, the dissemination of
cancer cells is mainly by means of lymphatic metastasis (6–10). Thus, a
crucial target for anti-metastatic agents is tumor
lymphangiogenesis. Lymphangiogenesis is a complex multi-stage
process including proliferation, survival, migration and tube
formation of endothelial cells, which depends largely on the
signaling of vascular endothelial growth factor C (VEGF-C) and VEGF
receptor 3 (VEGFR3) (11,12). VEGF-C is a member of the
platelet-derived growth factor family (11). VEGFR3 is a tyrosine kinase receptor
that is expressed in human lymphatic endothelial cells (HLECs)
(13). After the binding of VEGF-C to
VEGFR3, downstream pathways, including PI3K/AKT, ERK and STAT3, are
activated, leading to the growth of endothelial cells or to
proliferation, survival, migration, and tube formation of
endothelial cells and subsequent lymphangiogenesis and metastasis.
Thus, expression of VEGF-C is a key for determining whether a tumor
has metastasized (14).
Hedyotis diffusa Willd. (HDW) is a well-known
member of the Rubiaceae family of tropical herbs, shrubs, and
trees. HDW is known for its properties of heat-clearing and
detoxification (in Chinese, Qing Re Jie Du), promotion of blood
circulation and the removal of blood stasis (in Chinese, Huo Xue
Hua Yu); therefore, it is widely used as a toxin-clearing herb in
traditional Chinese medicine (15–17).
According to a previous report, an ethanol extract of HDW (EEHDW)
was found to inhibit the growth of colon cancer by inhibiting cell
proliferation, tumor angiogenesis and metastasis; and it promoted
cell apoptosis via regulation of multiple signaling pathways
(18–21). However, the mechanisms underlying the
anti-metastatic effect of HDW have not been elucidated, especially
regarding lymphangiogenesis. In the present study, by using various
CRC cell lines and applying a VEGF-C-stimulated HLEC model, the
effects of EEHDW on tumor metastasis and lymphangiogenesis were
investigated.
Materials and methods
Preparation of an ethanol extract of
Hedyotis diffusa Willd
The preparation of EEHDW was carried out using the
same procedure as previously described (22). Briefly, EEHDW powder was dissolved
using 100% DMSO. The concentration of EEHDW was 500 mg/ml and was
stored at −20°C. The same volume of DMSO was administered to all
control groups in this study.
Cell culture
Human colorectal carcinoma cell lines HCT116 (Cell
Bank of the Chinese Academy of Sciences, Shanghai, China), HCT-8
(Nanjing Keygen Biotech, Jiangsu, China) and HLECs (JNO-19268;
Jennio Biotech Company, Guangdong, China) were cultured in
RPMI-1640 (cat. no. C11875500BT, Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) medium containing 10% (v/v) fetal bovine serum
(FBS; cat. no. 10099-141, Thermo Fisher Scientific, Inc.), 1%
penicillin-streptomycin (cat. no. SV30010, Thermo Fisher
Scientific, Inc.), and then cultured in a 37°C humidified incubator
with 5% CO2.
Administration of EEHDW and exogenous
VEGF-C
CRC cells were seeded into 6-well plates
(2.5×105 cells/well) for 12 h, and were subsequently
treated with EEHDW for the indicated times as specified for the
MTT, colony formation, flow cytometry, Transwell, tube formation
and western blot assays. HLECs were grown until achieving ~60%
confluency in complete medium and then the medium was replaced with
FBS-free medium overnight. The cells were then placed into another
complete medium which contained 2% FBS; and then the cells were
treated with 5 ng/ml VEGF-C (cat. no. CYT-527, Prospec-Tany
TechnoGene, Ltd., East Brunswick, NJ, USA) and/or various doses of
EEHDW for the indicated times.
Cell viability assay
MTT assays were used to detect cell viability.
Firstly, cells were plated into a 96-well (1×105
cells/well) overnight, and then the cells were treated with EEHDW
(0, 0.125, 0.25, 0.5 and 1, 2 mg/ml) and/or exogenous VEGF-C (5
ng/ml) for 24 h; then MTT (0.5 mg/ml) was added to each well (100
µl/well) and incubated for 4 h. Subsequently, all wells were
treated with DMSO (100 µl/well). An ELISA reader (Infinite M200
PRO; Tecan Austria GmbH, Austria) was used to read the absorbance
(570 nm). This experiment was repeated eight times.
Colony formation assay
The procedures were conducted in the same way as
previously described (15). Briefly,
after pretreatment with EEHDW (0, 0.125, 0.25, 0.5 and 1 mg/ml) for
24 h, cells at a density of 1,000 per group were seeded into a
6-well plate, and cultured for 10 days for the formation of
colonies. Then the colonies formed were fixed with 4% formaldehyde
for 10 min, followed by visualization treatment with 0.3% crystal
violet staining for 15 min. After removing excess crystal violet by
rinsing the plate with PBS, the visible colonies were counted by
ImageJ software, and the rate of colony formation was calculated in
each group with the control group set as 100%. This experiment was
repeated three times.
Wound healing assay
CRC cells were seeded into a 6-well plate with 2 ml
of medium in each well (5×105 cells/well). After 24 h,
the incubated cells were scraped away vertically in each well with
a P200 pipette tip. Under a phase-contrast inverted microscope at a
magnification of ×100, for each well three images along the scraped
line were randomly selected. Then similarly, after the cells
underwent another 24 h of incubation in the indicated
concentrations of EEHDW (0, 0.25, 0.5 and 1 mg/ml), a new set of
images were captured. By comparing these two groups of images, the
reduction in the number of cells within the scraped area indicated
the cell migration rate. ImageJ software was used to analysis the
images in this assay. This experiment was repeated three times.
Assessment of cell migration by
Transwell assay
Transwell cell culture chambers (cat. no. 3422;
Corning Life Sciences, Corning, NY, USA) were used and the
Transwell inserts were placed into a 24-well plate where each well
contained chemoattractant solution. The cells for migration assay
were prepared by being seeded into a 6-well plate and incubated for
24 h after treatment with EEHDW (0, 0.125, 0.25, 0.5 and 1 mg/ml).
The incubated cells were then diluted into a density of
5×104 cells (HCT116), 7.5×104 cells (HCT-8)
and 5×104 cells (HLECs) and pipetted into the inserts
containing suspension solution with 0.2 ml RPMI-1640 medium. After
the cells were incubated for 24 h, the upper-side of the
polycarbonate membrane was scraped, leaving the underside of the
membrane with migrated cells, which were stained with crystal
violet for 15 min at room temperature. To count the number of the
migrated cells, three random areas on each membrane were chosen and
counted under a phase-contrast microscope (Leica, Germany) at a
magnification of ×200. This experiment was repeated three
times.
ELISA assays
The secretion level of VEGF-C was measured using
ELISA kits (cat. no. TAE-577h-c; Tianjin Aoric Bio-Technology Co.,
Tianjin, China) according to the manufacturer's instructions. The
wells in a coated microwell plate were filled with 50 µl of each
concentration of standard or 25 µl test samples, and then the test
sample plates were filled with 25 µl biotin. All wells were filled
with 50 µl enzyme conjugation liquid and incubated for 1 h at 37°C.
After 5 times of washes with washing liquid, TMB1 and TMB2 were
added to the wells, and incubation was carried out for 20 min at
room temperature. Finally, the wells were added with stopping
liquid and the absorbance was measured at 450 nm. This experiment
was repeated eight times.
Cell cycle assays by flow
cytometry
Cell cycle distribution was assessed in the HLECs
after treatment with the indicated concentrations of EEHDW (0,
0.125, 0.25 and 0.5 mg/ml). Cell cycle progression was estimated
using a Propidium Iodide (PI) kit (cat. no. KGA512; KeyGen Biotech
Co., Nanjing, China) with fluorescence-activated cell sorting
(ModFitLT version 3.0; Verity Software House, Inc., Topsham, ME,
USA) according to the manufacturer's instructions.
Cell apoptosis assays by flow
cytometry
A total of 2×105 HLECs cells were seeded
into 6-plates in 2 ml medium and treated with indicated
concentrations of EEHDW (0, 0.125, 0.25 and 0.5 mg/ml) or 5 ng/ml
VEGF-C for 24 h. The apoptosis in HLECs after treatment with EEHDW
was estimated using an Annexin V-FITC/PI kit (cat. no. KGA108,
KeyGen Biotech Co.) with fluorescence-activated cell sorting
caliber (FACSCalibur™; BD Biosciences, San Jose, CA, USA). The
treatment method was carried out according to the manufacturer's
instructions.
Tube formation assays
The assay was conducted as previously described
(10). Briefly, after treatment with
EEHDW (0.125, 0.25 and 0.5 mg/ml) and/or exogenous VEGF-C (5 ng/ml)
for 24 h, HLECs were diluted into a density of 2×105
cells in 200 µl medium per group, before being seeded into a 1:1
ECMatix (ECM625; Millipore, Billerica, MA, USA) gel (v/v)-coated
48-well plate. The cells then were incubated for 8 h. Then by using
a phase-contrast microscope at a magnification of ×40, the series
of tube-like structures were examined and photographed.
Western blot assays
HCT116, HCT-8 and HLECs cells were seeded at a
density of 2.5×105 cells/well into 6-well plates in 2 ml
complete medium, and were treated with various concentrations of
EEHDW (0, 0.125, 0.25, 0.5 and 1 mg/ml) for a total of 24 h at
37°C. The treated cells were washed with PBS and scraped off into a
tube, then lysed using lysis buffer containing protease and
phosphatase inhibitor cocktails on ice for 15 min. Following
high-speed centrifugation (12,000 × g) for 20 min at 4°C,
supernatant containing the sample proteins was collected. The
concentration of proteins was determined using the bicinchoninic
acid (BCA) assay (cat. no. 23227; Thermo Fisher Scientific, Inc.),
and the total protein concentrations were determined. After
separating the total proteins into 50 µg and resolving them in 10%
SDS-PAGE gels, electro-blotting was carried out. After this, the
separated proteins were subsequently transferred onto NC membranes
(cat. no. HATF00010; Millipore, Billerica, MA, USA) which were
blocked and probed with primary antibodies: VEGF-C, VEGFR-3 and
β-actin (cat. nos. 2445, 2485, 4967, respectively; dilution
1:1,000; Cell Signaling, Beverly, MA, USA), PI3K, AKT, ERK and
STAT3 (cat. nos. 13329-1-AP, 10176-2-AP, 16443-1-AP and 10253-2-AP,
respectively; dilution 1:2,000; Proteintech, USA), p-PI3K, p-AKT,
p-ERK and cyclin D1 (cat. nos. sc-12929, sc-135650, sc-16982 and
sc-753, respectively; dilution 1:1,000; Santa Cruz Biotechnology,
Santa Cruz, CA, USA), CDK4, MMP2, MMP9 and p-STAT3 (cat. nos.
ab137675, ab37150, ab38898 and ab76315, respectively; dilution
1;1,000; Abcam) overnight at 4°C. On the second day, the
appropriate HRP-conjugated secondary antibody (cat. no. E030120;
EarthOx, Millbrae, CA, USA) was added and SuperSignal West Pico
Chemiluminescent Substrate was used to detect the signal. Image
Lab™ Software (version 3.0) was used for densitometric
analysis/quantification of the western blot (Bio-Rad Laboratories
Inc., Hercules, CA, USA).
Statistical analysis
One-way ANOVA and SPSS software (version 18.0; SPSS,
Inc., Chicago, IL, USA) were used to analyze all the data in this
study. LSD and Dunnet's were used as post-hoc tests. Data are
expressed as mean ± standard deviation (SD). P<0.05 was
considered as indicative of a statistically significant result.
Results
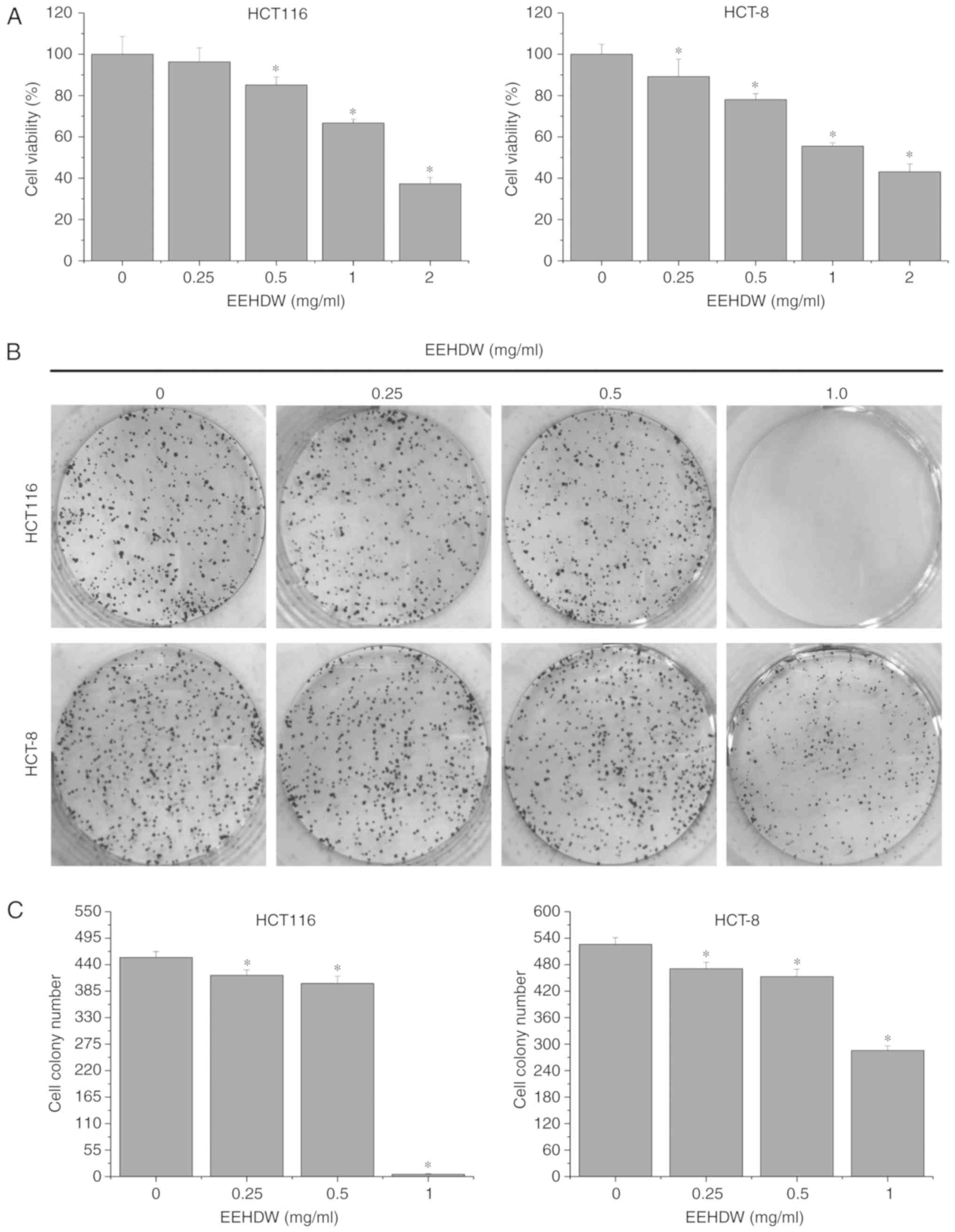
EEHDW inhibits the growth of CRC cell
lines
We used MTT assays to determine the effect of EEHDW
on the viability of HCT116 and HCT-8 cell lines. After their
treatment with EEHDW at 0, 0.25, 0.5, 1.0 and 2.0 mg/ml for 24 h,
the cell viability was reduced from 100% (control) to 37.32±3.08%
(2 mg/ml EEHDW) (HCT116) and from 100% (control) to 43.10±3.80% (2
mg/ml EEHDW) (HCT-8) (Fig. 1A)
(P<0.05). For verification, we performed a colony formation
assay to examine the effect of EEHDW on the survival of CRC cells.
As shown in Fig. 1B and C, the
survival rates of the EEHDW-treated cells decreased with increasing
dose in a dose-dependent manner, relative to the survival rate of
the control EEHDW-untreated group.
EEHDW inhibits the migration of CRC
cell lines
Wound-healing assays were used to investigate the
effect of EEHDW on the migratory ability of CRC cells. During the
process, 24 h after wounding, we observed the migration of the
untreated HCT116 and HCT-8 cells (control groups) into the wounded
(clear) area of the cell monolayer. Compared with the control group
(100%), the percentages of the wound area in the HCT116 cells after
treatment with 0, 0.25, 0.5 and 1.0 mg/ml EEHDW were 51.92±5.56,
64.55±6.37, 72.38±4.95 and 98.43±7.21%, respectively (P<0.05);
the percentages of the wound area in the HCT-8 cells were
21.18±3.14, 38.31±5.24, 45.89±5.98 and 50.91±6.09%, respectively
(P<0.05) (Fig. 2A and B), which
confirmed that the migration rates of both cell lines were
significantly inhibited by the EEHDW treatment. We also used a
Transwell assay to detect the effect, as shown in Fig. 2C and D, and found that EEHDW
significantly inhibited the migration of the CRC cells.
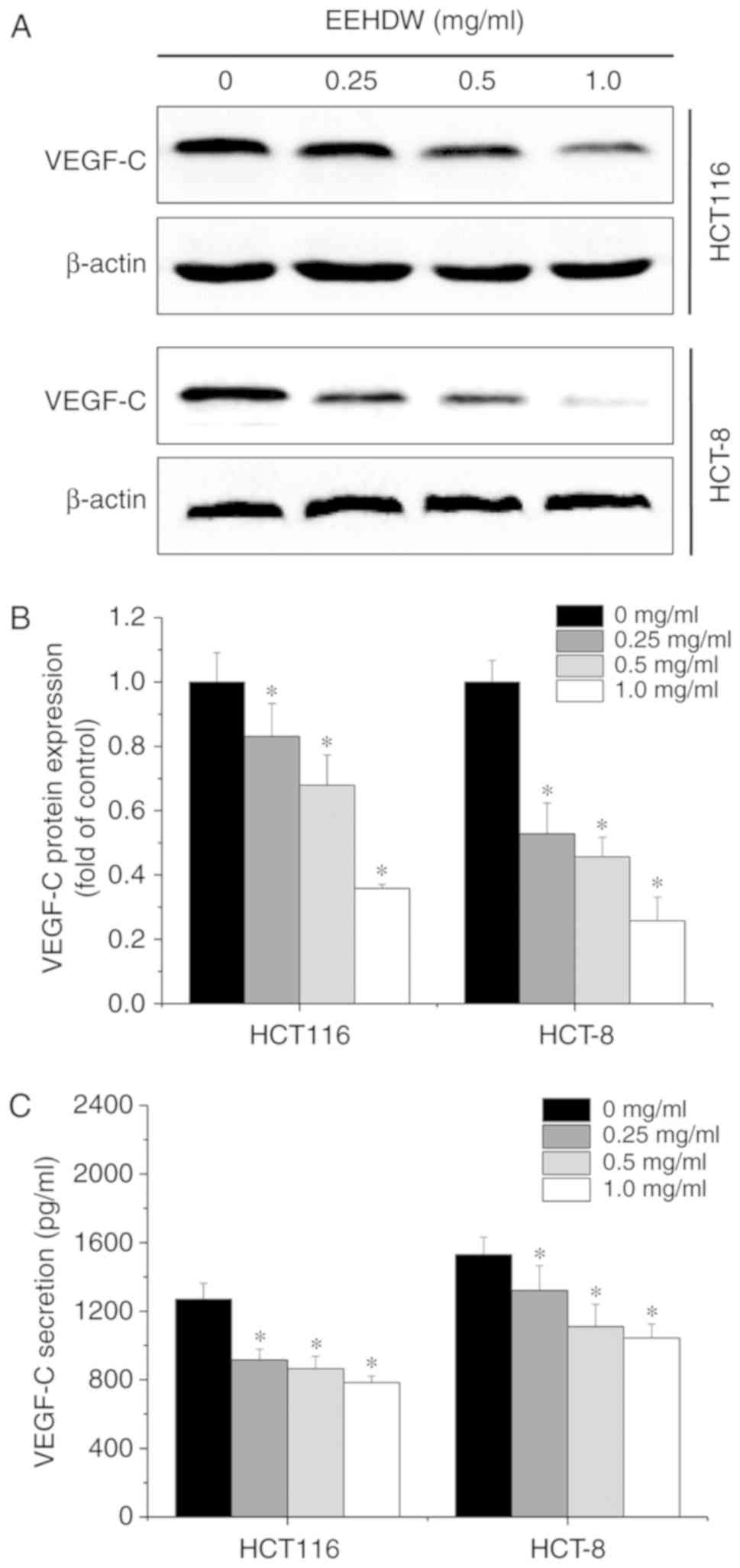
EEHDW downregulates the expression and
secretion of VEGF-C in the CRC cell lines
To understand the potential mechanism involved in
the EEHDW-mediated inhibition of CRC cell migration, the expression
and secretion levels of VEGF-C were assessed in the CRC cells
following treatment with 0, 0.25, 0.5 and 1.0 mg/ml EEHDW by
western blot analysis and ELISA assay. We found that EEHDW
significantly inhibited the expression (Fig. 3A and B) and secretion (Fig. 3C) of VEGF-C (P=0.00).
EEHDW reduces the growth of
VEGF-C-stimulated HLECs
The viability of HLECs was increased to 106.45±6.59%
at 24 h after VEGF-C stimulation relative to the viability of the
control cells (P<0.05), whereas viability was significantly
decreased from 94.87±2.91 to 45.64±2.46% after EEHDW treatment
(from 0.125 to 1.0 mg/ml) (Fig. 4A).
Using a colony formation assay, we found that after VEGF-C
stimulation, the survival rate of the HLECs was increased. After
EEHDW treatment, the survival rate decreased dose-dependently
relative to the rate in the untreated control cells (Fig. 4B). Cell colony formation assay
indicates the cell survival ability; therefore, in our study, this
data only confirmed that EEHDW decreased the ability of survival.
We also performed cell cycle assay to detect the effect of EEHDW on
cell cycle distribution. However, the distribution of cells in the
G0/G1, S, G2/M phases following treatment with 0, 0.125, 0.25 and
0.5 mg/ml EEHDW had no statistically significant difference
(Fig. 4C). Thus, the effect of EEHDW
on the significant reduction in cell proliferation is influenced by
the changes in cell survival ability. Moreover, we used Annexin
V/PI staining and fluorescence-activated cell sorting analysis to
investigate the effect of EEHDW on the apoptosis of HLECs after
VEGF-C stimulation. The results showed that after EEHDW treatment
at 0.125, 0.25 and 0.5 mg/ml, there were no significant differences
in the percentage of cells undergoing apoptosis in the
EEHDW-treated groups and the control group without EEHDW treatment
(Fig. 4D). These results indicated
that EEHDW reduced the growth of VEGF-C-stimulated HLECs but had no
proapoptotic effect in the dose range of 0.125 to 0.5 mg/ml
EEHDW.
 | Figure 4.Effect of EEHDW on the growth of
VEGF-C-stimulated HLECs. (A) Cell viability in the VEGF-C (5
ng/ml)-stimulated HLECs following treatment with 0, 0.125, 0.25,
0.5 and 1.0 mg/ml EEHDW was determined by MTT assay. *P<0.05 vs.
the control group; #P<0.05 vs. the VEGF-C-stimulated
group. (B) Cell growth ability was measured in VEGF-C-stimulated
HLECs following treatment with 0, 0.125, 0.25 and 0.5 mg/ml EEHDW
by colony formation assay. (C) Cell cycle distribution in
VEGF-C-stimulated HLECs after treatment with 0, 0.125, 0.25 and 0.5
mg/ml EEHDW was detected by PI staining with flow cytometric
analysis. (D) Apoptosis in VEGF-C-stimulated HLECs after treatment
with VEGF-C and EEHDW was detected by Annexin V-FITC/PI staining
with flow cytometry analysis. VEGF-C, vascular endothelial growth
factor C; HLECs, human lymphatic endothelial cells; HDW,
Hedyotis diffusa Willd.; EEHDW, ethanol extract of HDW. |
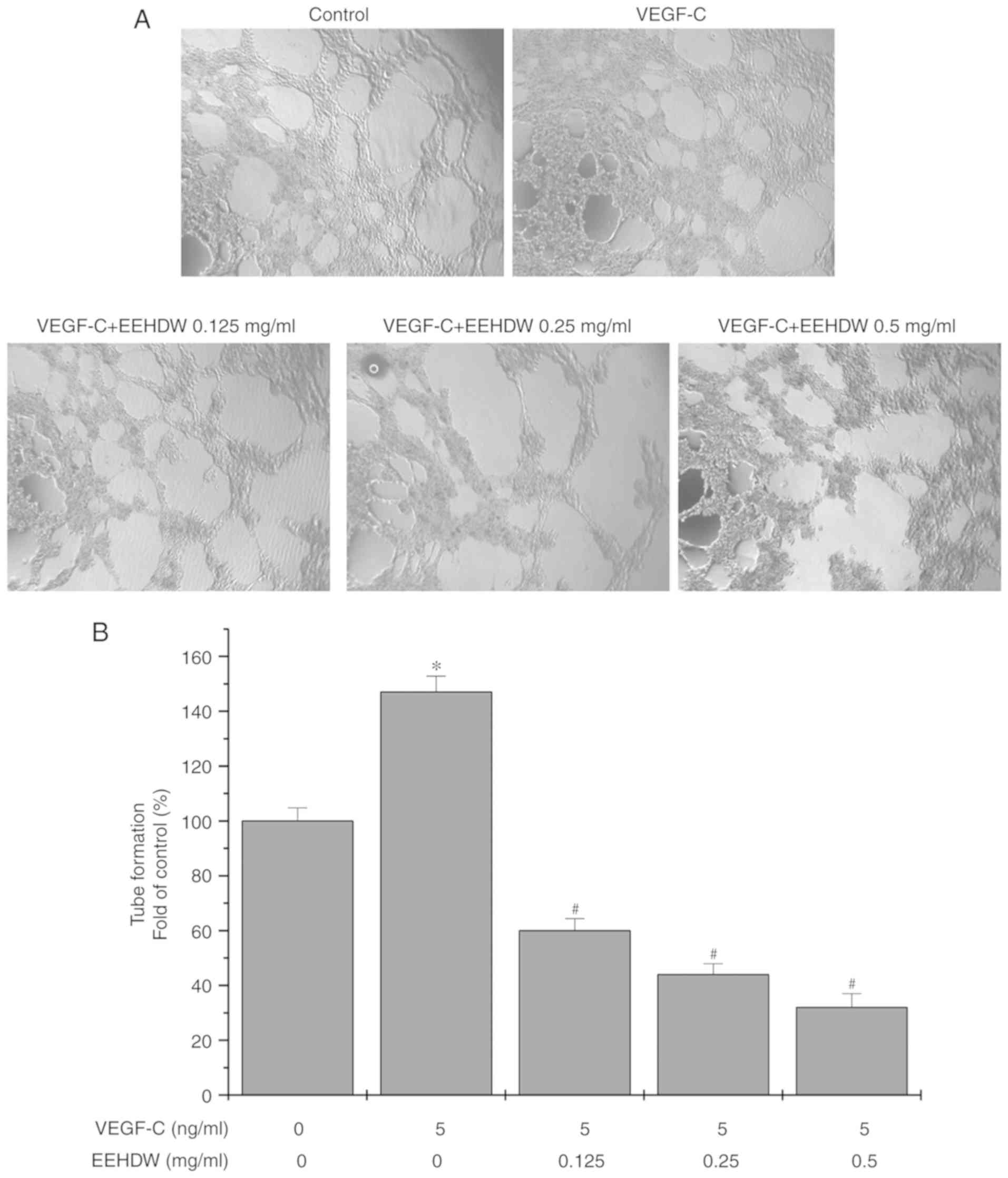
EEHDW inhibits the migration and tube
formation of VEGF-C-stimulated HLECs
The migration abilities of HLECs after VEGF-C
stimulation were enhanced, whereas this enhanced effect was
significantly attenuated by treatment with 0.125, 0.25 and 0.5
mg/ml of EEHDW (Fig. 5). Similarly,
the rate of tube formation of HLECs significantly increased with
exogenous VEGF-C stimulation, relative to the rate in the control
cells without VEGF-C treatment, whereas the rate of tube formation
of HLECs was significantly decreased after treatment with 0.125,
0.25 and 0.5 mg/ml of EEHDW (Fig.
6).
EEHDW suppresses the expression of
MMP2, MMP9, cyclin D1 and CDK4 in VEGF-C-stimulated HLECs
During cancer metastasis, matrix metalloproteinase
(MMP)2, MMP9, cyclin D1 and cyclin-dependent kinase 4 (CDK4) are
important factors for cell proliferation and migration (23,24).
Therefore, we detected the expression of MMP2, MMP9, cyclin D1 and
CDK4 after treatment with 0.125, 0.25 and 0.5 mg/ml of EEHDW. As
shown in Fig. 7, we found that
expression levels of MMP2, MMP9, cyclin D1 and CDK4 were
upregulated after VEGF-C stimulation but downregulated after EEHDW
treatment.
 | Figure 7.Effect of EEHDW on the expression of
MMP2, MMP9, cyclin D1 and CDK4 in VEGF-C (5 ng/ml)-stimulated
HLECs. (A) Expression levels of MMP2, MMP9, cyclin D1 and CDK4 in
HLECs after treatment with VEGF-C (5 ng/ml) and EEHDW (0, 0.125,
0.25 and 0.5 mg/ml) were determined by western blot analysis.
β-actin was used as an internal control. (B) Quantitative analysis
of the western blot in (A). *P<0.05 vs. the control group;
#P<0.05 vs. the VEGF-C-stimulated group. MMP, matrix
metalloproteinase; CDK4, cyclin-dependent kinase 4; VEGF-C,
vascular endothelial growth factor C; HLECs, human lymphatic
endothelial cells; HDW, Hedyotis diffusa Willd.; EEHDW,
ethanol extract of HDW. |
EEHDW suppresses multiple signaling
pathways in the VEGF-C-stimulated HLECs
As important as VEGF-C is to the proliferation and
migration of HLECs (11,13), the expression of VEGFR-3 is also
closely related to these processes. Additionally, multiple
signaling pathways, including the PI3K/AKT, ERK and STAT3 pathways,
can be upregulated as well as some factors, such as MMPs, cyclins,
and relevant dependent kinases (14).
To further investigate the anti-lymphangiogenesis effects of EEHDW,
we examined the expression of related proteins after EEHDW
treatment. We found that the expression level of VEGFR3 increased
by 1.35±0.03 times after VEGF-C stimulation, whereas the level
decreased from 1.35±0.03 times to 0.72±0.02 times with EEHDW
treatment. The expression levels p-PI3K/PI3K, p-AKT/AKT, p-ERK/ERK
and p-STAT3/STAT3 were increased by 1.31±0.03, 1.11±0.05, 1.81±0.07
and 1.35±0.02 times, respectively, after VEGF-C stimulation,
whereas the levels decreased to 0.86±0.05, 0.73±0.04, 0.72±0.05 and
0.33±0.01 times, respectively, following EEHDW treatment (Fig. 8).
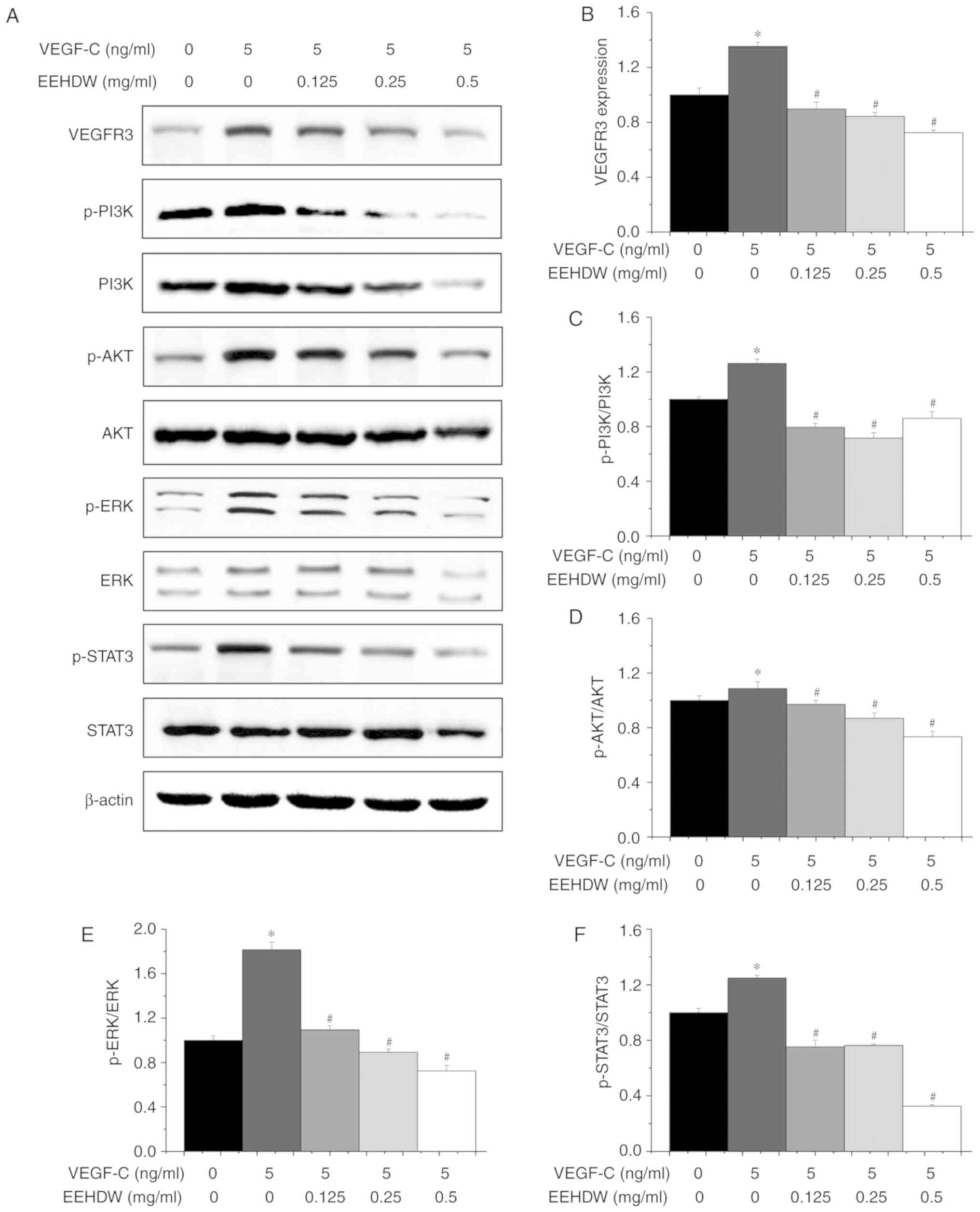 | Figure 8.Effect of EEHDW on multiple signaling
pathways in VEGF-C-stimulated HLECs. (A) Expression levels of
VEGFR3, PI3K, p-PI3K, AKT, p-AKT, ERK, p-ERK, STAT3 and p-STAT3 in
HLECs after treatment with VEGF-C (5 ng/ml) and EEHDW (0, 0.125,
0.25 and 0.5 mg/ml) were determined by western blot analysis.
β-actin was used as an internal control. (B-F) Quantitative
analyses of the western blot in (A). *P<0.05 vs. the control
group; #P<0.05 vs. the VEGF-C-stimulated group.
VEGF-C, vascular endothelial growth factor C; HLECs, human
lymphatic endothelial cells; HDW, Hedyotis diffusa Willd.;
EEHDW, ethanol extract of HDW. |
Discussion
Many colorectal cancer (CRC) patients succumb to
disease due to tumor metastasis (3).
Many cell processes, including tumor cell migration, angiogenesis
and lymph angiogenesis, promote such tumor metastasis, and a key
process is lymph angiogenesis (25,26).
Preclinical evidence suggests that the spread of cancer can be
blocked by the inhibition of cancer-mediated lymphangiogenesis
(27). Recently, many studies have
provided new clues to the development of lymphatic vessels and the
mechanisms underlying lymph node metastasis.
Reportedly, VEGF-C, which is a lymphangiogenic
marker, determines the development of lymphangiogenesis and lymph
metastasis (28). Furthermore, the
role of VEGF-C is important in many human cancers, including CRC
and lung cancer (29). Previous
research (10) suggest that lymphatic
vessel growth and metastasis can be promoted by the complex of
VEGF-C and VEGFR3, abundantly expressed in human lymphatic
endothelial cells (HLECs).
A new therapeutic strategy for CRC is the targeting
of lymphangiogenesis and VEGF-C. However, in clinical practice,
there are many disadvantages, including drug resistance,
metastatsis and tumor recurrence. Recently, cancer treatment using
medicinal herbs has drawn increasing attention due to their
significant efficacy and few side effects. Hedyotis diffusa
Willd. (HDW), an annual herb, has long been used in clinical
practice for its anti-inflammatory, anti-oxidant and anticancer
effects, and previous studies of our research group found that an
ethanol extract of HDW (EEHDW) inhibited proliferation, migration,
invasion and promoted apoptosis in vivo and inhibited CRC
growth in vivo via inhibition of SHH-mediated tumor
angiogenesis (10,18,30), and
other scholars have also studied the toxicological safety of HDW
(31). We also conducted animal
experiments to evaluate whether EEHDW is toxic and has adverse side
effects on animal body weight, intestinal tissue, liver and other
organs. The results demonstrated that EEHDW showed no toxicity and
side effects in regards to animal body weight, intestinal tissue,
liver and other organs compared with the control group (unpublished
data).
In the present study, it was verified that EEHDW
could reduce viability, survival ability, and migration in CRC cell
lines, which indicated the significant capability of EEHDW in
regards to anti-metastasis (Figs. 1
and 2). To explain the mechanism
further, we investigated VEGF-C expression and secretion levels in
CRC cells. The results showed that EEHDW significantly
downregulated VEGF-C expression and secretion (Fig. 3), an indication that a potential
mechanism of the anti-metastatic effects of EEHDW may lie in the
inhibition of lymphangiogenesis-related VEGF-C expression. Since
CRC-secreted VEGF-C can promote the formation of lymphatic vessels
in HLECs, we established an in vitro model by using
exogenous VEGF-C to stimulate HLECs. We found that the viability of
the cells and their ability to migrate and form tubes were
significantly enhanced after stimulation with 5 ng/ml of exogenous
VEGF-C (Figs. 4 and 5). In contrary, these cell behaviors were
attenuated after EEHDW treatment. Yet, these effects were not the
result of cell apoptosis, as we showed that EEHDW did not affect
apoptosis in HLECs (Fig. 4D). The
present study also showed that EEHDW did not affect the cell cycle
distribution of HLECs. Additionally, the expression of VEGFR-3, a
cognate receptor to VEGF-C, in HLECs was increased after
stimulation by exogenous VEGF-C but was notably downregulated by
EEHDW treatment.
In addition, various downstream transcription
factors (e.g. those in cancer) can be activated by VEGF-C/VEGFR-3.
We found that EEHDW prominently inhibited the expression of MMP2,
MMP9, cyclin D1 and CDK4 (Fig. 7).
MMP2 and MMP9 are two important factors involved in cell migration,
whereas exogenous VEGF-C stimulation increased their expression. In
tumor tissue, injury to the extracellular matrix can be caused by
the overexpression of MMP2 and MMP9 finally leading to the
migration of HLECs (32). Previously,
EEHDW has been shown to prominently downregulate the expression of
cyclin D1 and CDK4, two key factors in cell proliferation, and
overexpression of the cyclin D1/CDK4 complex enhanced cell
proliferation (33). Overall, these
results show that the suppressive effect of EEHDW on lymph
angiogenesis via downregulation of VEGF-C may be one of the
molecular-level mechanisms that explains how EEHDW inhibits cell
metastasis in CRC. In a word, our objective of the present study
was to evaluate whether EEHDW can inhibit VEGF-C-mediated
lymphangiogenesis in CRC. The results showed that the
VEGF-C-stimulated cell migration and tube formation ability were
attenuated by EEHDW, and the VEGF-C-stimulated relevant protein
expression was also attenuated by EEHDW. All of these results
support our final conclusion that EEHDW can inhibit VEGF-C-mediated
lymphangiogenesis in CRC. It is universally acknowledged that
VEGF-C is currently recognized as the strongest factor involved in
lymphangiogenesis and its receptor is VEGFR3. Thus, only after the
combination of VEGF-C and VEGFR3 can the corresponding biological
effects occur (11–13). This accepted theory does not need to
be verified again in this study. But to further strengthen our
theory, it may be appropriate to address the mimicking assays, such
as, conducting RNA interference assay using siRNA for VEGFR3 in
HLEC cells, and analyze the downstream signaling molecules. Thus,
this is considered as a limitation of the present study and future
research is warranted.
In addition, the VEGF-C/VEGFR3 pathway can activate
its downstream pathways, including the PI3K/AKT, ERK, and STAT3
pathways (34,35). PI3K, a key factor during many cellular
processes, can mediate signals from receptor tyrosine kinases.
Then, AKT is phosphorylated and further activates downstream
molecules, consequently regulating cell growth and gene
transcription. The PI3K/AKT signaling pathway plays a key role in
cell survival. As demonstrated by our results, the protein
expression ratios of p-PI3K/PI3K, and p-AKT/AKT were increased by
VEGF-C but decreased by EEHDW, which indicated that the PI3K/AKT
pathway is involved in lymphangiogenesis. Additionally, ERK, part
of the MAPK pathway, has been identified as a cell growth-related
pathway. In the present study, we found that after stimulation with
VEGF-C, the expression ratio of p-ERK/ERK was increased, whereas
after treatment with EEHDW, the protein expression ratio was
decreased (Fig. 8). Additionally, in
cutaneous melanoma, interaction between the PI3K/AKT and ERK
pathways has been previously demonstrated (36,37).
Importantly, these study findings suggest that
activation of the PI3K/AKT, ERK, and STAT3 pathways occur in
parallel, with a wide range of evidence on their interconnectivity.
Between these three pathways, multiple crosstalk points have been
detected and activated coordinately, which determine the fate of
cells. Not surprisingly, both negative and positive influences of
the PI3K/AKT, ERK, and STAT3 pathways have been identified during
different stages of signal propagation. Our findings indicate that
the PI3K/AKT, ERK, and STAT3 pathways could provide a promising
target for cancer therapy (Fig.
8).
In conclusion, EEHDW inhibited VEGF-C-mediated
lymphangiogenesis in CRC by the suppression of multiple (PI3K/AKT,
ERK, STAT3) signaling pathways and appears to be a promising
multi-potent anticancer agent for the clinical treatment of
CRC.
Acknowledgements
Not applicable.
Funding
This study was sponsored by the Project of Funding
for the Training of Young and Middle-aged ‘Backbone’ Personnel of
Fujian Provincial Health and Family Planning Commission (Fujian,
China; grant no. 2016-ZQN-67), and Scientific Research Foundation
of Traditional Chinese Medicine of Fujian Provincial Health and
Family Planning Commission (Fujian, China; grant no.
2017FJZYZY203).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HL and JL conceived and designed the experiments.
HY, ZL and JP conducted the MTT, colony formation, wound healing
and Western blot assays and analysis of the data. ZL and YC
conducted the migration, Annexin V/PI and tube formation assays and
analysis of the data. HY and JL wrote the manuscript. All authors
read and approved the final manuscript, and agree to be accountable
for all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no financial or commercial
conflict of interest.
References
|
1
|
Rose L: Financial incentives for At-home
colorectal cancer screening tests. JAMA Netw Open. 2:e1911682019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel R, Desantis C and Jemal A:
Colorectal cancer statistics, 2014. CA Cancer J Clin. 64:104–117.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dang YZ, Li P, Li JP, Zhang Y, Zhao LN, Li
WW, Wei LC and Shi M: Efficacy and toxicity of IMRT-based
simultaneous integrated boost for the definitive management of
positive Lymph nodes in patients with cervical cancer. J Cancer.
10:1103–1109. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee YH, Kung PT, Wang YH, Kuo WY, Kao SL
and Tsai WC: Effect of length of time from diagnosis to treatment
on colorectal cancer survival: A population-based study. PLoS One.
14:e02104652019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Deng J, Cui J, Jiang N, Zhang R, Zhang L,
Hao X and Liang H: STAT3 regulation the expression of VEGF-D in
HGC-27 gastric cancer cell. Am J Transl Res. 6:756–767.
2014.PubMed/NCBI
|
|
7
|
Chen C, He W, Huang J, Wang B, Li H, Cai
Q, Su F, Bi J, Liu H, Zhang B, et al: LNMAT1 promotes lymphatic
metastasis of bladder cancer via CCL2 dependent macrophage
recruitment. Nat Commun. 9:38262018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim HJ, Jo MJ, Kim BR, Kim JL, Jeong YA,
Na YJ, Park SH, Lee SY, Lee DH, Kim BH, et al: Overexpression of
Romo1 is an unfavorable prognostic biomarker and a predictor of
lymphatic metastasis in non-small cell lung cancer patients.
OncoTargets Ther. 11:4233–4246. 2018. View Article : Google Scholar
|
|
9
|
Zheng Y, Song D, Xiao K, Yang C, Ding Y,
Deng W and Tong S: LncRNA GAS5 contributes to lymphatic metastasis
in colorectal cancer. Oncotarget. 7:83727–83734. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin J, Feng J, Jin Y, Yan Z, Lai Z and
Peng J: Pien Tze Huang suppresses VEGF-C-mediated lymphangiogenesis
in colorectal cancer. Oncol Rep. 6:3568–3576. 2016. View Article : Google Scholar
|
|
11
|
Podemska-Jedrzejczak Z, Malinska A,
Sujka-Kordowska P, Nowicki M, Puslecki M, Jemielity M and Perek B:
Vascular restenosis in coronary artery bypass grafting might be
associated with VEGF-C/VEGFR-3 signaling pathway. Heart Vessels.
33:1106–1120. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li C, Zhu M, Lou X, Liu C, Chen H, Lin X,
Ji W, Li Z and Su C: Transcriptional factor OCT4 promotes
esophageal cancer metastasis by inducing epithelial-mesenchymal
transition through VEGF-C/VEGFR-3 signaling pathway. Oncotarget.
8:71933–71945. 2017.PubMed/NCBI
|
|
13
|
Liu ZY, Qiu HO, Yuan XJ, Ni YY, Sun JJ,
Jing W and Fan YZ: Suppression of lymphangiogenesis in human
lymphatic endothelial cells by simultaneously blocking VEGF-C and
VEGF-D/VEGFR-3 with norcantharidin. Int J Oncol. 41:1762–1772.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Deng H, Zhou T, Mo X, Liu C and Yin Y:
Low-density lipoprotein promotes lymphatic metastasis of esophageal
squamous cell carcinoma and is an adverse prognostic factor. Oncol
Lett. 17:1053–1061. 2019.PubMed/NCBI
|
|
15
|
Dai Y, Tong R, Guo H, Yu T and Wang C:
Association of CXCR4, CCR7, VEGF-C and VEGF-D expression with lymph
node metastasis in patients with cervical cancer. Eur J Obstet
Gynecol Reprod Biol. 214:178–183. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu X, Wu J, Zhang D, Wang K, Duan X and
Zhang X: A network pharmacology approach to uncover the multiple
mechanisms of Hedyotis diffusa Willd. on colorectal cancer.
Evid Based Complement Alternat Med. 2018:65170342018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang C, Zhou X, Wang Y, Wei D, Deng C, Xu
X, Xin P and Sun S: the antitumor constituents from Hedyotis
diffusa Willd. Molecules. 22:E21012017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen WJ, Jin YY, Yang H, Wei LH and Lin
JM: Hedyotis diffusa Willd reduces migration and invasion
through inhibition of TGF-β-induced EMT in colorectal cancer cells.
Eur J Intre Med. 23:57–63. 2018. View Article : Google Scholar
|
|
19
|
Li Q, Lai Z, Yan Z, Peng J, Jin Y, Wei L
and Lin J: Hedyotis diffusa Willd inhibits proliferation and
induces apoptosis of 5-FU resistant colorectal cancer cells by
regulating the PI3K/AKT signaling pathway. Mol Med Rep. 17:358–365.
2018.PubMed/NCBI
|
|
20
|
Yan Z, Feng J, Peng J, Lai Z, Zhang L, Jin
Y, Yang H, Chen W and Lin J: Chloroform extract of Hedyotis
diffusa Willd inhibits viability of human colorectal cancer
cells via suppression of AKT and ERK signaling pathways. Oncol
Lett. 14:7923–7930. 2017.PubMed/NCBI
|
|
21
|
Lai Z, Yan Z, Chen W, Peng J, Feng J, Li
Q, Jin Y and Lin J: Hedyotis diffusa Willd suppresses
metastasis in 5-fluorouracil-resistant colorectal cancer cells by
regulating the TGF-β signaling pathway. Mol Med Rep. 16:7752–7758.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Feng J, Jin Y, Peng J, Wei L, Cai Q, Yan
Z, Lai Z and Lin J: Hedyotis diffusa willd extract
suppresses colorectal cancer growth through multiple cellular
pathways. Oncol Lett. 14:8197–8205. 2017.PubMed/NCBI
|
|
23
|
Li Y, Wang X, Wang X, Wan L, Liu Y, Shi Y,
Zhang L, Fang Z and Wei Z: PDCD4 suppresses proliferation,
migration, and invasion of endometrial cells by inhibiting
autophagy and NF-κB/MMP2/MMP9 signal pathway. Biol Reprod.
99:360–372. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cao L, Liu Y, Wang D, Huang L, Li F, Liu
J, Zhang C, Shen Z, Gao Q, Yuan W and Zhang Y: MiR-760 suppresses
human colorectal cancer growth by targeting BATF3/AP-1/cyclinD1
signaling. J Exp Clin Cancer Res. 37:832018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kodera Y, Katanasaka Y, Kitamura Y, Tsuda
H, Nishio K, Tamura T and Koizumi F: Sunitinib inhibits lymphatic
endothelial cell functions and lymph node metastasis in a breast
cancer model through inhibition of vascular endothelial growth
factor receptor 3. Breast Cancer Res. 13:R662011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sammarco G, Varricchi G, Ferraro V,
Ammendola M, De Fazio M, Altomare DF, Luposella M, Maltese L, Currò
G, Marone G, et al: Mast cells, angiogenesis and lymphangiogenesis
in human gastric cancer. Int J Mol Sci. 9:E21062019. View Article : Google Scholar
|
|
27
|
Huang C and Chen Y: Lymphangiogenesis and
colorectal cancer. Saudi Med J. 38:237–244. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang J, Huang Y, Zhang J, Wei Y, Mahoud S,
Bakheet AM, Wang L, Zhou S and Tang J: Pathway-related molecules of
VEGFC/D-VEGFR3/NRP2 axis in tumor lymphangiogenesis and lymphatic
metastasis. Clin Chim Acta. 461:165–171. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tao P, Wen H, Yang B, Zhang A, Wu X and Li
Q: miR-144 inhibits growth and metastasis of cervical cancer cells
by targeting VEGFA and VEGFC. Exp Ther Med. 15:562–568.
2018.PubMed/NCBI
|
|
30
|
Lin J, Wei L, Shen A, Cai Q, Xu W, Li H,
Zhan Y, Hong Z and Peng J: Hedyotis diffusa Willd extract
suppresses Sonic hedgehog signaling leading to the inhibition of
colorectal cancer angiogenesis. Int J Oncol. 42:651–656. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang J, Tang H, Zhang YM and Zhang TL:
Toxicological safety of Hedyotis diffusa. J Toxicol.
28:249–252. 2014.(In Chinese).
|
|
32
|
Naim A, Pan Q and Baig MS: Matrix
metalloproteinases (MMPs) in liver diseases. J Clin Exp Hepatol.
7:367–372. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jing ZT, Liu W, Wu SX, He Y, Lin YT, Chen
WN, Lin XJ and Lin X: Hepatitis B virus surface antigen enhances
the sensitivity of hepatocytes to Fas-mediated apoptosis via
suppression of AKT phosphorylation. J Immunol. 201:2303–2314. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xiang L, Xie G, Ou J, Wei X, Pan F and
Liang H: The extra domain A of fibronectin increases VEGF-C
expression in colorectal carcinoma involving the PI3K/AKT signaling
pathway. PLoS One. 7:e353782012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhu C, Qi X, Chen Y, Sun B, Dai Y and Gu
Y: PI3K/Akt and MAPK/ERK1/2 signaling pathways are involved in
IGF-1-induced VEGF-C upregulation in breast cancer. J Cancer Res
Clin Oncol. 137:1587–1594. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Peng X, Wang Z, Liu Y, Peng X, Liu Y, Zhu
S, Zhang Z, Qiu Y, Jin M, Wang R, et al: Oxyfadichalcone C inhibits
melanoma A375 cell proliferation and metastasis via suppressing
PI3K/Akt and MAPK/ERK pathways. Life Sci. 206:35–44. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou S and Sakamoto K: Pyruvic acid/ethyl
pyruvate inhibits melanogenesis in B16F10 melanoma cells through
PI3K/AKT, GSK3β, and ROS-ERK signaling pathways. Genes Cells.
24:60–69. 2019. View Article : Google Scholar : PubMed/NCBI
|