Introduction
Acute myeloid leukemia (AML) is a common
hematopoietic malignancy that mainly affects adults (1), with the incidence increasing with age
(2). AML was reported to be
responsible for 11,540 deaths in the United States in 2022,
accounting for ~48% of leukemia-related deaths (3). Despite improvements in survival rates
for younger patients with AML, due to advances in intensive
chemotherapy regimens and supportive care, survival rates for older
adults have not significantly improved in the past decade (4,5). The
prognosis for AML patients remains poor (6), with 5-year relative survival rates of
68% for patients under 20 years of age and only 8.2% for patients
over 65 years of age (3).
Recurrence of leukemia is one of the main causes of treatment
failure.
Signal transducer and activator of transcription 5
(STAT5) was initially identified as a PRL-activated sheep mammary
gland factor (7). There are two
predominant isoforms of STAT5, STAT5a and STAT5b, with the former
being more prevalent in the mammary gland and the latter in the
liver. The two transcription factors are >90% homologous at the
protein level, with differences mainly located in the SH2 domain
and transactivation domains required for activation. A previous
study reported that STAT5a and STAT5b have different functions in
hematopoietic stem cells (HSC) and leukemia stem cells (LSCs), and
that STAT5b is the driving force behind the maintenance and
self-renewal of these cells (8). In
the hematopoietic environment, pluripotent hemopoietic stem cells
undergo continuous self-renewal, lineage commitment and terminal
differentiation to generate a sufficient number of fully developed
hematopoietic cells. The expression of STAT5 is crucial for the
maintenance and expansion of human stem/progenitor cells during
normal and leukemic hematopoiesis. STAT5 expression regulates the
self-renewal and differentiation of human stem/progenitor cells, as
well as the survival and proliferation of mature blood cells. As
LSCs can self-renew and cause the recurrence of hematopoietic
malignancies, their elimination is the primary therapeutic goal.
Furthermore, hematopoietic malignancies often exhibit enhanced
STAT5 signaling, which is usually due to STAT5b function
acquisition via gain-of-function mutations (GOF) or activation by
upstream oncogenic kinases (9–15).
STAT5 is an important transcription factor at the
hematopoietic level, and is involved in the self-renewal,
proliferation and apoptosis of numerous cytokines (16–18).
STAT5 acts as both a tumor suppressor and an oncogene that drives
disease progression in AML and other cancers. STAT5 signaling
dysregulation is frequently caused by constitutive activation due
to overexpression, increased receptor signaling or loss of negative
regulatory factors (19). This
dysregulation is commonly associated with the occurrence and
progression of hematologic malignancies, including AML, where ~70%
of patients show STAT5 activation in AML cells (20). STAT5 activation is a major driver in
the development and progression of certain types of cancer,
including AML and non-small cell lung cancer, which are associated
with high mortality rates (21–23).
Therefore, there is an urgent need to develop new therapeutic
strategies that inhibit STAT5 activation.
In AML, FMS related tyrosine kinase 3 internal
tandem duplication (FLT3-ITD) mutations constitutively activate the
FLT3 receptor, produce abnormal STAT5 signaling expression and
drive cell survival and proliferation. Therefore, understanding the
mechanism of STAT5 activation and expression could help to develop
new therapeutic strategies for STAT5-activated cancers, including
FLT3-ITD+ AML. For all patients with AML, ~30% are
affected by FLT3 gene mutations and ~25% have FLT3-ITDs (24). Furthermore, the JAK2-STAT5 signaling
pathway is the main downstream signaling pathway of FLT3. Elevated
STAT5 phosphorylation levels (25,26) in
samples from patients with AML have been previously reported to
activate FLT3 mutations, which leads to constitutive activation of
STAT5 signaling in AML (27).
Therefore, STAT5 maintains constitutive phosphorylation in
FLT3-ITD+ AML cells in the FLT3 signaling pathway
(28). FLT3-ITD is a negative
prognostic marker for AML (29,30).
Although FLT3 inhibitors have shown clinical
efficacy, chemotherapy resistance resulting from STAT5 activation
mutations remains a significant obstacle in the treatment of
FLT3-ITD+ AML (31).
Consequently, treatment outcomes for these patients are often
unsatisfactory. Recent progress in biomolecular drug targets
provides a promising approach to improve the survival rate of
FLT3-ITD leukemia patients. One potential alternative strategy is
to directly target STAT5. Targeting STAT5 is a promising treatment
approach for AML and other hematological malignancies, such as
AC-4-130 (32).
When cytokines or growth factors bind to receptors,
JAK is phosphorylated and activated, which leads to the
phosphorylation of downstream target proteins. Transcription factor
STAT is then recruited and phosphorylated, forming a dimer that
enters the nucleus and binds to target genes (33). This process regulates downstream
gene transcription and affects cell proliferation, differentiation
and apoptosis. Phosphorylated tyrosine residues serve as binding
sites for the SH2 domain in STATs. Homologous or heterodimer
formation occurs through phosphorylated tyrosine-SH2 interaction
and are immediately transferred to the nucleus. Direct inhibition
of STAT5 can be achieved by disrupting tyrosine phosphorylation,
dimerization, DNA binding and nuclear translocation. As
dimerization is a critical step in the regulation of STAT5
function, blocking this process is the most effective strategy for
directly inhibiting abnormal STAT5 signaling in hematopoietic
cancers (34,35). Therefore, the main focus of
designing and identifying selective STAT5 inhibitors is to
interrupt the formation of dimers by inhibiting STAT5
phosphorylation (36).
In the present study, a novel small-molecule
inhibitor targeting STAT5, topotecan hydrochloride (laboratory
compound library no. 108), was identified by screening the
laboratory compound library.
Materials and methods
Cell culture
All cells used in the present study were supplied by
the Zhengfang Yi laboratory at East China Normal University. THP-1
and OCI-AML3 cells were cultured in 80% RPMI-1640 medium (Gibco;
Thermo Fisher Scientific, Inc.), supplemented with 20% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin. MOLM13, HL60, NB4, PBMC and KG-1 cells
were cultured in RPMI-1640 medium supplemented with 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.) and 1% penicillin/streptomycin.
Human umbilical vein endothelial cells [HUVECs; cat. no.
iCell-h110; 5th passage; Saibaikang (Xiamen) Biotechnology Co.,
Ltd.] were cultured in primary endothelial cell basal medium [cat.
no. PriMed-iCell-002, Saibaikang (Xiamen) Biotechnology Co.,
Ltd.].
Reverse transcription-quantitative
(RT-q)PCR
RNA extraction from MOLM13 and KG1 cells was
performed using Trizol (Takara Bio, Inc.). cDNA synthesis was
performed using a PrimeScript model RT kit (Takara Bio, Inc.)
according to the manufacturer's methods. Thermocycling for RT-PCR
was as follows: 37°C for 30 min, 85°C for 5 sec, 16°C for 1 h.
Thermocycling for qPCR was as follows: 95°C for 10 min; 40 cycles
of 95°C for 30 sec, 58°C for 30 sec, 72°C for 30 sec and 95°C for 5
min; then 72°C for 6 min and 16°C for 5 min. RT-qPCR was performed
using three technical replicates on a QuantStudio3®
platform (Applied Biosystems; Thermo Fisher Scientific, Inc.) using
SYBR Premix Ex Taq (Takara Bio, Inc.) and normalized using the
2−ΔΔCq method (37) to
calculate mRNA expression levels. β-actin was used for
normalization. Primer sequences (Genewiz, Inc.) used were as
follows: β-actin forward (F), GTACGCCAACACAGTGCTG and reverse (R),
CGTCATACTCCTGCTTGCTG; cellular-myelocytomatosis viral oncogene
(CMYC) F, GTCAAGAGGCGAACACACAAC and R, TTGGACGGACAGGATGTATGC; and
STAT5B F, GAGGTGCGGCATTATTTATCCC and R, GCGGTCATACGTGTTCTGGAG.
Cell cycle analyses
Cells were treated with 0, 10, 30 or 100 nM
topotecan hydrochloride at 37°C for 24 h, cell suspensions were
centrifuged at 1,500 × g at 4°C for 3 min, then re-suspended with
pre-cooled PBS, centrifuged again and re-suspended in pre-cooled
75% ethanol and immobilized at 4°C overnight. The immobilized cells
were centrifuged at 1,500 × g at 4°C for 3 min then re-suspended
with pre-cooled PBS, centrifuged again and re-suspended in 1 µl
RNase (cat. no. EN0601; Thermo Fisher Scientific, Inc.) and 5 µl
propyl iodide (MilliporeSigma) was added to each PCR tube and
stained for 30 min 4°C. Transferred to flow tubes and analyzed
using a FACSCanto II (BD Diagnostics) flow cytometer and FlowJo
(version, 7.6; BD Diagnostics).
Apoptosis analysis
MOLM13 and KG1 cells were seeded at a density of
3.3×106 cells per 6 cm dish and treated with either DMSO
or a range of concentrations of topotecan hydrochloride at 37°C for
2 days. Cell cycle staining was performed using the Cell Death
Detection® kit (Nanjing KeyGen Biotech Co., Ltd.) and
membrane junction V-FITC Detection® kit (Nanjing KeyGen
Biotech Co., Ltd.) according to the manufacturer's protocol. After
staining, the cells were washed three times with pre-cooled PBS.
The cells were re-suspended in 1× binding buffer from the V-FITC
detection kit. The suspension was transferred to flow tubes and
analyzed using a FACSCanto II (BD Diagnostics) flow cytometer and
FlowJo (version, 7.6; BD Diagnostics).
Fluorescence polarization (FP)
assay
FP assays were performed according to a previously
described protocol (38). The
stability of the system was tested, and the optimum culture
temperature, time and strength were determined. The effect of DMSO
on the system was assessed to be negligible. Briefly, total protein
content was calculated and the protein concentration was diluted to
200 nM with FP buffer. A total of 8 µl FP buffer (1 mM HEPES, pH
7.5, 5 mM NaCl, 0.1% NP-40, 0.5 mM EDTA, 1 mM DTT), 200 nM His-STAT
protein diluent (STAT1-6) and 2 µl compound diluent was added to
each well and incubated for 60 min at 37°C. Subsequently, 10 nM
labeled peptide diluent was added to each well, away from light.
After incubation for 1 h at room temperature, measurements were
performed using a Cytation5 Cell Imaging Multi-Mode Reader (BioTek
Instruments, Inc.). The generated data was analyzed using GraphPad
Prism 7.0 (Dotmatics). The compound library (cat. no. L2110;
TargetMol Chemicals, Inc.) was then screened.
Molecular docking
The molecular docking of topotecan hydrochloride
with STAT5-SH2 domain protein was constructed using AutoDock Vina
1.2.2 software (https://vina.scripps.edu/).
Cellular thermal shift assay
(CETSA)
MOLM13 cells (3.3×106 cells/dish) were
plated and treated with medium containing DMSO or 10 µM topotecan
hydrochloride at 37°C for 1 h. Cells were collected, washed with
PBS three times, and then re-suspended with 1 ml PBS supplemented
with protease inhibitors, phosphatase inhibitors and PMSF (10 µl
each). The cell suspensions were transferred to PCR tubes and
heated at 40.0, 40.8, 42.2, 44.6, 47.4 or 49.5°C for 3 min using a
PCR instrument. After heating, the cells were transferred to PCR
tubes, freeze-thawed with liquid nitrogen for two rounds, and
centrifuged at 6,000 × g at 4°C for 20 min. The lysates were
diluted with 5× loading buffer and the protein samples were boiled
at 100°C for 15 min. Then the samples were assessed using western
blotting.
Cell proliferation assay
Cell proliferation assays were performed at 37°C.
AML cells were inoculated into 96-well plates with a density of
5×103 cells per well. HUVEC and PBMC cells were
inoculated with a density of 1×104 cells per well. The
cells were incubated overnight in a 37°C, 5% CO2
incubator, observed to assess adhesion and treated with 1 µM
topotecan hydrochloride, 50 µM AC-4-130 or 30 µM Pimozide) after
cell adhesion, for 72 h. A total of 20 µl MTS was added to each
well away from light and incubated in a 37°C incubator for 20–50
min. The optical density (OD) of each well was measured at 490 nm
using an Spectra Max 190 enzyme spectrometer (Molecular Devices
LLC), and the OD of all samples were recorded when the OD of the
control reached 0.8–1.0.
Western blotting
Western blotting was performed according to a
previously reported method (39).
Briefly, MOLM13, KG1 and NB4 cells were treated with topotecan
hydrochloride for 24 h, the cells were then collected and the
protein was extracted using RIPA (cat. no. P0013B; Beyotime
Institute of Biotechnology). The protein content was determined
using the BCA method and the protein samples were adjusted to 100
µg/30 µl. A total of 100 µg/lane protein was loaded onto 10%
SDS-PAGE gels. The blots were transferred to nitrocellulose
membranes which were blocked with 5% skim milk solution for 1 h at
room temperature. The membranes were incubated with antibodies
against STAT5 (1;500; cat. no. 25656T; Cell Signaling Technology,
Inc.), phosphorylated (p)-STAT5Y694 (1;500; cat. no.
4322T; Cell Signaling Technology, Inc.), CMYC (1:1,000; cat. no.
ab32072; Abcam) and GAPDH (1:1,000; cat. no. ab181602; Abcam)
overnight at 4°C. IRDye 680/800 (both 1:10,000; cat. nos. 926-32221
and 926-32210; LI-COR Biosciences) were used as the secondary
antibodies and samples were incubated with these antibodies for 1 h
at room temperature.
IL-3 and GM-CSF stimulating factor
Western blotting
MOLM13 cells were cultured in medium without fetal
bovine serum and starved for 24 h (GM-CSF) or 48 h (IL-3).
Different concentrations of IL-3 and GM-CSF were added to stimulate
the cells for 20 min. Then the cells were collected and western
blotting was performed, according to the aforementioned method, to
assess protein expression levels. Cells were starved for 24 or 48 h
in medium without fetal bovine serum and then treated with a range
of concentrations of topotecan hydrochloride for 24 h. Cells were
then stimulated with 5 ng/ml IL-3 and GM-CSF for 20 min and
subjected to western blotting, according to the aforementioned
method.
Subcutaneous AML xenograft model
Male NOD/SCID mice (n=20; 4–6 weeks) were purchased
from Jiangsu Huachuang Xinnuo Pharmaceutical Technology Co., Ltd.
(animal licence no. SCXK2020-0009) and raised in a sterile
environment. Mice were housed in a 12 h light/dark cycle at 20–26°C
and 40–70% relative humidity with ad libitum food and water.
MOLM13 cells (5×106) were suspended in PBS containing
20% Matrigel and injected into the underarm of the right forelimb
of the mice. Tumors grew to a volume of 150–250 mm3, and
the mice were then randomly divided into groups (n=5) as follows:
i) Control, ii) 1 mg/kg/day topotecan hydrochloride via gavage,
iii) 3 mg/kg/d topotecan hydrochloride via gavage and iv) positive
control (3 mg/kg/day Azactidine via intraperitoneal injection at
weeks 1, 3 and 4, 5 days a week). Body weight and tumor volume were
measured after 4 days, with the volume calculated using the
formula, volume=length × wdith2/2. Mice were sacrificed
by cervical dislocation when the tumor reached a volume of 2,000
mm3 and administration of the treatment continued until
mice were sacrificed. Solid tumors were removed for later western
blotting assay and immunohistochemical analysis. The heart, liver,
spleen, lung and kidney tissues of one mouse in each group were
collected for hematoxylin and eosin (H&E) staining
analysis.
In situ AML model
MOLM13 cells (1×106) which were purchased
already stably expressing luciferase (MOLM13-Luc; cat. no NM-B28-1;
Shanghai Model organisms) were injected into the tail veins of
NOD/SCID mice pre-irradiated with 2.5 Gy. After 4–5 days, the mice
were subjected to bioluminescence imaging using an IVIS Lumina III
Small animal live optical two-dimensional imaging system and then
divided into four groups, with animals evenly distributed based on
tumor sizes as indicated by the luciferase luminescence values The
mice were treated as follows: i) Control, ii) 1 mg/kg/day topotecan
hydrochloride via gavage, iii) 3 mg/kg/d topotecan hydrochloride
via gavage; and iv) positive control (3 mg/kg/day Azactidine via
intraperitoneal injection at weeks 1, 3 and 4, 5 days a week).
Tumor development were assessed weekly using the IVIS®
imaging platform (Xenogen Corp.). The weight of the mice was
measured every three days. The mice were sacrificed by cervical
dislocation when demonstrating signs of imminent death, such as
reduced mobility or temperature. Data were analyzed using the
Living Image 4.4 (PerkinElmer, Inc.) and Xenogen IVIS 100 (Xenogen
Inc.).
Hematoxylin and eosin (H&E)
staining
Tissue samples were fixed in 10% neutrally buffered
formaldehyde for one day at room temperature, dehydrated using an
increasing ethanol series and then embedded in paraffin. The
paraffin-embedded samples were sectioned at a thickness of 4 µm.
The sections were then subjected to H&E staining at room
temperature for 5 min per stain to visualize the nucleus and
cytoplasm. The stained sections were assessed for any
histopathological changes using a light microscope (Leica
Microsystems) and imaged.
Western blotting experiment of tumor
tissue
Previously collected tumor tissues were cut and
placed in tissue-crushing tubes, and 2–3 iron beads and RIPA buffer
were added to each tube. The tissue was homogenized using a tissue
crusher until a homogenous solution was obtained. The homogenate
was centrifuged at 6,037.2 × g at 4°C for 20 min, and the resulting
supernatant was transferred to microcentrifuge tubes. Protein
content in the supernatant was determined using a BCA kit, and
protein samples were prepared for western blotting analysis
according to the aforementioned method.
Immunohistochemistry (IHC)
Tumor tissue samples from both the xenograft and
in situ models were fixed in a 4% paraformaldehyde solution
overnight at 4°C and subsequently sectioned at a thickness of 4 µm.
The sections were then subjected to overnight staining using
primary antibodies against p-STAT5 (1:100; cat. no. ab32364;
Abcam), STAT5 (1:100; cat. no. ab230670; Abcam), Cleaved Caspase 3
(1:100; cat. no. AF7022; Affinity Biosciences) and Ki-67 (1:100;
cat. no. ab1667; Abcam) at 4°C. HRP labeled goat anti-rabbit/mouse
secondary antibody (cat no. PR30009; Proteintech Group, Inc.)
incubated at 37°C for 30 min. Anti-biotin protein-biotin peroxidase
complexes were then utilized according to the manufacturer's
instructions, followed by colorimetric detection using DAB
(3,3′-Diaminobenzidine). Finally, hematoxylin was used to
counterstain the sections at room temperature for 2 min, which were
then mounted with coverslips. Sections were imaged using an Olympus
BX53 biological microscope and images were assessed using Image-Pro
Plus 6.0 (media Cybernetics, Inc.).
Statistical analyses
Experiments were performed with ≥3 replicates and
statistical analyses were performed using Student's t-test or
one-way ANOVA followed by Dunnett's post-hoc test. The data are
presented as mean ± SD. The evaluations were performed using
Microsoft Excel 2019 (Microsoft Corporation) and GraphPad (version
7.0; Dotmatics). P<0.05 was considered to indicate a
statistically significant difference.
Results
Topotecan hydrochloride binds to and
inhibits STAT5
Previous studies have reported that STAT5b is the
driving force behind maintenance and self-renewal of HSC and LSCs
(8); therefore, STAT5b was assessed
in the present study. To screen small molecule inhibitors targeting
STAT5 function, a homogeneous method based on fluorescence
polarization (FP) in vitro was developed. Due to the high
homology of the STAT family, the STAT1, STAT2, STAT3, STAT4, STAT5b
and STAT6 proteins were first purified and used to confirm system
specificity. The dissociation constants (KD) of the STAT family
(STAT1-6) were determined and it was demonstrated that the
selective activity of the FP system against STAT5 was the best
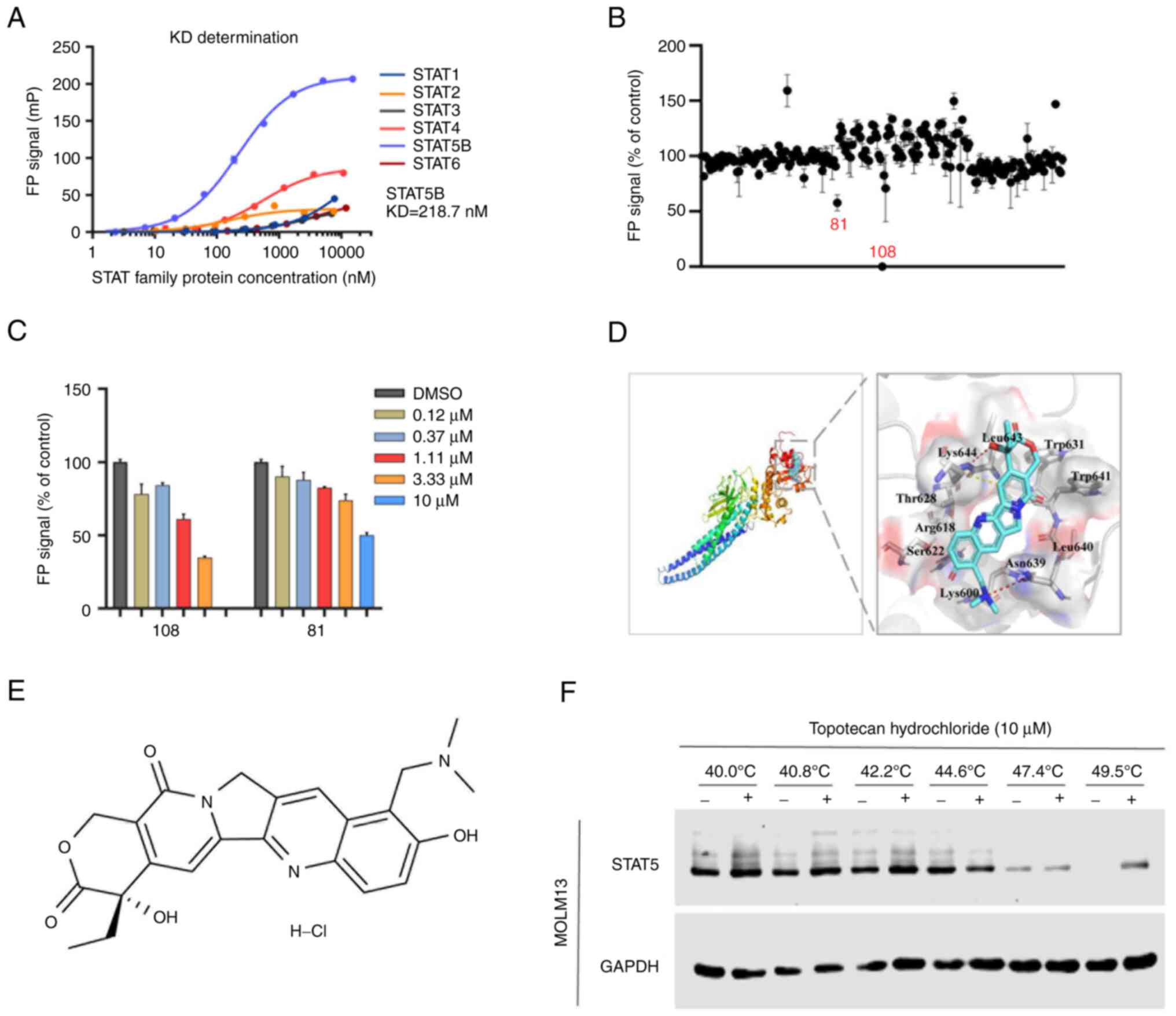
(STAT3 KD <50 nM; STAT4 KD <100 nM; STAT5 KD=218.7 nM)
(Fig. 1A), so the system was
considered to be specific to STAT5. After that, the stability of
the system was tested, and the optimal incubation temperature, time
and strength were determined. It was demonstrated that the
influence of DMSO on the system was negligible (Fig. S1A). Furthermore, incubation at room
temperature for 1 h was the best condition for the combination of
protein and peptide (Fig. S1C),
which confirmed the stability of the FP system (Fig. S1B).
Using the FP-STAT5 system, a compound library was
screened and two potential inhibitors, namely compounds 81 and 108,
with inhibitor constant values of 10 and 1–3 µM, respectively, were
identified. Compound 108 (topotecan hydrochloride) demonstrated the
most potent inhibitory activity (50% inhibition; 81, 10 µM; and
108, <10 µM) and was selected as the lead candidate for further
experimental investigation (Fig. 1B and
C).
To evaluate the binding pattern between topotecan
hydrochloride and the STAT5-SH2 domain, an in silico
molecular simulation docking experiment was performed. The
molecular model of interaction between topotecan hydrochloride and
STAT5-SH2 demonstrated that the hydroxyl part of the six-member
lactone ring of topotecan hydrochloride and the dis-substituted
amino part of the benzene ring were bonded to Asn639 and Lys644 of
the STAT5-SH2 domain by hydrogen bonds (Fig. 1D and E). To confirm whether
topotecan hydrochloride specifically targeted STAT5, CETSA was
performed. After treating cells with 10 µM topotecan hydrochloride
for 1 h, western blotting analysis demonstrated that the thermal
denaturation temperature of STAT5 in the control group treated with
DMSO was 47.4°C. However, after treatment with topotecan
hydrochloride, the thermal denaturation temperature increased to
49.5°C, with no apparent effect on the stability of GAPDH
expression (Fig. 1F). The increase
in thermal stability of the STAT5 target protein indicated that
topotecan hydrochloride specifically bound to STAT5. This indicated
that topotecan hydrochloride was an effective small molecule
inhibitor of STAT5.
Topotecan hydrochloride suppresses
STAT5 activation in AML
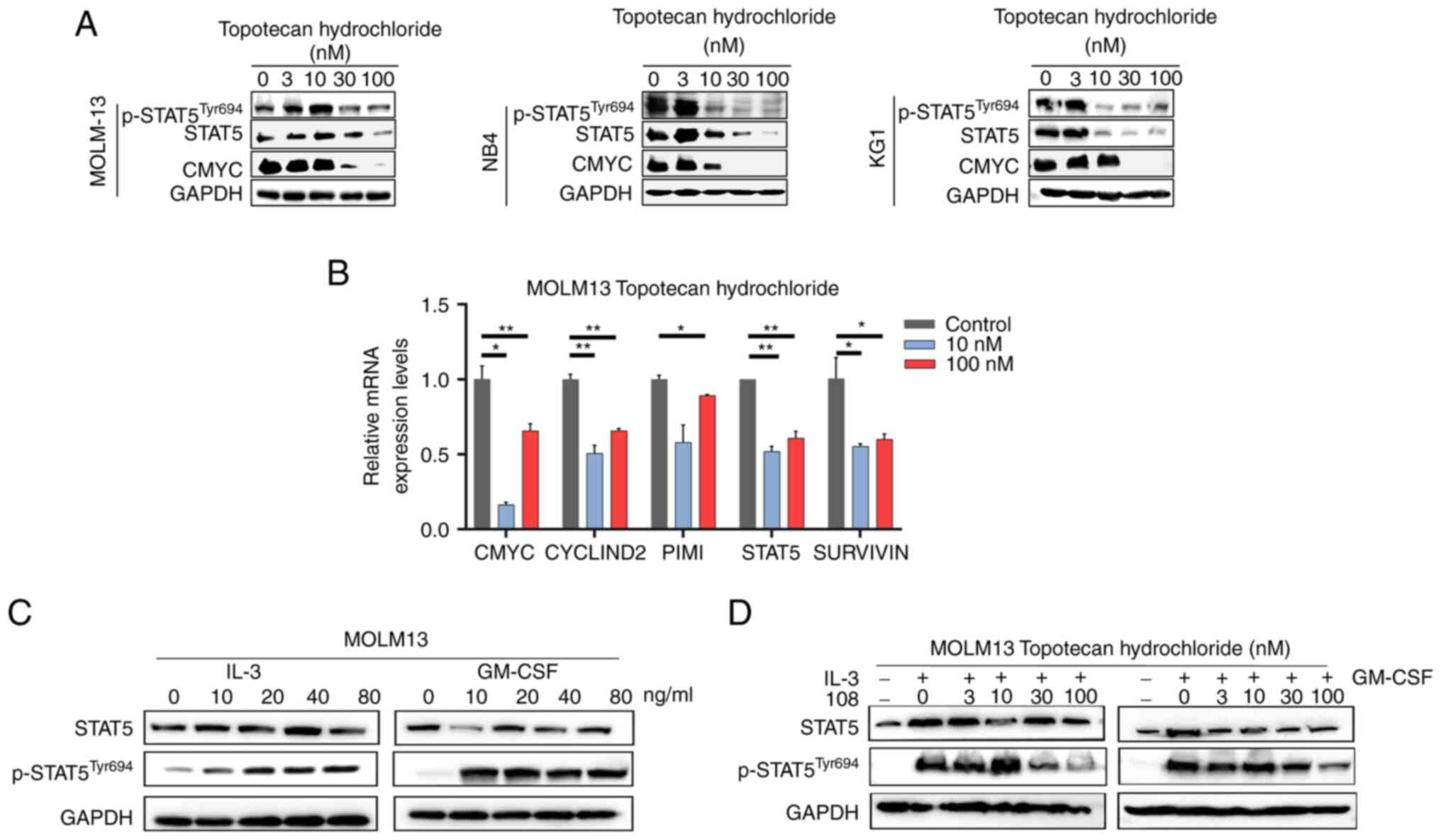
To assess the potential of topotecan hydrochloride
as a STAT5 inhibitor, its inhibitory effect on STAT5 activation and
downstream signaling were assessed in AML cells, including the
MOLM13 (FLT3-ITD+), NB4 and KG1 cell lines. Following
treatment with a range of concentrations of topotecan hydrochloride
for 24 h, western blotting demonstrated that topotecan
hydrochloride effectively blocked STAT5 phosphorylation in the
three cell lines, which indicated its potential as a STAT5
inhibitor (Fig. 2A). In addition,
as the concentration of topotecan hydrochloride increased, the
protein expression level of the downstream STAT5 gene, CMYC,
markedly decreased, which indicated that it could block the
downstream STAT5 signaling pathway. Moreover, RT-qPCR assays using
MOLM13 cells demonstrated that topotecan hydrochloride
significantly reduced the mRNA expression levels of STAT5-specific
target genes, such as CMYC, CYCLIND2, PIMI and STAT5. Notably, a
significant decrease in the mRNA expression levels of SURVIVIN
suggested that topotecan hydrochloride could induce apoptosis
(Fig. 2B). In summary, the results
of the present study indicated that topotecan hydrochloride has the
potential to inhibit STAT5 activation, block downstream signaling
and induce apoptosis in AML cells.
IL-3 and granulocyte-macrophage colony stimulating
factor (GM-CSF) are known to activate STAT5 via receptor-dependent
pathways, inducing the expression of STAT5 target genes through
JAK2 and STAT5 signaling pathways (40,41).
The present study demonstrated that with the addition of IL-3 and
GM-CSF cytokines, the phosphorylation of STAT5 in MOLM13 cells
increased notably as the concentrations of IL-3 and GM-CSF
increased (Fig. 2C). Cells were
treated with topotecan hydrochloride for 24 h and stimulated with
IL-3 and GM-CSF for 20 min, it was observed that topotecan
hydrochloride effectively inhibited STAT5 phosphorylation even when
IL-3 and GM-CSF activated STAT5 expression, which suggested that it
could block STAT5 activation (Fig.
2D). The results demonstrated that topotecan hydrochloride
could specifically reduce STAT5 phosphorylation and downstream gene
expression, thereby inhibiting STAT5 activation in AML cells.
Topotecan hydrochloride induces
apoptosis and cell cycle arrest in AML cells
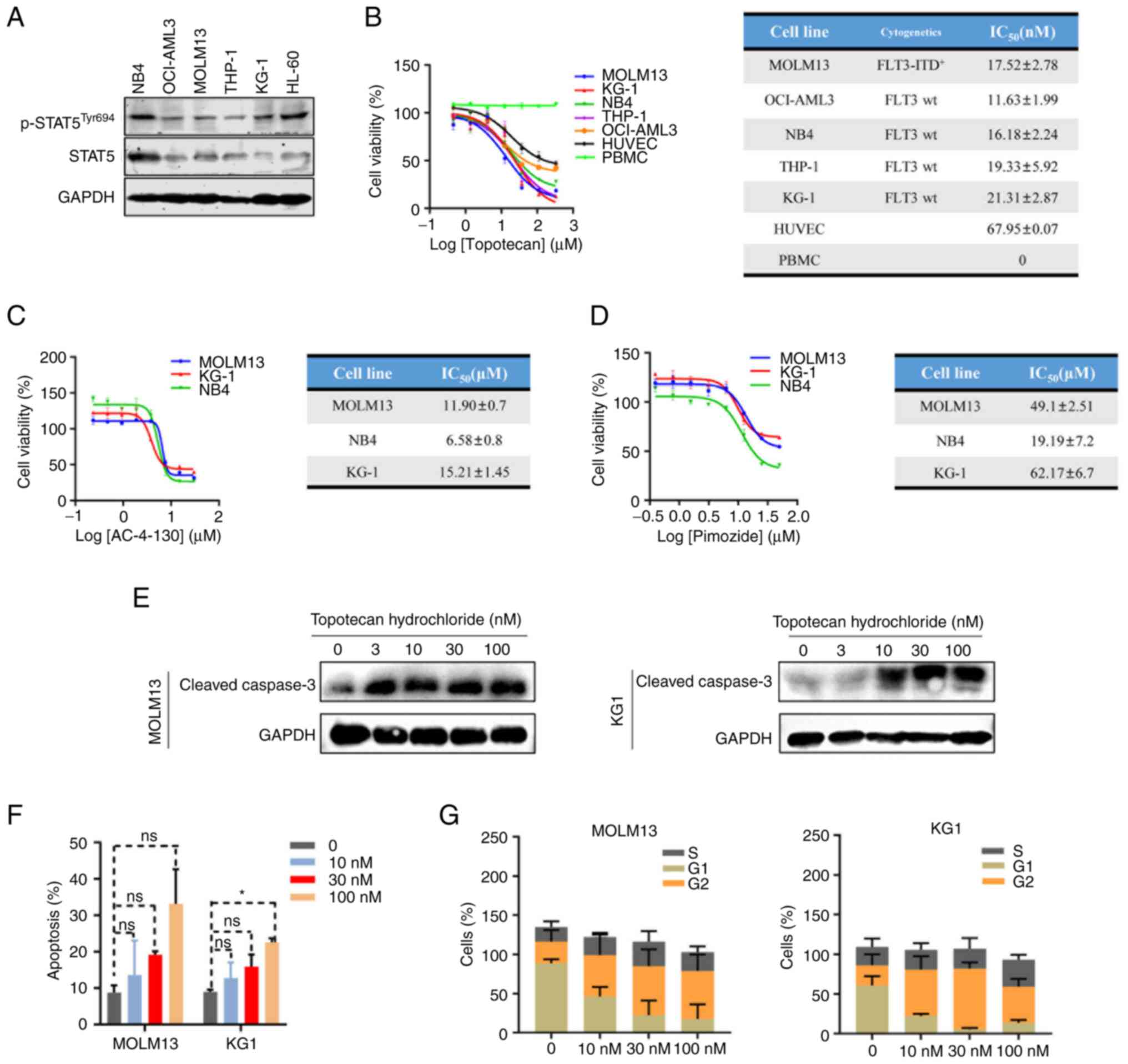
MOLM13 (FLT-ITD+), KG1, HL-60 and NB4
cells demonstrated high protein expression levels of p-STAT5
(Fig. 3A), MOLM13 and KG1 (as the
most commonly used in the literature (32,42)
cells were selected for further study. The MTS cell viability assay
demonstrated that topotecan hydrochloride had a strong inhibitory
activity against AML cells, with IC50 values ranging
from 11–21 nM (Fig. 3B). Topotecan
hydrochloride demonstrated poor cell activity in control cell lines
(HUVECs) and had no inhibitory effect on normal monocytes (PBMC),
which indicated that topotecan hydrochloride specifically inhibited
the proliferation of STAT5-expressing AML cells (Fig. 3B). The results demonstrated that
topotecan hydrochloride effectively inhibited AML cell growth by
targeting STAT5.
AML cells were treated with the existing STAT5
inhibitors AC-4-130 and Pimozide, and MTS cell viability was
determined. The results demonstrated that the IC50
values for AC-4-130 were 6.58–15.21 µM and those for Pimozide were
19.19–62.17 µM. The two inhibitors had poor cellular activity and
topotecan hydrochloride demonstrated a ~1,000-fold greater cellular
activity compared with them (Fig. 3C
and D).
In MOLM13 and KG1 cells, expression of Cleaved
Caspase 3, the apoptosis marker, markedly increased in an
apparently concentration-dependent manner, which suggested that
topotecan hydrochloride could induce apoptosis (Fig. 3E). The cell cycle and apoptosis of
MOLM13 and KG1 cells treated with topotecan hydrochloride were
analyzed, flow cytometry demonstrated that topotecan hydrochloride
induced apoptosis and cell cycle arrest, with a marked decrease in
the number of cells in G1 phase and a marked increase in G2/M phase
(Figs. 3F and G, and S2A and B).
These results suggested that topotecan hydrochloride
inhibited the proliferation of AML cells by inducing cell cycle
arrest and apoptosis. In summary, topotecan hydrochloride targeted
AML cells that expressed STAT5 and inhibited their proliferation
through specific targeting of STAT5 pathways.
Topotecan hydrochloride inhibit AML
tumor development in vivo
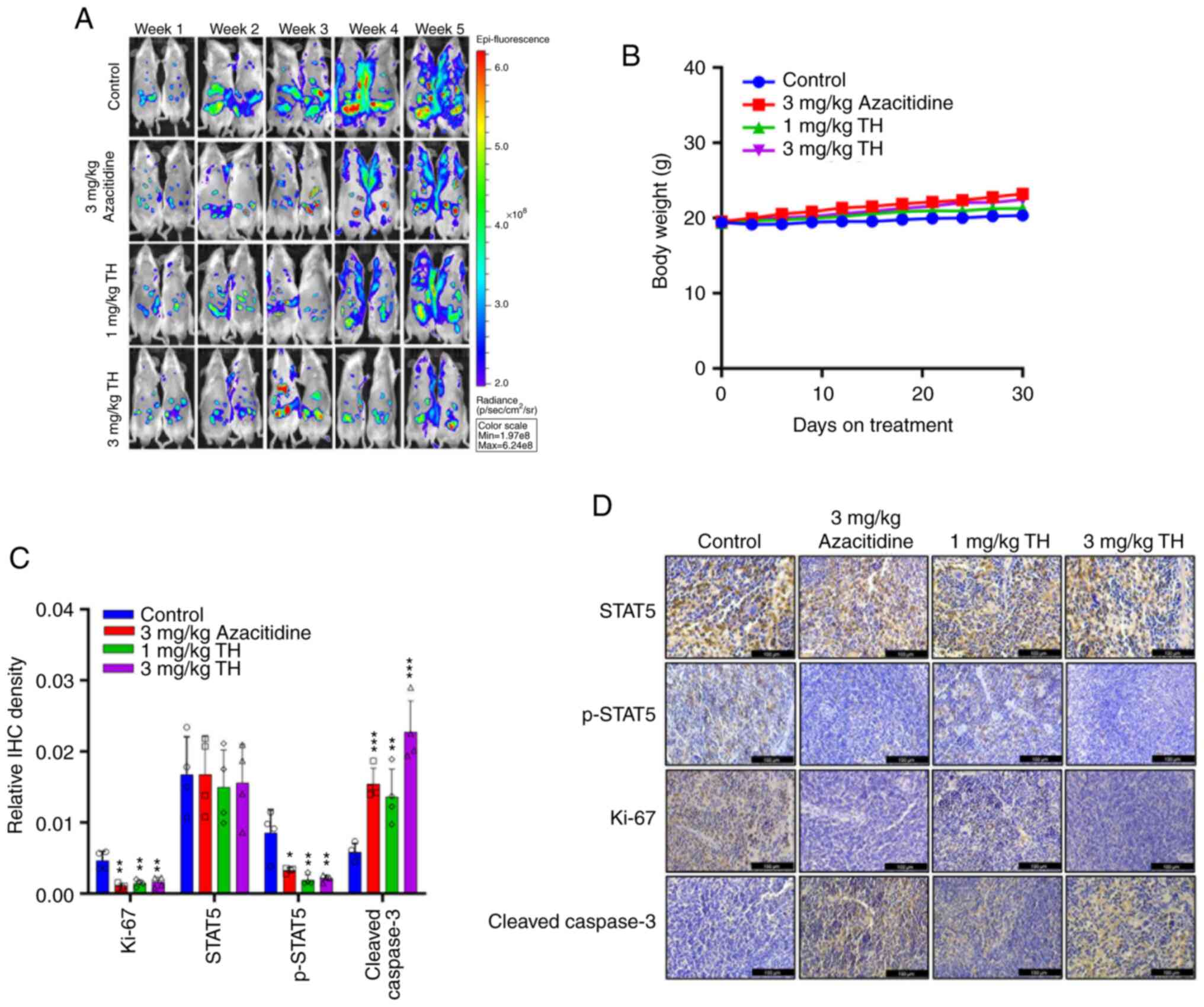
To assess the anti-tumor effect of topotecan
hydrochloride in vivo, a subcutaneous tumor growth xenograft
model using MOLM13 was established. The results demonstrated that
after 30 days of treatment, the 1 mg/kg and 3 mg/kg topotecan
hydrochloride groups demonstrated a significant reduction in tumor
volume and inhibition of AML tumor growth, compared with the
control (Fig. 4A and B). Upon
killing the mice, the tumor was collected, imaged and weighed to
record the tumor mass. A notable trend of reduced tumor volume and
tumor mass was apparent. Throughout the experiment, the mice
well-tolerated different doses of topotecan hydrochloride, and body
weight was maintained (Fig. 4C). In
comparison with the control group, H&E staining of the heart,
liver, spleen, lungs and kidneys indicated no notable differences
in the topotecan hydrochloride treatment group (Fig. S3). IHC results demonstrated that
topotecan hydrochloride significantly inhibited
STAT5Tyr694 phosphorylation (Fig. 4D and E). Increasing drug
concentration was observed to markedly increase Cleaved Caspase 3
levels, and topotecan hydrochloride significantly inhibited Ki-67
expression in tumor xenograft models, which indicated that
topotecan hydrochloride induced apoptosis of AML cells in
vivo. Overall, these results suggested that topotecan
hydrochloride could significantly inhibit AML tumor growth, induce
tumor cell apoptosis and inhibit STAT5 phosphorylation in
vivo.
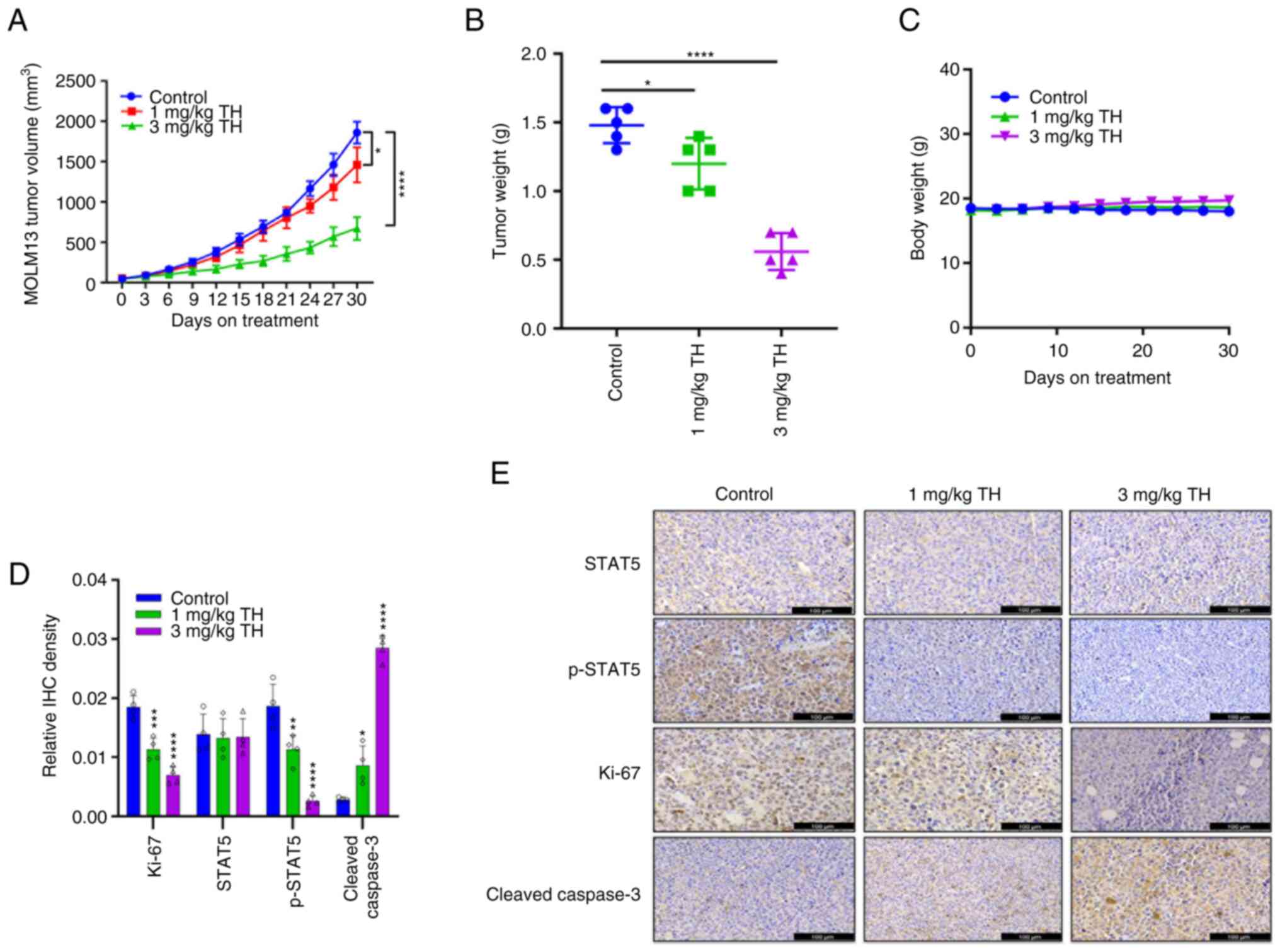 | Figure 4.Topotecan hydrochloride inhibits
acute myeloid leukemia tumor growth in vivo. MOLM13 cells
were subcutaneously administered at a density of 5×106
cells/mouse. After the tumor volume grew to 100–150 mm3,
the animals were randomly grouped and treated according to group
administration. Equal amounts of solvent (n=5), 1 mg/kg/day
topotecan hydrochloride (n=5), 3 mg/kg/day topotecan hydrochloride
(n=5), or 3 mg/kg/day Azacitidine (n=5) were injected
intraperitoneally once a day for 5 days a week at weeks 1, 3 and 4.
(A) The tumor volume was calculated as (length ×
width2)/2. (B) After the experiments, all the tumors
were weighed. (C) Body weight was measured every three days. IHC
analysis was performed on the different treatment groups for
p-STAT5, STAT5, Ki-67 and Cleaved Caspase 3. The results were
quantified (D) and imaged (E). Scale bar=100 µm. Data are presented
as mean ± standard deviation. *P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. TH, topotecan hydrochloride;
IHC, immunohistochemistry; p, phosphorylated. |
Topotecan hydrochloride inhibits
leukemic metastasis in vivo
Topotecan hydrochloride was demonstrated to be
effective in an AML subcutaneous tumor growth xenograft model;
therefore, a MOLM13/Luc metastasis model was used to assess the
effectiveness of topotecan hydrochloride in inhibiting leukemia
metastasis and tumor growth in vivo. Azacytidine is a
positive control agent and is a first-line chemotherapy agent in
elderly patients with AML who are not suitable for intensive
therapy (43), which was approved
in the European Union in 2017 for the treatment of adult AML.
MOLM13 cells expressing luciferase were injected
into NOD/SCID mice through the caudal vein. Tumor formation in
vivo was observed by two rounds of bioluminescence imaging 4–5
days after inoculation, tumor metastasis was monitored once a week
after inoculation. The topotecan hydrochloride treatment group (1
and 3 mg/kg) markedly inhibit leukemia metastasis (Fig. 5A) and markedly reduced tumor growth
and tumor volume, and the anti-tumor effect of 3 mg/kg topotecan
hydrochloride was notably greater than that of Azactidine. The body
weight of mice in both the administration and control groups
demonstrated a slight, steady increase (Fig. 5B). In comparison with the control
group, H&E staining of the spleen demonstrated no significant
difference in the effects of topotecan hydrochloride on the organs,
which indicated that topotecan hydrochloride had low toxicity and
good safety in mice at a dose of 1–3 mg/kg (Fig. S4). IHC demonstrated markedly
decreased protein expression levels of STAT5, significantly
decreased p-STAT5 and Ki-67 expression levels, and significantly
increased Cleaved Caspase 3 in the 3 mg/kg topotecan hydrochloride
group compared with the control (Fig.
5C and D), which suggested that topotecan hydrochloride
inhibited phosphorylation and promoted apoptosis of AML cells in
vivo.
Discussion
The death rate from AML increased every year between
2017–2022, and the recurrence of AML is one of the reasons for
treatment failure. There is an urgent need to develop new
therapeutic strategies to improve the prognosis of AML patients
(3). In recent years, the
development of small-molecule targeted drugs has become a promising
strategy for AML treatment, particularly for older patients (>68
years) who cannot tolerate high-intensity chemotherapy (6). This approach offers new treatment
options for this population. Although inhibition of the JAK/STAT
signaling pathway is a promising strategy for inhibiting tumor
growth, targeting this protein can be challenging (44).
In the present study, topotecan hydrochloride, a new
STAT5 inhibitor, was identified using FP system screening, and
through computer docking and CETSA experiments, it was demonstrated
that topotecan hydrochloride directly combined with STAT5.
Topotecan hydrochloride has good activity in cells at the nanomolar
level. Topotecan hydrochloride selectively inhibits the activation
and phosphorylation of STAT5 in AML cells and blocks the formation
of dimers, which inhibits the growth and proliferation of AML. In
addition, it was demonstrated that topotecan hydrochloride showed
good anti-tumor activity in mice xenograft model via inhibition of
STAT5 signaling.
It has previously been reported that the action of
STAT5 small molecule inhibitors against AML proliferation is not
good, at the micromolar level (45–47),
however, topotecan hydrochloride demonstrated good activity in AML
cells, with a 1,000-fold increase in cell activity compared with
other STAT5 inhibitors (AC-4-130 and Pimozide). Furthermore,
topotecan hydrochloride was demonstrated to impede the
phosphorylation of STAT5 and hinder dimer formation in AML cells,
including FLT3-ITD+ AML cells, which suggested its
potential as an effective inhibitor of AML resistance and
recurrence. However, there are still certain questions that need
further study. Firstly, the specific binding sites of topotecan
hydrochloride to STAT5 need to be elucidated to gain a better
understanding of the molecular mechanisms involved. Proteins with
different domain fragments should be purified and used to confirm
the specific binding position of topotecan hydrochloride and STAT5
through methods such as microscale thermophoresis and surface
plasmon resonance. Secondly, given that FLT3-ITD+ AML
cells are more sensitive to STAT5 inhibition, the specific
inhibitory mechanism of STAT5 on FLT3-ITD+ AML cells
should be studied in the later stages of cell growth. Thirdly, the
mechanism of drug resistance in AML should be further assessed.
The design and development of STAT5 inhibitors could
lay a foundation for further development of FLT3-ITD+
AML compounds with clinical value. STAT5 inhibitors not only
represent a new therapeutic approach, but also indicate the
potential undefined functions of STAT5 in AML cells. Overall, the
present study demonstrated an advance in the development of
treatments for AML and highlighted the potential of targeted
therapies to combat drug resistance and improve patient
outcomes.
Supplementary Material
Supporting Data
Acknowledgements
Thanks to the Zhengfang Yi Laboratory of East China
Normal University for its technical support for the performance of
certain experiments.
Funding
This work was supported by grants from the National Natural
Science Foundation of China (grant no. 81872418) and the Science
and Technology Commission of Shanghai Municipality (grant no.
21S11902000).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL, BT, ZS and YM designed the work, acquired data
and interpreted the results. GL performed the statistical analysis.
JL and ZS drafted the manuscript. JL, BT and YM confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
All in vivo experiments were approved by the
Animal Ethics Committee of Shanghai Fengxian District Central
Hospital (approval no. 6600).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Döhner H, Weisdorf DJ and Bloomfield CD:
Acute myeloid leukemia. N Engl J Med. 373:1136–1152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
National Cancer Institute (NCI), . Cancer
stat facts: Leukemia-acute myeloid leukemia. NCI; Bethesda, MD:
2020
|
|
3
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shah A, Andersson TML, Rachet B, Björkholm
M and Lambert PC: Survival and cure of acute myeloid leukaemia in
England, 1971–2006: A population-based study. Br J Haematol.
162:509–516. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thein MS, Ershler WB, Jemal A, Yates JW
and Baer MR: Outcome of older patients with acute myeloid leukemia:
An analysis of SEER data over 3 decades. Cancer. 119:2720–2727.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou J and Chng WJ: Identification and
targeting leukemia stem cells: The path to the cure for acute
myeloid leukemia. World J Stem Cells. 6:473–484. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wakao H, Gouilleux F and Groner B: Mammary
gland factor (MGF) is a novel member of the cytokine regulated
transcription factor gene family and confers the prolactin
response. EMBO J. 13:2182–2191. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kollmann S, Grausenburger R, Klampfl T,
Prchal-Murphy M, Bastl K, Pisa H, Knab VM, Brandstoetter T, Doma E,
Sperr WR, et al: A STAT5B-CD9 axis determines self-renewal in
hematopoietic and leukemic stem cells. Blood. 138:2347–2359. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pham HTT, Maurer B, Prchal-Murphy M,
Grausenburger R, Grundschober E, Javaheri T, Nivarthi H, Boersma A,
Kolbe T, Elabd M, et al: STAT5BN642H is a driver mutation for T
cell neoplasia. J Clin Invest. 128:387–401. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bandapalli OR, Schuessele S, Kunz JB,
Rausch T, Stütz AM, Tal N, Geron I, Gershman N, Izraeli S, Eilers
J, et al: The activating STAT5B N642H mutation is a common
abnormality in pediatric T-cell acute lymphoblastic leukemia and
confers a higher risk of relapse. Haematologica. 99:e188–e192.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kontro M, Kuusanmäki H, Eldfors S,
Burmeister T, Andersson EI, Bruserud O, Brümmendorf TH, Edgren H,
Gjertsen BT, Itälä-Remes M, et al: Novel activating STAT5B
mutations as putative drivers of T-cell acute lymphoblastic
leukemia. Leukemia. 28:1738–1742. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Küçük C, Jiang B, Hu X, Zhang W, Chan JK,
Xiao W, Lack N, Alkan C, Williams JC, Avery KN, et al: Activating
mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK
cells. Nat Commun. 6:60252015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rajala HLM, Eldfors S, Kuusanmäki H, van
Adrichem AJ, Olson T, Lagström S, Andersson EI, Jerez A, Clemente
MJ, Yan Y, et al: Discovery of somatic STAT5b mutations in large
granular lymphocytic leukemia. Blood. 121:4541–4550. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kiel MJ, Velusamy T, Rolland D,
Sahasrabuddhe AA, Chung F, Bailey NG, Schrader A, Li B, Li JZ, Ozel
AB, et al: Integrated genomic sequencing reveals mutational
landscape of T-cell prolymphocytic leukemia. Blood. 124:1460–1472.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nicolae A, Xi L, Pittaluga S, Abdullaev Z,
Pack SD, Chen J, Waldmann TA, Jaffe ES and Raffeld M: Frequent
STAT5B mutations in γδ hepatosplenic T-cell lymphomas. Leukemia.
28:2244–2248. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ihle JN: The Stat family in cytokine
signaling. Curr Opin Cell Biol. 13:211–217. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Smithgall TE, Briggs SD, Schreiner S,
Lerner EC, Cheng H and Wilson MB: Control of myeloid
differentiation and survival by Stats. Oncogene. 19:2612–2618.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Coffer PJ, Koenderman L and de Groot RP:
The role of STATs in myeloid differentiation and leukemia.
Oncogene. 19:2511–2522. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Halim CE, Deng S, Ong MS and Yap CT:
Involvement of STAT5 in oncogenesis. Biomedicines. 8:3162020.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dellomo AJ, Abbotts R, Eberly CL,
Karbowski M, Baer MR, Kingsbury TJ and Rassool FV: PARP1 PARylates
and stabilizes STAT5 in FLT3-ITD acute myeloid leukemia and other
STAT5-activated cancers. Transl Oncol. 15:1012832022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Scherr M, Chaturvedi A, Battmer K,
Dallmann I, Schultheis B, Ganser A and Eder M: Enhanced sensitivity
to inhibition of SHP2, STAT5, and Gab2 expression in chronic
myeloid leukemia (CML). Blood. 107:3279–3287. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nieborowska-Skorska M, Wasik MA, Slupianek
A, Salomoni P, Kitamura T, Calabretta B and Skorski T: Signal
transducer and activator of transcription (STAT)5 activation by
BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains
of BCR/ABL and is required for leukemogenesis. J Exp Med.
189:1229–1242. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
de Groot RP, Raaijmakers JA, Lammers JW,
Jove R and Koenderman L: STAT5 Activation by BCR-Abl Contributes to
Transformation of K562 Leukemia Cells. Blood. 94:1108–1112. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Spiekermann K, Bagrintseva K, Schwab R,
Schmieja K and Hiddemann W: Overexpression and constitutive
activation of FLT3 induces STAT5 activation in primary acute
myeloid leukemia blast cells. Clin Cancer Res. 9:2140–2150.
2003.PubMed/NCBI
|
|
25
|
Ikezoe T, Kojima S, Furihata M, Yang J,
Nishioka C, Takeuchi A, Isaka M, Koeffler HP and Yokoyama A:
Expression of p-JAK2 predicts clinical outcome and is a potential
molecular target of acute myelogenous leukemia. Int J Cancer.
129:2512–2521. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Venugopal S, Bar-Natan M and Mascarenhas
JO: JAKs to STATs: A tantalizing therapeutic target in acute
myeloid leukemia. Blood Rev. 40:1006342020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen CY, Tsay W, Tang JL, Shen HL, Lin SW,
Huang SY, Yao M, Chen YC, Shen MC, Wang CH and Tien HF: SOCS1
methylation in patients with newly diagnosed acute myeloid
leukemia. Genes Chromosomes Cancer. 37:300–305. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang S, Fukuda S, Lee Y, Hangoc G, Cooper
S, Spolski R, Leonard WJ and Broxmeyer HE: Essential role of signal
transducer and activator of transcription (Stat)5a but not Stat5b
for Flt3-dependent signaling. J Exp Med. 192:719–728. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xu B, Tian H and Zhou SY: Detection of
FLT3 gene and FLT3/ITD gene mutation in chronic myeloid leukemia
and its significance. Ai Zheng. 23:1218–1221. 2004.(In Chinese).
PubMed/NCBI
|
|
30
|
Kiyoi H, Naoe T, Nakano Y, Yokota S,
Minami S, Miyawaki S, Asou N, Kuriyama K, Jinnai I, Shimazaki C, et
al: Prognostic implication of FLT3 and N-RAS gene mutations in
acute myeloid leukemia. Blood. 93:3074–3080. 1999.PubMed/NCBI
|
|
31
|
Perl AE, Altman JK, Cortes J, Smith C,
Litzow M, Baer MR, Claxton D, Erba HP, Gill S, Goldberg S, et al:
Selective inhibition of FLT3 by gilteritinib in relapsed or
refractory acute myeloid leukaemia: A multicentre, first-in-human,
open-label, phase 1–2 study. Lancet Oncol. 18:1061–1075. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wingelhofer B, Maurer B, Heyes EC,
Cumaraswamy AA, Berger-Becvar A, de Araujo ED, Orlova A, Freund P,
Ruge F, Park J, et al: Pharmacologic inhibition of STAT5 in acute
myeloid leukemia. Leukemia. 32:1135–1146. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wingelhofer B, Neubauer HA, Valent P, Han
X, Constantinescu SN, Gunning PT, Müller M and Moriggl R:
Implications of STAT3 and STAT5 signaling on gene regulation and
chromatin remodeling in hematopoietic cancer. Leukemia.
32:1713–1726. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Elumalai N, Berg A, Rubner S, Blechschmidt
L, Song C, Natarajan K, Matysik J and Berg T: Rational development
of Stafib-2: A selective, nanomolar inhibitor of the transcription
factor STAT5b. Sci Rep. 7:8192017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Haftchenary S, Luchman HA, Jouk AO, Veloso
AJ, Page BD, Cheng XR, Dawson SS, Grinshtein N, Shahani VM, Kerman
K, et al: Potent targeting of the STAT3 protein in brain cancer
stem cells: A promising route for treating glioblastoma. ACS Med
Chem Lett. 4:1102–1107. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Brachet-Botineau M, Polomski M, Neubauer
HA, Juen L, Hédou D, Viaud-Massuard MC, Prié G and Gouilleux F:
Pharmacological inhibition of oncogenic STAT3 and STAT5 signaling
in hematopoietic cancers. Cancers (Basel). 12:2402020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang X, Sun Y, Pireddu R, Yang H, Urlam
MK, Lawrence HR, Guida WC, Lawrence NJ and Sebti SM: A novel
inhibitor of STAT3 homodimerization selectively suppresses STAT3
activity and malignant transformation. Cancer Res. 73:1922–1933.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
He Y, Peng S, Wang J, Chen H, Cong X, Chen
A, Hu M, Qin M, Wu H, Gao S, et al: Ailanthone targets p23 to
overcome MDV3100 resistance in castration-resistant prostate
cancer. Nat Commun. 7:131222016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee J, Seong S, Kim JH, Kim K, Kim I,
Jeong BC, Nam KI, Kim KK, Hennighausen L and Kim N: STAT5 is a key
transcription factor for IL-3-mediated inhibition of RANKL-induced
osteoclastogenesis. Sci Rep. 6:309772016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sheng W, Yang F, Zhou Y, Yang H, Low PY,
Kemeny DM, Tan P, Moh A, Kaplan MH, Zhang Y and Fu XY: STAT5
programs a distinct subset of GM-CSF-producing T helper cells that
is essential for autoimmune neuroinflammation. Cell Res.
24:1387–1402. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Guo Z, Wang A, Zhang W, Levit M, Gao Q,
Barberis C, Tabart M, Zhang J, Hoffmann D, Wiederschain D, et al:
PIM inhibitors target CD25-positive AML cells through concomitant
suppression of STAT5 activation and degradation of MYC oncogene.
Blood. 124:1777–1789. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fenaux P, Mufti GJ, Hellstrom-Lindberg E,
Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz
G, List A, et al: Efficacy of azacitidine compared with that of
conventional care regimens in the treatment of higher-risk
myelodysplastic syndromes: A randomised, open-label, phase III
study. Lancet Oncol. 10:223–232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cook AM, Li L, Ho Y, Lin A, Li L, Stein A,
Forman S, Perrotti D, Jove R and Bhatia R: Role of altered growth
factor receptor-mediated JAK2 signaling in growth and maintenance
of human acute myeloid leukemia stem cells. Blood. 123:2826–2837.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hoelbl A, Schuster C, Kovacic B, Zhu B,
Wickre M, Hoelzl MA, Fajmann S, Grebien F, Warsch W, Stengl G, et
al: Stat5 is indispensable for the maintenance of bcr/abl-positive
leukaemia. EMBO Mol Med. 2:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Walz C, Ahmed W, Lazarides K, Betancur M,
Patel N, Hennighausen L, Zaleskas VM and Van Etten RA: Essential
role for Stat5a/b in myeloproliferative neoplasms induced by
BCR-ABL1 and JAK2(V617F) in mice. Blood. 119:3550–3560. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yan D, Hutchison RE and Mohi G: Critical
requirement for Stat5 in a mouse model of polycythemia vera. Blood.
119:3539–3549. 2012. View Article : Google Scholar : PubMed/NCBI
|