Introduction
Head and neck squamous cell carcinoma (HNSCC) is a
cancer of the squamous epithelium of the oral cavity, larynx,
pharynx and nasal cavity (1). HNSCC
is the seventh leading cause of human malignancy, accounting for
890,000 new diagnoses and 450,000 cancer related-deaths per year
worldwide. According to GLOBOCAN 2020, the global incidence of
HNSCC has been increasing in recent years, and this trend is
partially attributed to the growing prevalence of human
papillomavirus (HPV)-related oropharyngeal carcinoma (2,3). In
Italy, HNSCC accounts for ~3% of all malignancies, most observed in
the male population (3). The major
risk factors for HNSCC are smoking, alcohol abuse, and HPV
(1). Treatments of HNSCC include
surgery, radiotherapy and chemotherapy (4). However, the prognosis of HNSCC is
inauspicious due to recurrent or metastatic HNSCC; in this case,
curative options are very limited (1,2,4).
Liquid biopsy is an important tool in molecular oncology as it is
an excellent source of biomolecules, particularly circulating
cell-free DNA (ccfDNA) released into the bloodstream from cell
secretion or as a result of apoptosis and necrosis (5,6). In
total, <1% of the ccfDNA is circulating tumor DNA (ctDNA)
characterized by cancer hallmarks, such as mutations or aberrant
gene methylation; their detection can serve as molecular indicators
for diagnosis, prognosis, and identification of early recurrence
(7,8). Alterations in the DNA methylation
profile are known to occur early during cancer development, and
hypermethylation of the promoter region of tumor suppressor genes
is involved in cancer onset and progression (9,10). DNA
methylation is a stable covalent modification that mainly occurs at
the 5C position of cytosine in CpG dinucleotides to form the
5-methylcytosine and can be detected in bio-fluids by PCR-based
methods (11). DNA hypermethylation
of septin 9 (SEPT9) and short stature homeobox 2
(SHOX2) has been previously described in tissues and in
ccfDNA from plasma of patients with HNSCC using qPCR assay
(12). SEPT9 belongs to the
septin family and is involved in cytokinesis and cell cycle control
(13). DNA methylation of
SEPT9 has been found in different tumors and its increased
methylation levels have been detected during the progression of
cells to malignancy (14–17). In colon mucosa, SEPT9
methylation levels were found gradually increasing in tissues from:
Controls-not-advanced adenomas-advanced adenomas-invasive
adenocarcinoma (14,15). Accordingly, a significant reduction
of SEPT9 protein levels was identified in adenoma and tumor tissues
(15). Similarly, SEPT9
hypermethylation was observed in breast cancer (BC) tissues, but
not in healthy breast tissues, and was inversely correlated with
SEPT9 mRNA expression in BC cell lines and tissues (16). In BC cells, DNA hypermethylation was
revealed to inhibit the expression of SEPT9, which, in turn,
altered the formation of structured filaments and increased the
migratory potential of tumor cells by promoting cancer progression
(13,18,19).
Hypermethylation of SHOX2 gene has been identified in
several malignancies (20). The
SHOX2 gene is a member of the SHOX gene family and
encodes for a protein containing a 60-amino acid DNA-binding
domain, suggesting its role as a transcriptional regulator. The
exact molecular mechanism of SHOX2 or the role of
SHOX2 hypermethylation during carcinogenesis has not been
determined (21). However, numerous
studies have clearly evidenced the strong association between
SHOX2 hypermethylation and cancer progression (14,22,23).
In colon mucosa, SHOX2 methylation levels gradually
increased during the progression from the non-cancerous stage to
the adenoma and adenocarcinoma stages (14). Similarly, SHOX2 methylation
was found to be absent or low in non-malignant brain tissues and
pilocytic astrocytomas, at intermediate values in lower-grade
gliomas, and high in glioblastomas (23). In lung adenocarcinoma, SHOX2
methylation levels gradually increased in accordance with disease
stage (from stage 0-II) and cancer invasiveness (22). Hypermethylation of circulating
SHOX2 and SEPT9 has been detected in several human
cancers, and they are considered promising circulating tumor liquid
biopsy biomarkers (24). Methods
used to detect DNA methylation are usually based on qPCR. The
commonly used droplet digital PCR (ddPCR) technology provides
greater sensitivity and absolute quantification of the template
than conventional qPCR systems (25–28).
The target templates are partitioned into 20,000 water-in-oil
droplets produced by a ‘generator’, each representing a nano-sized
PCR environment (29). The
PCR-positive and PCR-negative droplets are automatically counted by
a ‘reader’ to provide absolute quantification of the target DNA in
digital form (30,31). To the best of the authors'
knowledge, epigenetic studies in liquid biopsies of patients with
HNSCC using ddPCR are still very limited and the combined
SEPT9 and SHOX2 methylation analysis by ddPCR is
lacking (32). In the present
study, a ddPCR-based-assay was developed for absolute
quantification of SEPT9 and SHOX2 methylation levels
in ccfDNA. The ability of ddPCR-based assays to detect very low
copies of methylated SEPT9 and SHOX2 was
demonstrated. Finally, the feasibility of measuring SEPT9
and SHOX2 DNA methylation levels in the plasma of 20
patients with HNSCC, before curative treatment (surgery,
radiotherapy, chemotherapy) and during different follow-up time
points of the same patients with intervals of 3 months, was
revealed. The present study is preliminary research of a larger
project of liquid biopsies in HNSCC (‘Identify’ project) to assess
whether DNA methylation levels of SHOX2 and SEPT9 may
vary during treatment.
Materials and methods
Plasma samples from patients with
HNSCC
All patients enrolled in the present study (n=20)
were recruited from the Unit of Otorhinolaryngology-Head and Neck
Surgery, ASST Spedali Civili, Department of Medical and Surgical
Specialties, Radiological Sciences and Public Health, University of
Brescia (Brescia, Italy). Clinical and pathological characteristics
are reported in Table I for each
patient. All patients with HNSCC met the following criteria: i)
Histologically confirmed squamous cell carcinoma of the oral
cavity, oropharynx, hypopharynx or larynx; ii) clinical stage I–IV
according to the VIII edition of the American Joint Committee on
Cancer (AJCC) staging system (33)
iii) aged ≥18 years and written informed consent provided.
Peripheral blood samples were collected in EDTA-coated tubes. All
recruited patients were screened for HPV-related disease by
determining the HPV status genotyping by PANA RealTyper HPV kit
CE/IVD (cat. no. PNAM-5001; HLB PANAGENE). The p16 protein
expression was assessed using the CINtec p16 histological test
(cat. no. 06695256001; Roche Diagnostics) with strong and
widespread nuclear and cytoplasmic staining in at least 70% of
cells used as a reference for positivity (Fig. S1). All these clinical
characteristics were determined from the medical records of the
patients; therefore they were not investigated as part of the
present study. The screenings were assessed routinely in HNSCC
clinic management. Peripheral blood (10 ml/patient) from patients
with HNSCC was collected before the start of the first treatment
(T0) and at intervals after the first treatment,
including surgery, radiotherapy and chemotherapy (T1=3
months, T2=6 months, T3=12 months after
treatment). Collecting liquid biopsies from patients that received
the same type of treatment would have taken much longer. Plasma was
obtained by centrifugation of peripheral blood at 200 × g for 10
min at 4°C in an accuSpin Micro21 centrifuge (Thermo Fisher
Scientific, Inc.). The plasma was transferred to a new tube and
stored at −80°C until DNA extraction. The study was approved by the
Ethics Committee of Spedali Civili of Brescia (Protocol Identify,
Ethics Committee approval no. NP 4551).
 | Table I.Clinical characteristics of HNSCC
patients enrolled in the study. |
Table I.
Clinical characteristics of HNSCC
patients enrolled in the study.
| ID of patient | Tumor site | Staging (VIII ed.
AJCC) | aHPV (SCC oropharynx) | Treatment type | Status of disease
(at last FU) | Time point of FU
with blood sample collected |
|---|
| BS002 | Larynx | III |
| Surgery + adj | Right neck lymph
node recurrence + pulmonary metastasis at 6 months of FU | 6 months
(T2) |
| BS003 | Oral cavity | II |
| Surgery + adj | Local recurrence at
8 months of FU | 6 months
(T2) |
| BS006 | Oral cavity | III |
| Surgery + adj | NED (FU 18
months) | 12 months
(T3) |
| BS007 | Oral cavity | II |
| Surgery | NED (FU 18
months) | 12 months
(T3) |
| BS008 | Oropharynx | II | p16+,
HPV DNA+ | RT-CHT | Tumor persistence
at T1 | Pre-treatment
(T0) |
| BS009 | Oral cavity | II |
| Surgery + adj | NED (FU 18
months) | 12 months
(T3) |
| BS010 | Larynx | II |
| CHT neo + RT | NED (FU 12
months) | 6 months
(T2) |
| BS011 | Oral cavity | III |
| Surgery + adj | Second primary
tumor (SCC of the right tonsil) at 15 months of FU | 12 months
(T3) |
| BS013 | Oropharynx | II |
p16+ | RT-CHT | NED (FU 18
months) | 12 months
(T3) |
| BS014 | Larynx | III |
| Surgery + adj | Progressive disease
with pulmonary metastasis at 4 months of FU | Pre-treatment
(T0) |
| BS015 | Hypopharynx | III |
| Surgery + adj | Pulmonary
metastasis at 9 months of FU | 6 months
(T2) |
| BS016 | Larynx | II |
| Surgery + adj | NED (FU 18
months) | 12 months
(T3) |
| BS017 | Oropharynx | I |
p16+ | RT-CHT | NED (FU 15
months) | 12 months
(T3) |
| BS018 | Oropharynx | II |
p16+ | RT-CHT | NED (FU 15
months) | 12 months
(T3) |
| BS019 | Oropharynx | IV | NEG | RT-CHT | NED (FU 15
months) | 12 months
(T3) |
| BS020 | Oropharynx | II |
p16+ | RT-CHT | NED until
T2, then lost at FU | 6 months
(T2) |
| BS021 | Oral cavity | III |
| Surgery + adj | NED (FU 18
months) | 12 months
(T3) |
| BS029 | Larynx | II |
| Surgery | NED (FU 15
months) | 6 months
(T2) |
| BS023 | Oropharynx | I | p16+,
HPV DNA+ | RT-CHT | NED (FU 12
months) | 6 months
(T2) |
| BS024 | Oropharynx | I |
p16+ | RT-CHT | NED (FU 12
months) | 6 months
(T2) |
ccfDNA isolation from plasma and
bisulfite conversion
According to the manufacturer's instructions, ccfDNA
was isolated from 2 ml of plasma using MagMAX Cell-Free DNA
isolation kit (cat. no. A29319; ThermoFisher Scientific, Inc.).
Purified ccfDNA was eluted in a 30-µl volume, and 1 µl ccfDNA was
used for ccfDNA quantification using Qubit Fluorometer and Qubit
dsDNA HS (High Sensitivity) Assay kit (cat. no. Q32854; Thermo
Fisher Scientific, Inc.). Following the manufacturer's
instructions, the remaining ccfDNA (29 µl) was used for bisulfite
conversion using EZ DNA Methylation-Lightning kit (cat. no. D5030;
Zymo Research Corp). A total of 500 ng of a methylated and
non-methylated human DNA standard (Human Methylated &
Non-methylated DNA Set; cat. no. D5014; Zymo Research Corp.) were
converted with bisulfite as positive controls. Subsequently, 13 and
10 µl of bisulfite-converted DNA were obtained from ccfDNA and
methylated and non-methylated DNA, respectively, and stored at
−80°C until their use.
Methylation-specific ddPCR (MS-ddPCR)
assays
The MS-ddPCR assays were optimized according to the
principles of MS-PCR (34,35) to detect the methylation levels of
SEPT9 and SHOX2. MS-ddPCR experiments were performed
using QX200™ ddPCR System (Bio-Rad Laboraties, Inc.) (36,37).
The MS-ddPCR reaction mix consisted of the 2X ddPCR Supermix for
Probes, and locus-specific primers and probes. For the SEPT9 assay,
the primers and probe sequences were designed using Beacon Designer
(Premier Biosoft International). Two sets of primers and probes
were obtained and correspond to the bisulfite-modified methylated
or unmethylated sequence: The set with primers and probe with the
fluorescent FAM reporter for methylated SEPT9 (named
SEPT9-M) and the set with primers and probe with HEX reporter for
unmethylated SEPT9 (named SEPT9-U). For SHOX2, the
assay was designed by Beacon Designer to detect bisulfite-modified
methylated SHOX2 using a FAM-labelled probe set (named
SHOX2) and to detect, after bisulfite conversion, a CpG-free region
in the actin beta (ACTB) gene using a HEX-labelled probe set
(named ACTB) (38). The complete
list of all primer and probe sequences is provided in Table SI. The PCR mix was prepared in a
22-µl reaction volume containing 11 µl 2X ddPCR Supermix for Probes
(no dUTP) (cat. no. 186-3024; Bio-Rad Laboratories, Inc.), 0.55 µl
20X PCR probe assay specific for the methylated loci (SEPT9-M or
SHOX2) and 0.55 µl 20X PCR probe assay specific for the
unmethylated SEPT9 (SEPT9-U) or ACTB, and
bisulfite-treated DNA, as a template. Each ddPCR assay mixture (20
µl) was loaded into a disposable droplet generator cartridge (cat.
no. 1864008; Bio-Rad Laboratories, Inc.). Subsequently, 70 µl of
droplet generation oil for probes (cat. no. 1863005; Bio-Rad
Laboratories, Inc.) was loaded into each of the eight oil wells.
The cartridge was then placed inside the QX200 droplet generator
(Bio-Rad Laboratories, Inc.). After droplet generation was
completed, the droplets were transferred to a 96-well PCR plate
(cat. no. 12001925; Bio-Rad Laboratories, Inc.) using a
multichannel pipette. The plate was heat-sealed with foil and
placed in a T100 Thermal Cycler (Bio-Rad Laboratories, Inc.).
Thermal cycling conditions were as follows: 95°C for 10 min, 40
cycles at 94°C for 30 sec and 52°C (for the SEPT9 assay) or 57°C
(for the SHOX2 assay) for 1 min (ramp rate reduced to 2%), with a
final step at 98°C for 10 min and a 4°C indefinite hold. QuantaSoft
software version 1.7.4 (Bio-Rad Laboratories, Inc.) was used to
verify the number of total droplets and positive droplets for
methylated SEPT9 or SHOX2 in the FAM channel and for
the unmethylated SEPT9 or ACTB in the HEX channel.
The SEPT9 methylation level was calculated as a percentage:
Concentration (copies/µl) for SEPT9-M/(concentration
(copies/µl) for SEPT9-M + concentration (copies/µl) for SEPT9-U).
In addition, due to the lack of primers/probe set for unmethylated
SHOX2, the SHOX2 methylation level was calculated as
the ratio: Concentration (copies/µl) for SHOX2/concentration
(copies/µl) ACTB.
Establishing the efficiency of
MS-ddPCR assays
Methylated and non-methylated human DNA standards
(Zymo Research Corp.) converted with bisulfite were used to verify
the efficiency of MS-ddPCR assays in detecting SEPT9 and
SHOX2 methylation. By following the same experimental
workflow used by Yu et al (39), two-fold serial dilutions of fully
methylated DNA were prepared with water. A series of samples
containing 20,000, 10,000, 5,000, 2,500, 1,250, 625, 312.5, 156.25,
78.125 and 0 pg of standard bisulfite-converted DNA was assessed
for SEPT9 and SHOX2 by MS-ddPCR assays, as
aforementioned. The range of the standard curve comprised the
expected yield of DNA isolated from 1–2 ml of plasma. To verify the
ability of MS-ddPCR to discriminate methylated DNA from the DNA
background, 20 ng of total DNA containing the following percentages
of fully methylated DNA (99, 90, 70, 50, 30, 10 and 1%) were tested
(39). A negative template control
(NTC) containing all components of the reaction except for the DNA
template was included in each experiment.
MS-quantitative PCR (MS-qPCR)
Commercial 100% methylated and non-methylated human
DNA standards converted with bisulfite were used to verify the
efficiency of MS-qPCR assays in detecting SEPT9 and
SHOX2 methylation. Samples containing 20,000, 10,000, 5,000,
2,500, 1,250, 625, 312.5, 156.25, 78.125 and 0 pg of standard
bisulfite-converted DNA were tested for SEPT9 and
SHOX2 by MS-qPCR assays, as aforementioned. Furthermore, to
verify the ability of MS-qPCR to discriminate methylated DNA from
the DNA background, 20 ng of total DNA containing the following
percentages of fully methylated DNA (99, 90, 70, 50, 30, 10 and 1%)
were tested. The qPCR reaction (20 µl/well) contained 10 µl of
Taq-Man 2X Universal PCR Master Mix (Thermo Fisher Scientific,
Inc.), 0.5 µl 20X PCR probe assay specific for the methylated loci
(SEPT9-M or SHOX2), and 0.5 µl 20X PCR probe assay specific for
unmethylated SEPT9 (SEPT9-U) or ACTB, and bisulfite-treated DNA, as
a template. The PCR reactions were incubated at 95°C for 10 min,
followed by 40 cycles at 95°C for 15 sec and 52°C (for SEPT9 assay)
or 57°C (for SHOX2 assay) for 1 min. PCRs were performed in
triplicate using the QuantStudio 3 Real-Time PCR system (Thermo
Fisher Scientific, Inc.).
Detection of SEPT9 and SHOX2
methylation levels in ccfDNA of patients with HNSCC by
MS-ddPCR
To assess the methylation levels of SEPT9 and
SHOX2 in the plasma of patients with HNSCC, 6 µl of
bisulfite-converted ccfDNA were used for both MS-ddPCR assays.
Multiplex ddPCR assays and relative analysis were performed as
aforementioned. Each experiment included the positive control wells
for the methylated and unmethylated loci containing 4 µl (20 ng) of
fully methylated DNA (Zymo Research Corp.) converted with bisulfite
and 4 µl (20 ng) of completely unmethylated DNA (Zymo Research
Corp.) converted with bisulfite. NTC wells were also included.
Statistical analysis
Statistical analysis was carried out using GraphPad
Prism 7.0 software (Dotmatics). The linear regression between the
calculated percentage of the DNA methylation levels of SEPT9
and SHOX2 and the percentage of input methylated DNA was
performed to establish the efficiency of MS-ddPCR assays. The
experiments were performed in triplicate. One-way ANOVA or two-way
ANOVA, followed by Tukey's post hoc test, was used to compare the
mean values of methylation levels for SEPT9 and SHOX2
in ccfDNA among the different follow-up time points. The histograms
are presented as the mean values ± standard error of the mean
(SEM). The mean values of SEPT9 and SHOX2 methylation
levels at each time point (T0, T1,
T2, T3) were used to determine the trend,
shown as the red line, for longitudinal analysis of SEPT9
and SHOX2 methylation levels during the follow-up of
patients with HNSCC. P<0.05 was considered to indicate a
statistically significant difference.
Results
Establishing the efficiency of
MS-ddPCR assays for the detection of SEPT9 DNA methylation
In the present study, two multiplex assays were used
for measuring the methylation levels of SEPT9 and
SHOX2 using ddPCR technology, defined as MS-ddPCR. MS-ddPCR
for SEPT9 consisted of i) a TaqMan probe-based assay
designed with FAM reporter to detect the methylated
bisulfite-converted DNA (SEPT9-M) and ii) a TaqMan probe-based
assay with HEX reporter to detect the unmethylated
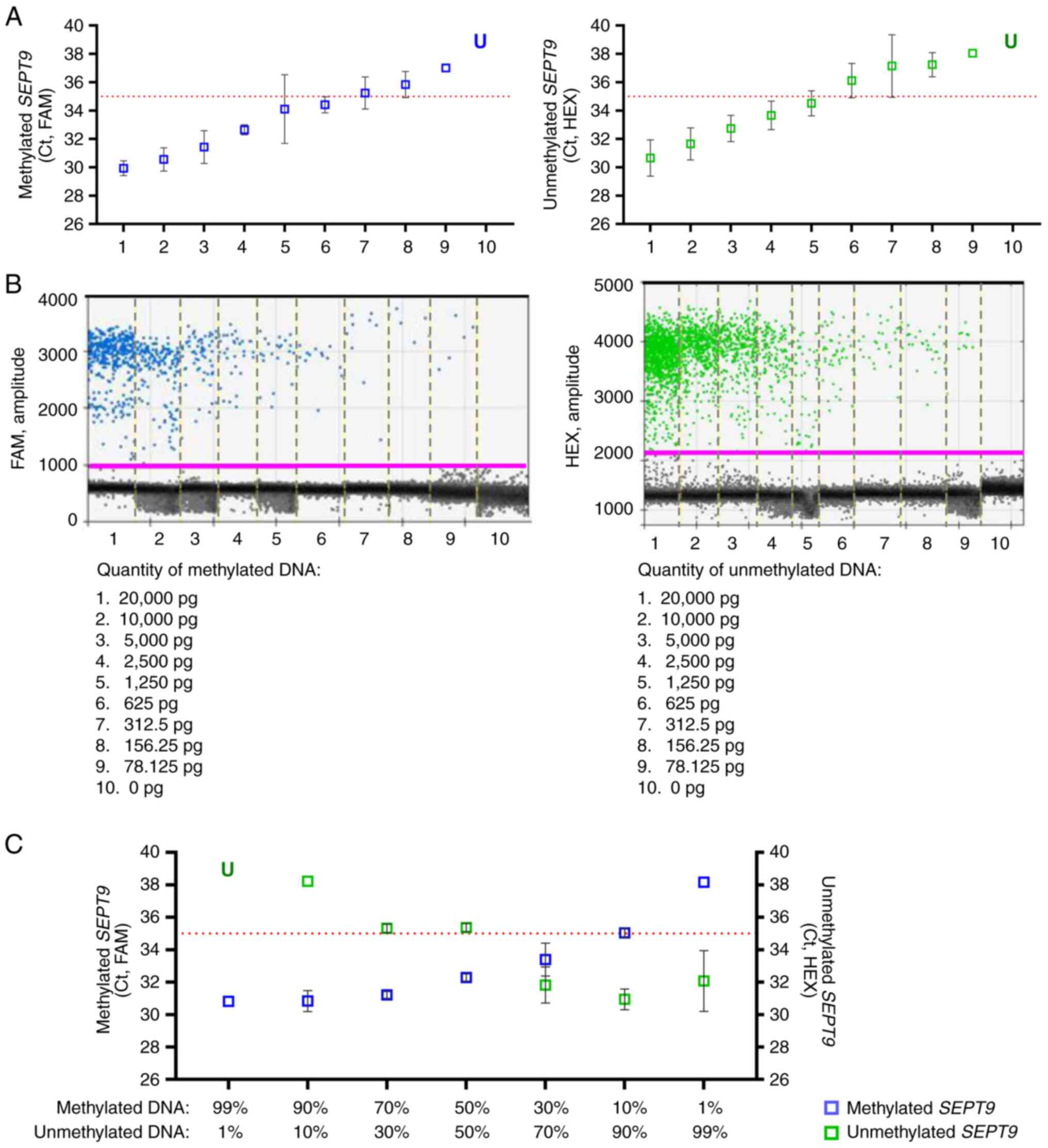
bisulfite-converted DNA (SEPT9-U) (Fig.
1A). The sensitivity and specificity of the assays were
assessed using commercial methylated DNA and unmethylated DNA after
bisulfite conversion. The two-dimensional (2D) amplitude plot
showed that the SEPT9-M set detected only the methylated template
(Fig. 1B, positive droplets in
blue, left) and, by contrast, the SEPT9-U set detected only the
unmethylated template (Fig. 1B,
positive droplets in green, right) in multiplex ddPCR experiments.
Next, the performance of the MS-ddPCR assay was evaluated by
considering its ability to detect the SEPT9 DNA methylation
levels in samples with low amounts of DNA input, and in the
presence of an unmethylated DNA background. MS-ddPCR for
SEPT9 displayed a dose-dependent trend, and the methylation
level was detectable using a starting input of commercial
bisulfite-treated DNA as low as 78.125 pg (Fig. 1C). To assess the ability of the
assay to detect methylated SEPT9 molecules in an
unmethylated DNA background, the methylated DNA with unmethylated
DNA was diluted at different percentages (99, 90, 70, 50, 30, 10
and 1%) and multiplex MS-ddPCR was performed on 20 ng of the
bisulfite-treated DNA mixtures. The concentration of the methylated
target (copies/µl, in blue) and that of the unmethylated target
(copies/µl, in green) decreased and increased, respectively,
according to the percentage of methylated DNA. The SEPT9-M and
SEPT9-U assays detected up to 1% methylated SEPT9 and
unmethylated SEPT9, with a concentration of 0.14 and 1.5
copies/µl, respectively (Fig. 1D).
The level of methylated SEPT9 (expressed as percent, %) was
calculated as described in the Materials and methods section. The
standard curve demonstrated good linearity between the level of
methylated SEPT9 (expressed as percent, %) and the
percentage of commercial bisulfite-treated methylated DNA loaded in
each reaction (R2=0.92; Fig.
1E).
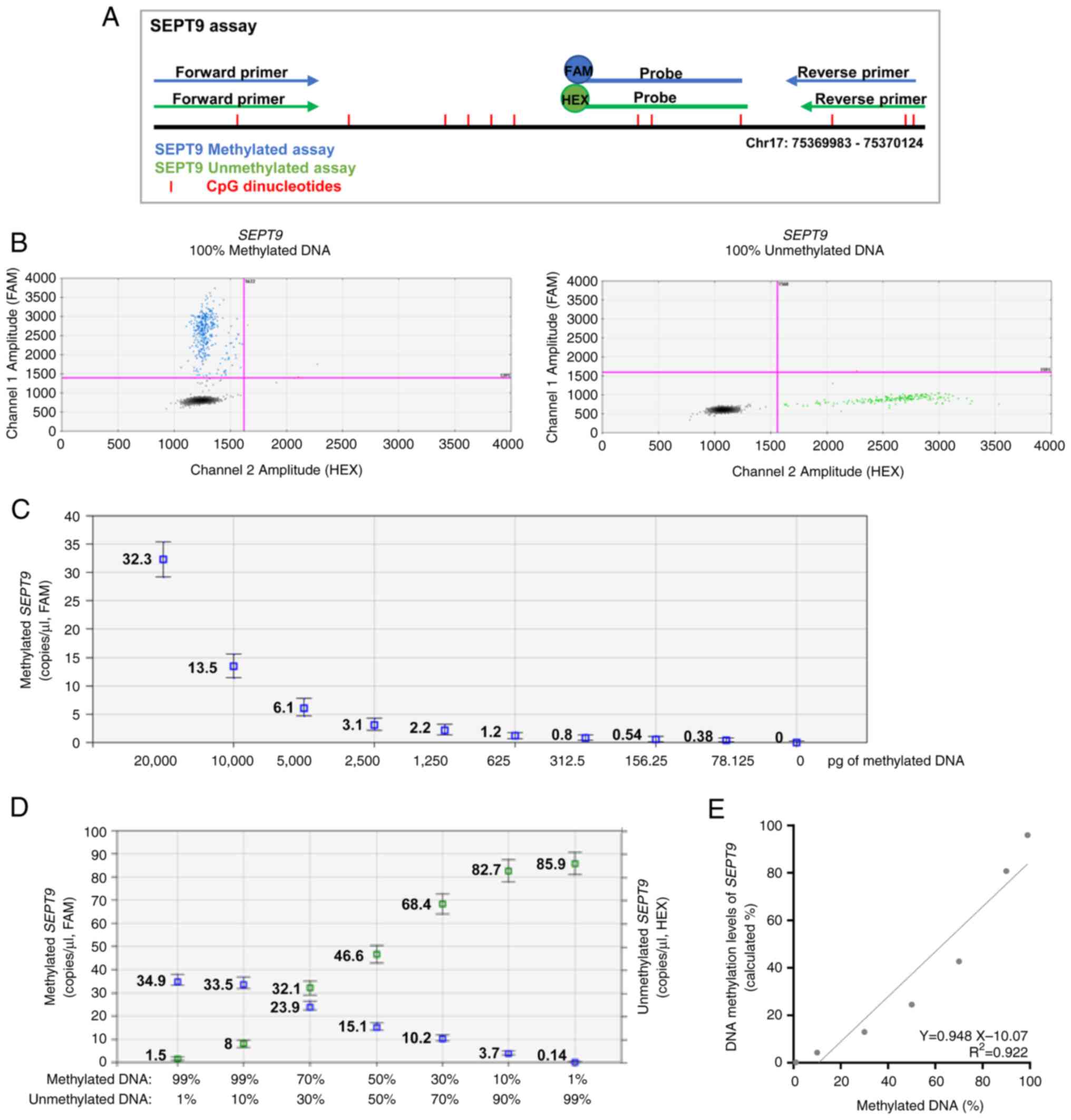 | Figure 1.Efficiency of MS-ddPCR assays for the
detection of SEPT9 DNA methylation. (A) Schematic
representation of the MS-ddPCR assay used to detect the methylation
levels of SEPT9. Multiplex ddPCR for the analysis of
SEPT9 methylation was performed on bisulfite-converted DNA
using the set specific for methylated DNA (in blue) and the set
specific for unmethylated DNA (in green). A methylation-specific
probe was designed with the FAM fluorescence dye, and an
unmethylation-specific probe was designed with the HEX fluorescence
dye. Vertical red lines represent the CpG dinucleotides; blue
arrows and lines are the primers and probe, respectively, used for
the detection of methylated SEPT9; green arrows and lines
are the primers and probe, respectively, used for the detection of
unmethylated SEPT9; the type of fluorescence dye is
indicated as FAM or HEX. (B) An example of a 2D amplitude plot of
the multiplex assay for SEPT9 using commercial methylated
DNA (left) and unmethylated DNA (right) converted with bisulfite. A
threshold was manually set for FAM and HEX dyes to select positive
droplets. Positive droplets for methylated SEPT9 were blue
(Channel 1, FAM); positive droplets for unmethylated SEPT9
were green (Channel 2, HEX); negative droplets were dark grey. (C)
Two-fold serial dilutions of commercial 100% methylated DNA
converted with bisulfite were prepared. ddPCR detected the
methylated SEPT9 as low as 78 pg of input methylated DNA.
(D) Samples containing commercial methylated DNA and unmethylated
DNA in different percentages (20 ng of total input DNA for each
well) were prepared to verify the ability of an MS-ddPCR assay to
detect methylated SEPT9 molecules in an unmethylated DNA
background. Concentrations (copies/µl) were reported for the
specific assay for methylated SEPT9 (in blue) and the
specific assay for unmethylated SEPT9 (in green). (E) A
standard quantification curve was obtained using the SEPT9
methylation level detected in the function of the percentage values
of fully methylated DNA loaded in each reaction. The SEPT9
methylation level was calculated as a percentage: Concentration
(copies/µl) for FAM/[concentration (copies/µl) for FAM +
concentration (copies/µl) for HEX]. SEPT9, septin 9;
MS-ddPCR, methylation-specific droplet digital PCR; ddPCR, droplet
digital PCR. |
To evaluate the efficiency of qPCR in detecting the
DNA methylation levels of SEPT9, the sensitivity and
specificity in the same conditions were assessed. The SEPT9-M set
reached the detection limit of qPCR (cycle threshold, Ct >35)
with 625 pg of input methylated DNA, and the SEPT9-U set with 1,250
pg of input unmethylated DNA (Fig.
2A). As revealed in Fig. 2B,
ddPCR was able to detect positive droplets up to 78.125 pg of input
DNA for both sets. Analysis of the ability of the assay to detect
methylated SEPT9 molecules in an unmethylated DNA
background, revealed that qPCR detected up to 30% of methylated
SEPT9 but the threshold cycles for 10 and 1% of methylated
SEPT9 were above the cutoff (Ct >35) (Fig. 2C). These results indicated the
higher sensitivity and specificity of ddPCR-based assays than
qPCR.
Establishing the efficiency of
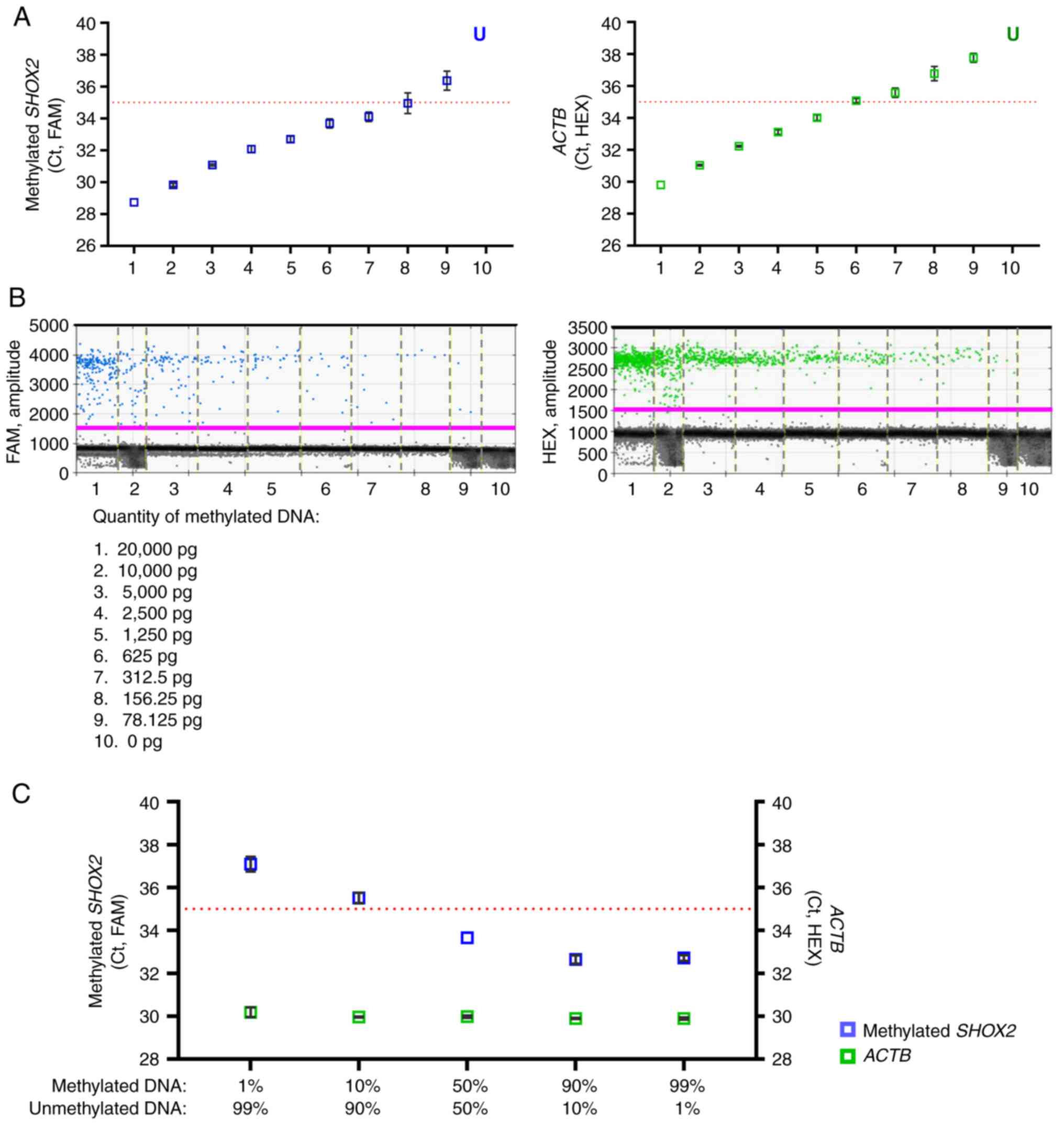
MS-ddPCR assays for the detection of SHOX2 DNA methylation
MS-ddPCR for SHOX2 consisted of i) a TaqMan
probe-based assay labeled with FAM reporter for methylated
SHOX2 and ii) a TaqMan probe-based assay labeled with HEX
reporter for a region CpG-free in the ACTB gene (Fig. 3A). The specificity of SHOX2 assays
was tested by following the procedures described for SEPT9.
Only methylated DNA treated with bisulfite was amplified using the
SHOX2 assay (Fig. 3B, positive
droplets in blue). As expected, the ACTB assay amplified methylated
and unmethylated DNA (Fig. 3B,
positive droplets in green). The MS-ddPCR assay for SHOX2
displayed a dose-dependent trend and could detect methylated
SHOX2 as low as 78.125 pg of commercial bisulfite-treated
DNA (Fig. 3C). The concentration of
ACTB (copies/µl, in green) remained stable with values
between 69.1 and 78.2 copies/µl; meanwhile, the concentration of
methylated SHOX2 (copies/µl, in blue) increased accordingly
to the percentage of input methylated DNA with good linearity
(R2=0.98; Fig. 3D and
E).
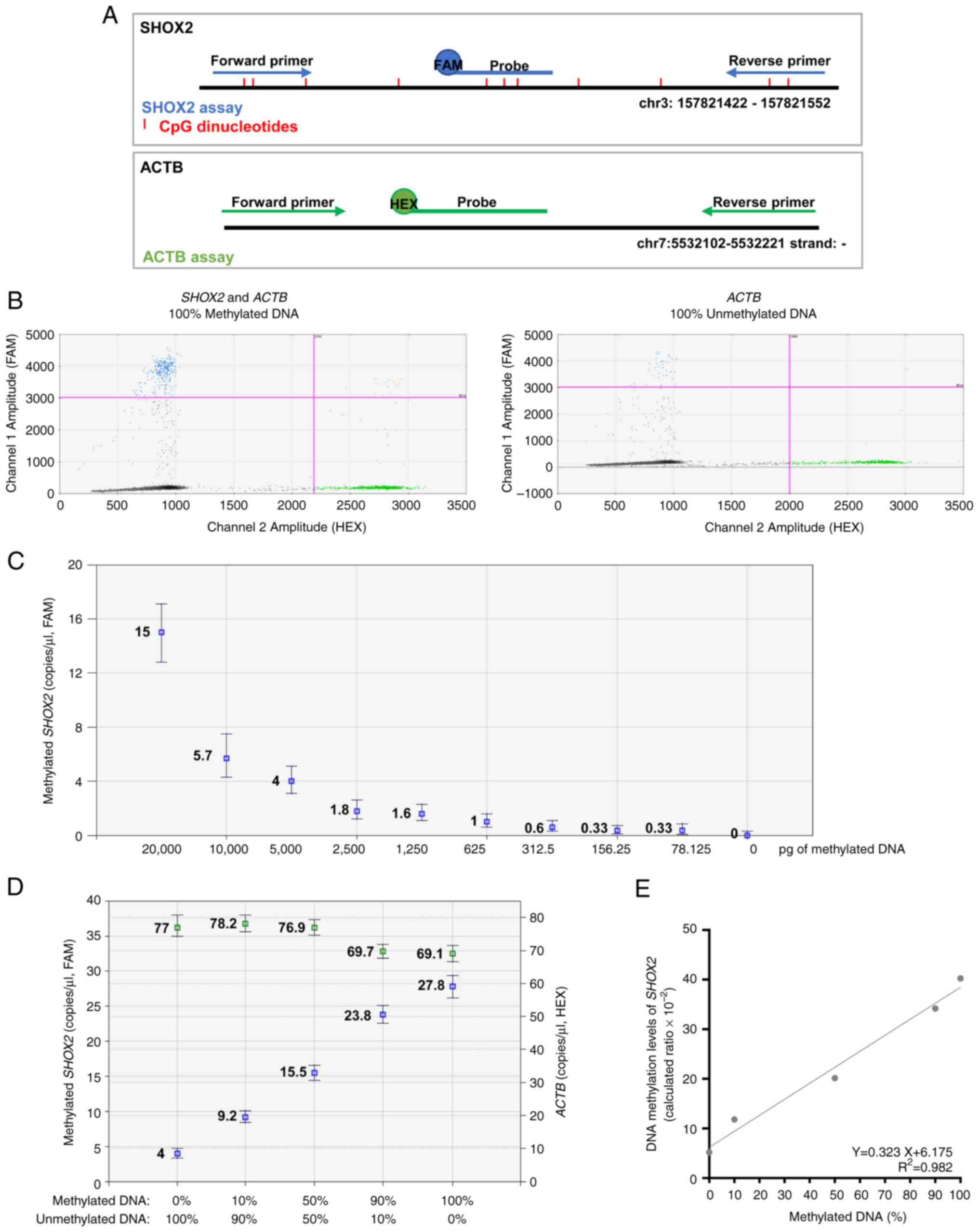 | Figure 3.Efficiency of MS-ddPCR assays for the
detection of SHOX2 DNA methylation. (A) Schematic
representation of an MS-ddPCR assay used to detect the methylation
levels of SHOX2. The assay was designed to detect methylated
SHOX2 using methylation-specific primers, a probe (in blue),
and a CpG-free region in the ACTB on bisulfite-converted
DNA. A methylation-specific probe was designed with the FAM
fluorescence dye, and the ACTB-specific probe was designed with the
HEX fluorescence dye. The vertical red lines represent the CpG
dinucleotides; the blue arrows and line are the primers and probe,
respectively, used for the detection of methylated SHOX2;
the green arrows and line are the primers and probe, respectively,
used for the detection of ACTB; the type of fluorescence dye
is indicated as FAM or HEX. (B) Example of a 2D amplitude plot of
the multiplex assay for SHOX2 using commercial methylated
DNA (left) and unmethylated DNA (right) converted with bisulfite. A
threshold was manually set for FAM and HEX dyes to select positive
droplets. Positive droplets for methylated SHOX2 were blue
(Channel 1, FAM), positive droplets for ACTB (sequence without CpG)
were green (Channel 2, HEX), and negative droplets were dark grey.
(C) Two-fold serial dilutions of commercial 100% methylated DNA
converted with bisulfite were prepared. ddPCR detected the
methylated SHOX2 as low as 78 pg of input methylated DNA.
(D) Samples were prepared containing commercial methylated DNA and
unmethylated DNA in different percentages (20 ng of total input DNA
for each well) to verify the ability of the SHOX2 assay to
detect methylated SHOX2 molecules in an unmethylated DNA
background. Concentrations (copies/µl) were reported for the assay
specific for methylated SHOX2 (in blue) and the assay
specific for ACTB (in green). (E) A standard quantification curve
was obtained using the SHOX2 methylation level detected in
the function of the percentage values of fully methylated DNA
loaded in each reaction. The SHOX2 methylation level was
calculated as a ratio: Concentration (copies/µl) for
FAM/concentration (copies/µl) for HEX. MS-ddPCR,
methylation-specific droplet digital PCR; SHOX2, short
stature homeobox 2; ACTB, actin beta. |
To evaluate the efficiency of qPCR in detecting DNA
methylation levels of SHOX2, the sensitivity and specificity
in the same conditions were assessed. The SHOX2 set reached the
detection limit of qPCR (Ct >35) with 156.25 pg and the ACTB set
with 1,250 pg of input methylated DNA (Fig. 4A). As shown in Fig. 4B, ddPCR was able to detect positive
droplets up to 78.125 pg of input DNA for both sets. Analysis of
the ability of the assay to detect methylated SHOX2
molecules in unmethylated DNA background showed that qPCR detected
up to 50% of methylated SHOX2 with threshold cycles below
the cut-off (Ct=35) (Fig. 4C).
These results indicated the higher sensitivity and specificity of
ddPCR-based assays compared with qPCR.
Methylation levels of SEPT9 and SHOX2
in ccfDNA from the plasma of patients with HNSCC
Using the MS-ddPCR technology, the methylation
levels of SEPT9 and SHOX2 in the plasma of 20
patients with HNSCC were assessed (Table I). The SEPT9 and SHOX2
methylation levels in the plasma of each patient before the
treatment (T0) and at 3-month intervals during follow-up
(T1=3 months and T2=6 months after treatment)
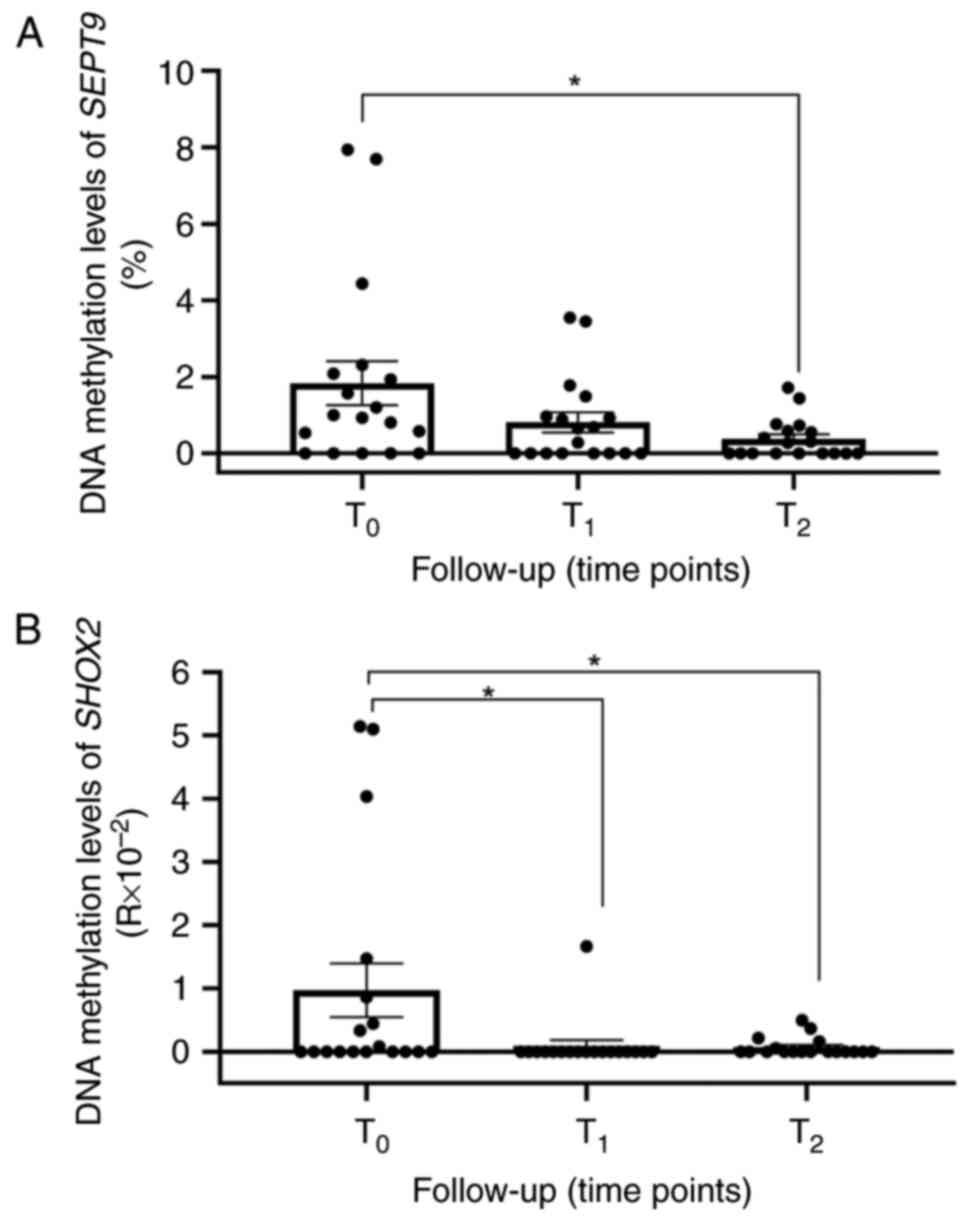
were analyzed. Considering all the patients with 2 time points of
follow-up (n=18; BS008 and BS014 developed distant metastasis or
tumor persistence and thus they were excluded from the subsequent
analysis), methylation of SEPT9 was detectable in 13 (72%)
patients at T0 (Fig.
5A). The mean methylation level of SEPT9 (mSEPT9)
decreased during follow-up, showing a reduction at T1
(mean mSEPT9 T0=1.84±2.44, mean mSEPT9
T1=0.81±1.12; fold change of 0.4) and a significant drop
at T2 (P<0.05; mean mSEPT9
T2=0.377±0.519; fold change of 0.2 vs. T0). A
total of 8 (44%) patients displayed SHOX2 methylation
(mSHOX2) in ccfDNA at T0 (mean mSHOX2
T0=0.97±1.798), and a significant decrease in the mean
methylation levels of SHOX2 at T1 and
T2 follow-up time points (Fig. 5B; P<0.05; mean mSHOX2
T1=0.093±0.39, mean mSHOX2
T2=0.072±0.146; fold change of 0.09 and 0.07,
respectively) was obtained. Of these 8 patients, 5 exhibited a
concomitant SEPT9 methylation in ccfDNA at
T0.
Longitudinal variations of methylated
SEPT9 and SHOX2 in ccfDNA from the plasma of patients with
HNSCC
Among the 18 patients who were followed up
longitudinally, 10 reached the time point of 12 months after the
treatment (T3) at the time of the writing of the present
study. For the SEPT9 analysis, 2 out of 18 patients were not
included due to undetectable methylation levels at all time points.
By monitoring the longitudinal methylation levels of SEPT9,
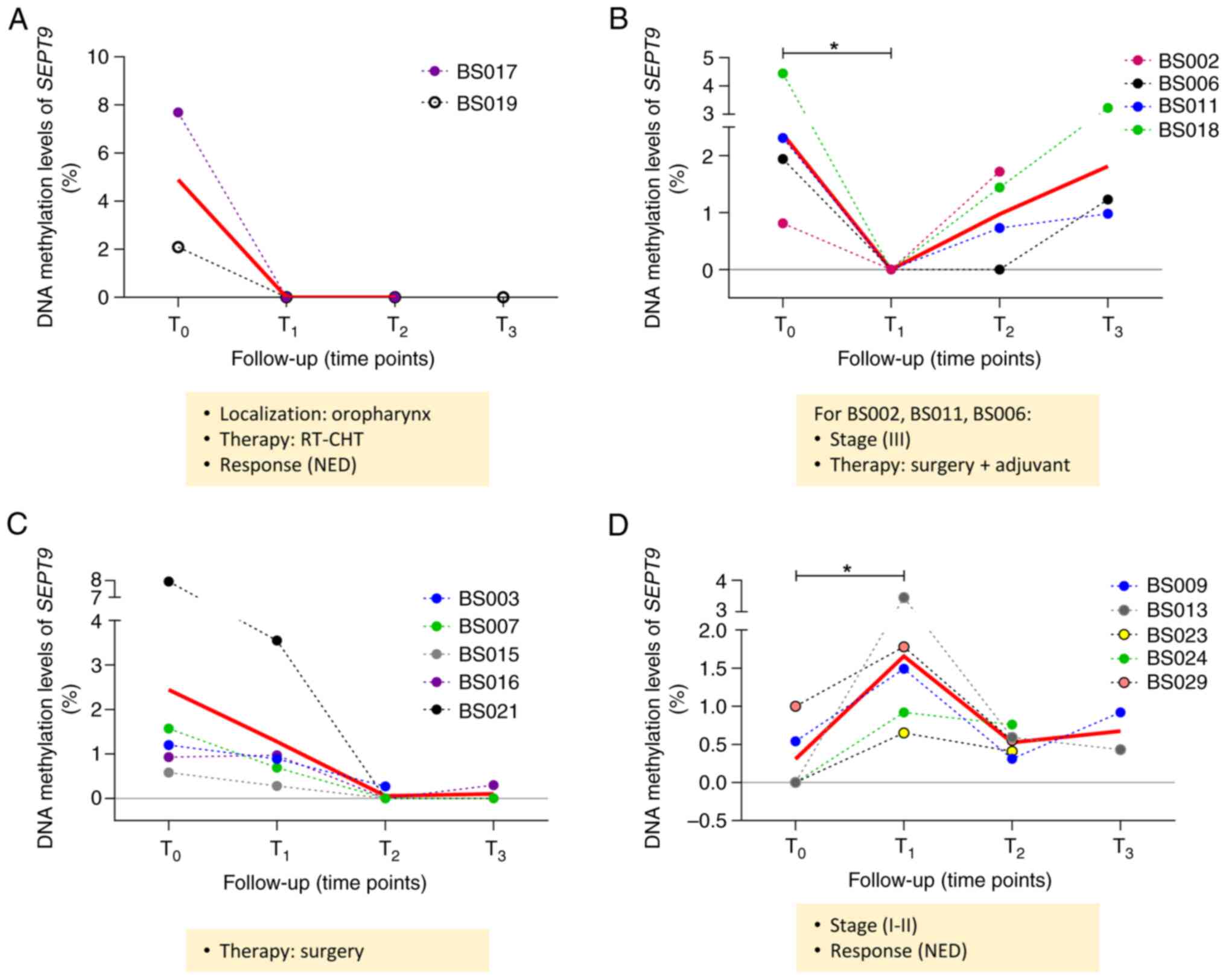
four different groups of patients were depicted according to
SEPT9 methylation levels during follow-up. As shown in
Fig. 6A, a decreasing trend was
observed for the first group of patients (BS017 and BS019), with a
mean of mSEPT9 in plasma from 4.89 at T0 to 0 at
post-treatment time points (T1 and T2). Both
of these patients presented oropharyngeal cancer, received the same
type of therapy (radiotherapy and chemotherapy), and had no
evidence of disease (NED) at T2/T3. A total
of 4 patients exhibited a decrease in methylated SEPT9 in
plasma at T1 followed by an increase in methylation
levels at T2 (BS002, BS011 and BS018) or T3
(BS006) (mean mSEPT9 T0=2.4, mean mSEPT9
T1=0, mean mSEPT9 T2=0.97, mean
mSEPT9 T3=1.81; Fig.
6B). A total of 3 patients had the same cancer stage (III) and
received the same therapy (surgery followed by adjuvant treatment).
Furthermore, 5 patients exhibited a decreasing trend of
SEPT9 methylation levels at T1 and T2
(mean mSEPT9 T0=2.44, mean mSEPT9
T1=1.28, mean mSEPT9 T2=0.05, mean
mSEPT9 T3=0.47; Fig.
6C). All these patients underwent surgery resection. The last
group of 5 patients showed a significant increase in SEPT9
methylation levels at T1, followed by a decrease at
T2 (mean mSEPT9 T0=0.31, mean
mSEPT9 T1=1.66; P<0.05; mean mSEPT9
T2=0.52, mean mSEPT9 T3=0.67; Fig. 6D). All these patients had stage I or
II HNSCC with NED at T2/T3. All the patients
were divided according to the disease status: NED (n=13) and
patients with progressive disease (PD; n=6). In the NED group, a
significant decrease in the mean mSEPT9 was found at
T2 vs. T0 (Fig.
S2A). For SHOX2 analysis, 6 patients out of 18 were
excluded because the methylation levels were undetectable at all
time points. In the remaining patients, three different
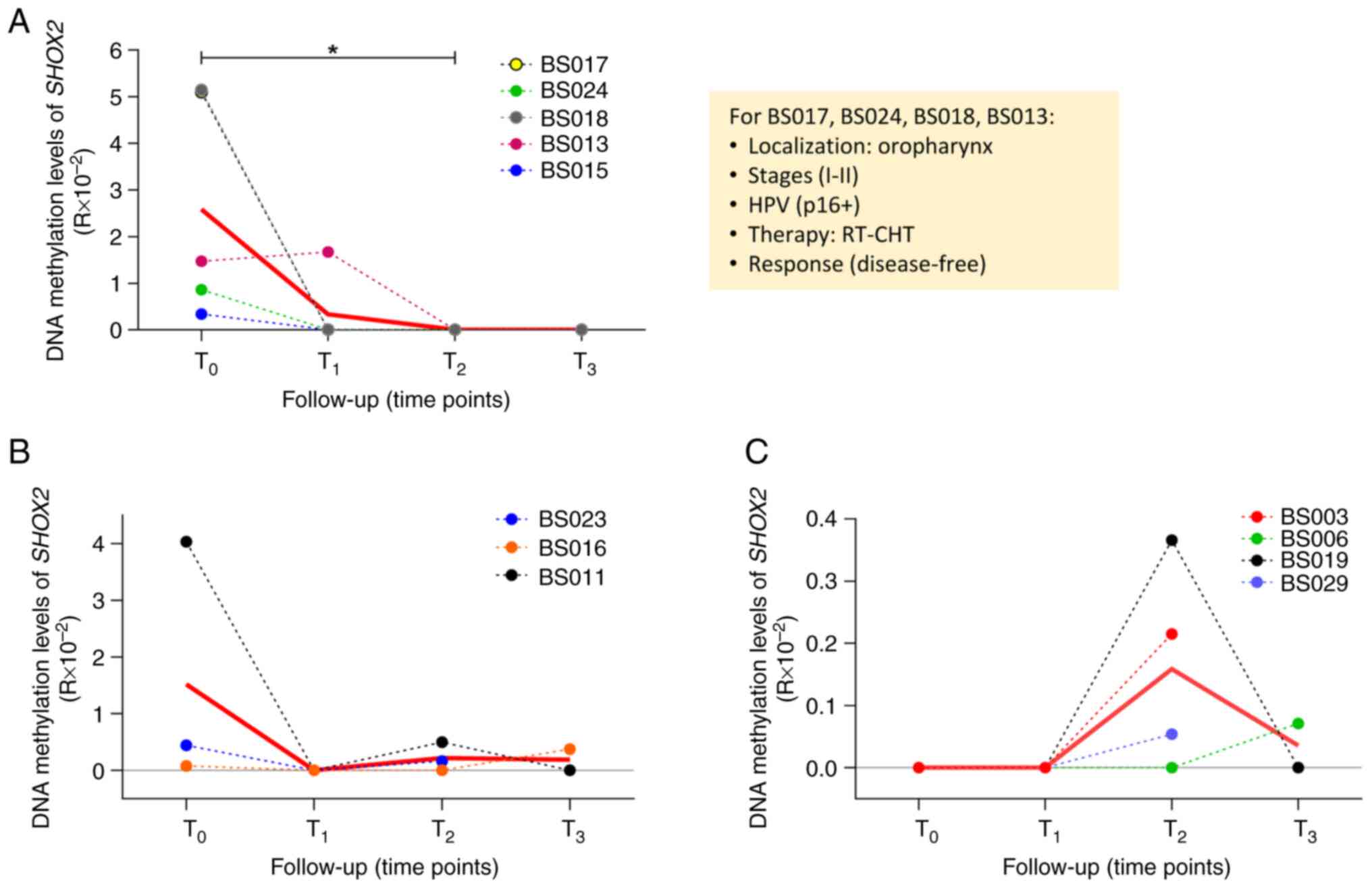
longitudinal trends were observed during follow-up (Fig. 7). In the first group, 5 patients
displayed a high methylation level of SHOX2 at T0
(mean mSHOX2 T0=2.58), followed by a decrease at
T1 (or T2 for BS013) (mean mSHOX2
T1=0.33; T0 vs. T2, P<0.05;
Fig. 7A). A total of 4 out of the 5
patients shared the following clinical characteristics: Tumor site
(oropharynx), cancer stage (I–II), HPV infection, therapy
(radiotherapy and chemotherapy), and NED. In the second group, 3
patients exhibited different methylation levels of SHOX2 at
T0 (mean mSHOX2 T0=1.52) followed by a
decrease to an undetectable level at T1 and a slight
increase at T2 (BS011 and BS023) or T3
(BS016) (mean mSHOX2 T2=0.22, mean mSHOX2
T3=0.19; Fig. 7B). In
the third group of patients, it was revealed that the methylation
levels of SHOX2 were absent at T0 and
T1 in 4 patients but they increased at T2
(BS003, BS019 and BS029) and T3 (BS006) (mean
mSHOX2 T2=0.16, mean mSHOX2
T3=0.03; Fig. 7C). The
patients in these two groups did not share any clinical
characteristics. No significant variations were found in the
methylation levels of SHOX2 among the different follow-up
time points in the NED and PD groups (Fig. S2B).
Discussion
The methylation levels of SEPT9 and
SHOX2 in ccfDNA are considered biomarkers of diagnosis,
staging, and prognosis for HNSCC and other malignancies (12,40,41).
It has been demonstrated that circulating levels of methylated
SEPT9 and SHOX2 are associated with some
clinicopathological features of patients with HNSCC, such as tumor
and nodal category, and high methylation levels were associated
with an increased risk of death (12). The concentration of ccfDNA ranges
from 1 to 15 ng/ml plasma in healthy individuals to 100 ng/ml
plasma in patients with cancer (42,43).
The total amount of ctDNA can also be <1% of total ccfDNA
(44), and these low concentrations
make detection challenging. Accurate and precise quantification of
the genomic alterations with prognostic and predictive values can
be of great importance for clinical management. Therefore, the
set-up of a ddPCR-based assay was considered useful and innovative
to improve the detection of the methylated SEPT9 and
SHOX2 circulating levels in the plasma of patients with
HNSCC. Additionally, ddPCR is a well-known end-point PCR method
that allows absolute quantification of the target template without
requiring standard curves. Several studies have previously reported
the advantages of ddPCR, including its high sensitivity and great
accuracy in assessing DNA methylation levels of low DNA input
samples (27,28,39,45).
However, for liquid biopsy, there is still limited data on the
levels of DNA methylated molecules of cancer-associated genes using
ddPCR (32,46). In the present study, the
methylation-specific assay with the ddPCR technology (MS-ddPCR or
MethyLight ddPCR) was combined to quantify the plasma amount of
methylated SEPT9 and SHOX2 in HNSCC. Specifically, to
detect the SEPT9 methylation levels in a multiplex ddPCR
reaction, two TaqMan probe-based assays labeled with FAM (SEPT9-M)
and HEX (SEPT9-U) were designed for the amplification of the
SEPT9 sequence in bisulfite-converted methylated and
unmethylated DNA, respectively. For SHOX2, an assay with a
FAM-labeled probe against the bisulfite-converted methylated
SHOX2 sequence was designed. Due to poor efficiency of
assays amplifying the unmethylated SHOX2, primers and a
HEX-labeled probe were used against a CpG-free sequence in the
ACTB gene to normalize data (38). Using a set of commercial fully
methylated and non-methylated DNA, the assays in the present study
detected up to 78 pg of methylated DNA and quantified up to 1% of
methylated DNA in a non-methylated DNA background. As
aforementioned, this amount and relative percentages may reflect
those detected in circulation. In the present study, the efficiency
of the SEPT9 and SHOX2 methylation assays were
evaluated using qPCR. The data revealed very low accuracy in
detecting small amounts of methylated DNA (as low as 625–312 pg)
and low percentages of methylated DNA (as low as 30–50%), making
ddPCR the ideal technology for quantifying very low levels of
methylated targets.
The ddPCR assay was then assessed on a discovery
cohort of 18 patients with HNSCC to determine the methylation
levels of SEPT9 and SHOX2 in plasma before the
initiation of therapies and during monitoring of treatment response
at three different follow-up time points. At the time of the
writing of the present study, plasma samples up to 1 year
(T3) after the end of treatment (surgical resection of
the tumor, chemotherapy and radiotherapy) with 3-months intervals
were collected. Most patients are still being monitored, and
methylation analysis will be performed at the available follow-up
time points.
A significant reduction of the mean methylation
plasma levels of SEPT9 and SHOX2 in patients at
T2 (SEPT9) and T1-T2
(SHOX2) monitoring times, including 3 (T1) and 6
(T2) months after the end of treatment, were found. In
this context, in a previous study, the post-therapeutic plasmatic
circulating SHOX2 and SEPT9 methylation ccfDNA levels
were decreased in patients with colorectal cancer with localized
disease, while there was no decrease in patients with distant
metastases (41). Furthermore, high
methylation levels of circulating SEPT9 and SHOX2
characterized patients with metastatic disease in prostate cancer
(47). In HNSCC, the baseline
positivity of SEPT9 and SHOX2 methylation in plasma
was identified in 15 patients (15/20, 75%), and methylation levels
decreased with systemic therapy (40). All the patients except one (BS002)
(n=17) were disease-free at the T2 monitoring time. In
the observational cohort of the present study, different trends
were observed in SEPT9 methylation levels by representing
the data according to the longitudinal quantification for each
patient during monitoring (as detailed in Results and shown in
Fig. 6). Among them, 5 patients
with I–II tumor stages treated differently (Fig. 6D and Table I) did not display tumor progression,
at least until T3 monitoring time, and exhibited a
decrease of SEPT9 methylation at T2. Furthermore,
3 patients with the same cancer stage (III) undergoing the same
treatment (surgery followed by adjuvant treatment) displayed a
significant decrease of SEPT9 methylation at T1
monitoring time followed by an increase at T2 (Fig. 6B). It appears that the changes of
the SEPT9 methylation level detectable at T2 may
be relevant, but it is necessary to expand the cohort to attribute
clinical significance to this observation. For SHOX2 (as
detailed in Results and shown in Fig.
7 and Table I) 5 patients
displayed a significant decrease in methylation levels at the
T2 monitoring time compared with T0. A total
of 4 patients out of 5 had the same clinicopathological features in
terms of tumor localization (oropharynx), tumor stage (I–II), HPV
p16 infection, type of treatment (chemotherapy and radiotherapy)
and they were all disease-free. Therefore, this could be promising
because it is known that high circulating levels of methylated
SHOX2 are correlated with a worse prognosis in patients with
HNSCC (48). The authors of the
present study are aware that the results obtained in this study are
derived from the analysis of circulating SEPT9 and
SHOX2 methylation levels in small groups of patients,
however promising longitudinal changes during the time points of
the follow-up of the patients were revealed. In an ongoing larger
study (Identify project), it will be thoroughly investigated
whether these variations are potentially associated with the
clinical features of the patients or response to treatments.
At present, at least to the best of the authors'
knowledge, three studies have evaluated the impact of circulating
SEPT9 and SHOX2 in post-therapeutic monitoring as
epigenetic biomarkers of prognosis and early diagnosis of tumor
recurrence in HNSCC. SEPT9 and SHOX2 methylation
levels determined by qPCR were revealed to be correlated with
diagnosis, prognosis, staging and monitoring of patients with HNSCC
(12). The impact of SEPT9
and SHOX2 DNA methylation in the diagnosis of HNSCC and
treatment response was evaluated by relative and quantitative
determinations using qPCR (24,40).
The main limitation of the present study is related
to the number of patients. It will be necessary to expand the
cohort to confidently define the translational implication of the
study. In addition, it is difficult at present to associate a
clinical significance to the longitudinal DNA methylation
variations detected in small groups of patients. Another limitation
may be the heterogeneity of the patients recruited thus far
according to major clinical features (such as tumor site, type of
systemic treatment, and tumor stage). There is a lack of an
association between the main risk factors for HNSCC (including
smoking, alcohol abuse, and HPV infection) and methylation levels
of circulating SEPT9 and SHOX2. Furthermore, the
methylation levels of SEPT9 and SHOX2 in solid
biopsies to compare with the corresponding circulating levels, were
not analyzed.
In conclusion, a sensitive assay based on ddPCR
technology was developed by the authors to detect the methylation
levels of circulating SEPT9 and SHOX2 DNA. At least
to the best of the authors' knowledge, the use of a performant
ddPCR assay for these two epigenetic markers has not yet been
developed. The use of ddPCR to detect small amounts of circulating
methylated SEPT9 and SHOX2 and monitor their dynamic
changes at multiple pre-established time points during clinical
monitoring represents an advancement in the HNSCC field. The
diagnostic accuracy of either methylated SEPT9 and
SHOX2 has been previously demonstrated leading to their use
as diagnostic biomarkers for lung cancer (Epi proLung) and
colorectal cancer (Epi proColon) (49). For future clinical practice, the
identification of the circulating methylation levels of
SEPT9 and SHOX2 in patients with HNSCC in the
post-treatment phase may allow for earlier diagnosis of
recurrence/second primary malignancy and the definition of a
personalized follow-up based on patient risk stratification.
Extensive validation of SEPT9 and SHOX2 as
circulating methylated biomarkers capable of stratifying groups of
patients with HNSCC based on homogenous clinicopathological
characteristics is necessary. For this purpose, the authors are
continuing with a multicenter study, to collect liquid biopsies
from eight different hospitals, to investigate the methylation
levels of SEPT9 and SHOX2 in a large cohort of
Italian patients.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
We would like to thank Dr Aashni Shah (quality
assurance manager and editorial consultant; Polistudium Srl, Milan,
Italy) and Ms Valentina Attanasio (English specialist; Polistudium
Srl, Milan, Italy) for the linguistic revision of the manuscript.
We woul also like to thank the family of Ms Claudia Massoni for the
support of the research.
Funding
The present study was supported by Fondazione Spedali Civili.
The research was also funded by CIB (Biotechnology Interuniversity
Consortium, Italy) grant no. 12/10/2020, and by the University of
Brescia (local grants; nos. 60/2021 and 60/2022). The funding
bodies played no role in the design of the study and in the
collection, analysis, and interpretation of data, or in the writing
of the manuscript.
Availability of data and materials
The data obtained and analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
PB, GDP and AS conceptualized the study. AS and IG
developed methodology. AS, IG and IAP conducted investigation. CA,
LL, DS, CG, SG, AP, DM, CP and PB acquired and interpreted data. AS
and IG wrote the original draft. AS, IG, IAP, GDP and PB wrote,
reviewed and edited the manuscript. AS and PB supervised the study.
PB, GDP and AS acquired funding. AS and IG confirm the authenticity
of all the raw data. All authors have read and approved the final
version of the manuscript. All authors have agreed to be
accountable for all aspects of the work in ensuring that questions
related to the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Spedali Civili of Brescia (Protocol Identify; Ethics
Committee approval no. NP 4551). Written informed consent was
obtained from each patient.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mody MD, Rocco JW, Yom SS, Haddad RI and
Saba NF: Head and neck cancer. Lancet. 398:2289–2299. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Barsouk A, Aluru JS, Rawla P, Saginala K
and Barsouk A: Epidemiology, risk factors, and prevention of head
and neck squamous cell carcinoma. Med Sci (Basel).
11:422023.PubMed/NCBI
|
|
4
|
Muzaffar J, Bari S, Kirtane K and Chung
CH: Recent advances and future directions in clinical management of
head and neck squamous cell carcinoma. Cancers (Basel). 13:3382021.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sánchez-Herrero E, Serna-Blasco R, Robado
de Lope L, González-Rumayor V, Romero A and Provencio M:
Circulating tumor DNA as a cancer biomarker: An overview of
biological features and factors that may impact on ctDNA analysis.
Front Oncol. 12:9432532022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Caputo V, Ciardiello F, Corte CMD, Martini
G, Troiani T and Napolitano S: Diagnostic value of liquid biopsy in
the era of precision medicine: 10 Years of clinical evidence in
cancer. Explor Target Antitumor Ther. 4:102–138. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kogo R, Manako T, Iwaya T, Nishizuka S,
Hiraki H, Sasaki Y, Idogawa M, Tokino T, Koide A, Komune N, et al:
Individualized circulating tumor DNA monitoring in head and neck
squamous cell carcinoma. Cancer Med. 11:3960–3968. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Khan KH, Cunningham D, Werner B,
Vlachogiannis G, Spiteri I, Heide T, Mateos JF, Vatsiou A, Lampis
A, Damavandi MD, et al: Longitudinal liquid biopsy and mathematical
modeling of clonal evolution forecast time to treatment failure in
the PROSPECT-C phase II colorectal cancer clinical trial. Cancer
Discov. 8:1270–1285. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Abeni E, Grossi I, Marchina E, Coniglio A,
Incardona P, Cavalli P, Zorzi F, Chiodera PL, Paties CT, Crosatti
M, et al: DNA methylation variations in familial female and male
breast cancer. Oncol Lett. 21:4682021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Abeni E, Salvi A, Marchina E, Traversa M,
Arici B and De Petro G: Sorafenib induces variations of the DNA
methylome in HA22T/VGH human hepatocellular carcinoma-derived
cells. Int J Oncol. 51:128–144. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Markou Α, Londra D, Tserpeli V, Kollias I,
Tsaroucha E, Vamvakaris I, Potaris K, Pateras I, Kotsakis Α,
Georgoulias V and Lianidou Ε: DNA methylation analysis of tumor
suppressor genes in liquid biopsy components of early stage NSCLC:
A promising tool for early detection. Clin Epigenetics. 14:612022.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schröck A, Leisse A, de Vos L, Gevensleben
H, Dröge F, Franzen A, Wachendörfer M, Schröck F, Ellinger J,
Teschke M, et al: Free-circulating methylated DNA in blood for
diagnosis, staging, prognosis, and monitoring of head and neck
squamous cell carcinoma patients: An observational prospective
cohort study. Clin Chem. 63:1288–1296. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun J, Zheng MY, Li YW and Zhang SW:
Structure and function of Septin 9 and its role in human malignant
tumors. World J Gastrointest Oncol. 12:619–631. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Semaan A, van Ellen A, Meller S, Bergheim
D, Branchi V, Lingohr P, Goltz D, Kalff JC, Kristiansen G, Matthaei
H, et al: SEPT9 and SHOX2 DNA methylation status and its utility in
the diagnosis of colonic adenomas and colorectal adenocarcinomas.
Clin Epigenetics. 8:1002016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wasserkort R, Kalmar A, Valcz G, Spisak S,
Krispin M, Toth K, Tulassay Z, Sledziewski AZ and Molnar B:
Aberrant septin 9 DNA methylation in colorectal cancer is
restricted to a single CpG island. BMC Cancer. 13:3982013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Matsui S, Kagara N, Mishima C, Naoi Y,
Shimoda M, Shimomura A, Shimazu K, Kim SJ and Noguchi S:
Methylation of the SEPT9-v2 promoter as a novel marker for the
detection of circulating tumor DNA in breast cancer patients. Oncol
Rep. 36:2225–2235. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jiang Y, Liu L, Xiang Q, He X, Wang Y,
Zhou D, Zou C, Chen Q, Peng M, He J, et al: SEPT9-v2, frequently
silenced by promoter hypermethylation, exerts anti-tumor functions
through inactivation of Wnt/β-catenin signaling pathway via
miR92b-3p/FZD10 in nasopharyngeal carcinoma cells. Clin
Epigenetics. 12:412020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Y, Song L, Gong Y and He B: Detection
of colorectal cancer by DNA methylation biomarker SEPT9: Past,
present and future. Biomark Med. 8:755–769. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Verdier-Pinard P, Salaun D, Bouguenina H,
Shimada S, Pophillat M, Audebert S, Agavnian E, Coslet S,
Charafe-Jauffret E, Tachibana T and Badache A: Septin 9_i2 is
downregulated in tumors, impairs cancer cell migration and alters
subnuclear actin filaments. Sci Rep. 7:449762017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou X, Lu X, Wu H, Liu J and Huang H:
Diagnostic performance of SHOX2 promoter methylation as biomarker
for lung cancer identification: A meta-analysis update. Thorac
Cancer. 12:3327–3332. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song L, Yu H and Li Y: Diagnosis of lung
cancer by SHOX2 gene methylation assay. Mol Diagn Ther. 19:159–167.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gao H, Yang J, He L, Wang W, Liu Y, Hu Y,
Ge M, Ding J and Ye Q: The diagnostic potential of SHOX2 and
RASSF1A DNA methylation in early lung adenocarcinoma. Front Oncol.
12:8490242022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang YA, Zhou Y, Luo X, Song K, Ma X,
Sathe A, Girard L, Xiao G and Gazdar AF: SHOX2 is a potent
independent biomarker to predict survival of WHO grade II–III
diffuse gliomas. EBioMedicine. 13:80–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
de Vos L, Gevensleben H, Schröck A,
Franzen A, Kristiansen G, Bootz F and Dietrich D: Comparison of
quantification algorithms for circulating cell-free DNA methylation
biomarkers in blood plasma from cancer patients. Clin Epigenetics.
9:1252017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hindson CM, Chevillet JR, Briggs HA,
Gallichotte EN, Ruf IK, Hindson BJ, Vessella RL and Tewari M:
Absolute quantification by droplet digital PCR versus analog
real-time PCR. Nat Methods. 10:1003–1005. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Camuzi D, Buexm LA, Lourenço SQC, Esposti
DD, Cuenin C, Lopes MSA, Manara F, Talukdar FR, Herceg Z, Ribeiro
Pinto LF and Soares-Lima SC: HPV infection leaves a DNA methylation
signature in oropharyngeal cancer affecting both coding genes and
transposable elements. Cancers (Basel). 13:36212021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wiencke JK, Bracci PM, Hsuang G, Zheng S,
Hansen H, Wrensch MR, Rice T, Eliot M and Kelsey KT: A comparison
of DNA methylation specific droplet digital PCR (ddPCR) and real
time qPCR with flow cytometry in characterizing human T cells in
peripheral blood. Epigenetics. 9:1360–1365. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Van Wesenbeeck L, Janssens L, Meeuws H,
Lagatie O and Stuyver L: Droplet digital PCR is an accurate method
to assess methylation status on FFPE samples. Epigenetics.
13:207–213. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hindson BJ, Ness KD, Masquelier DA,
Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY,
Hiddessen AL, Legler TC, et al: High-throughput droplet digital PCR
system for absolute quantitation of DNA copy number. Anal Chem.
83:8604–8610. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Manganelli M, Grossi I, Ferracin M,
Guerriero P, Negrini M, Ghidini M, Senti C, Ratti M, Pizzo C,
Passalacqua R, et al: Longitudinal circulating levels of
miR-23b-3p, miR-126-3p and lncRNA GAS5 in HCC patients treated with
sorafenib. Biomedicines. 9:8132021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
van Ginkel JH, Huibers MMH, van Es RJJ, de
Bree R and Willems SM: Droplet digital PCR for detection and
quantification of circulating tumor DNA in plasma of head and neck
cancer patients. BMC Cancer. 17:4282017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fung SYH, Chan KCA, Wong EWY, Ng CWK, Cho
R, Yeung ZWC, Lam JWK and Chan JYK: Droplet digital PCR of tumor
suppressor gene methylation in serial oral rinses of patients with
head and neck squamous cell carcinoma. Head Neck. 43:1812–1822.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huang SH and O'Sullivan B: Overview of the
8th edition TNM classification for head and neck cancer. Curr Treat
Options Oncol. 18:402017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ku JL, Jeon YK and Park JG:
Methylation-specific PCR. Methods Mol Biol. 791:23–32. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang Z, Bassil CF and Murphy SK:
Methylation-specific PCR. Methods Mol Biol. 1049:75–82. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Manganelli M, Grossi I, Corsi J,
D'Agostino VG, Jurikova K, Cusanelli E, Molfino S, Portolani N,
Salvi A and De Petro G: Expression of cellular and extracellular
TERRA, TERC and TERT in hepatocellular carcinoma. Int J Mol Sci.
23:61832022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Grossi I, Schiavone M, Cannone E, Grejdan
OA, Tobia C, Bonomini F, Rezzani R, Salvi A and De Petro G: Lasp1
expression is implicated in embryonic development of zebrafish.
Genes (Basel). 14:352022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vo TTL, Nguyen TN, Nguyen TT, Pham ATD,
Vuong DL, Ta VT and Ho VS: SHOX2 methylation in Vietnamese patients
with lung cancer. Mol Biol Rep. 49:3413–3421. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu M, Carter KT, Makar KW, Vickers K,
Ulrich CM, Schoen RE, Brenner D, Markowitz SD and Grady WM:
MethyLight droplet digital PCR for detection and absolute
quantification of infrequently methylated alleles. Epigenetics.
10:803–809. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
de Vos L, Jung M, Koerber RM, Bawden EG,
Holderried TAW, Dietrich J, Bootz F, Brossart P, Kristiansen G and
Dietrich D: Treatment response monitoring in patients with advanced
malignancies using cell-free SHOX2 and SEPT9 DNA methylation in
blood: An observational prospective study. J Mol Diagn. 22:920–933.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bergheim J, Semaan A, Gevensleben H,
Groening S, Knoblich A, Dietrich J, Weber J, Kalff JC, Bootz F,
Kristiansen G and Dietrich D: Potential of quantitative SEPT9 and
SHOX2 methylation in plasmatic circulating cell-free DNA as
auxiliary staging parameter in colorectal cancer: A prospective
observational cohort study. Br J Cancer. 118:1217–1228. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Crowley E, Di Nicolantonio F, Loupakis F
and Bardelli A: Liquid biopsy: Monitoring cancer-genetics in the
blood. Nat Rev Clin Oncol. 10:472–484. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Elazezy M and Joosse SA: Techniques of
using circulating tumor DNA as a liquid biopsy component in cancer
management. Comput Struct Biotechnol J. 16:370–378. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Stejskal P, Goodarzi H, Srovnal J, Hajdúch
M, van't Veer LJ and Magbanua MJM: Circulating tumor nucleic acids:
biology, release mechanisms, and clinical relevance. Mol Cancer.
22:152023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Postel M, Roosen A, Laurent-Puig P, Taly V
and Wang-Renault SF: Droplet-based digital PCR and next generation
sequencing for monitoring circulating tumor DNA: A cancer
diagnostic perspective. Expert Rev Mol Diagn. 18:7–17. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Luo H, Wei W, Ye Z, Zheng J and Xu RH:
Liquid biopsy of methylation biomarkers in cell-free DNA. Trends
Mol Med. 27:482–500. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Krausewitz P, Kluemper N, Richter AP,
Büttner T, Kristiansen G, Ritter M and Ellinger J: Early dynamics
of quantitative SEPT9 and SHOX2 methylation in circulating
cell-free plasma DNA during prostate biopsy for prostate cancer
diagnosis. Cancers (Basel). 14:43552022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Huang X, Duijf PHG, Sriram S, Perera G,
Vasani S, Kenny L, Leo P and Punyadeera C: Circulating tumour DNA
alterations: Emerging biomarker in head and neck squamous cell
carcinoma. J Biomed Sci. 30:652023. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Beltrán-García J, Osca-Verdegal R,
Mena-Mollá S and García-Giménez JL: Epigenetic IVD tests for
personalized precision medicine in cancer. Front Genet. 10:6212019.
View Article : Google Scholar : PubMed/NCBI
|