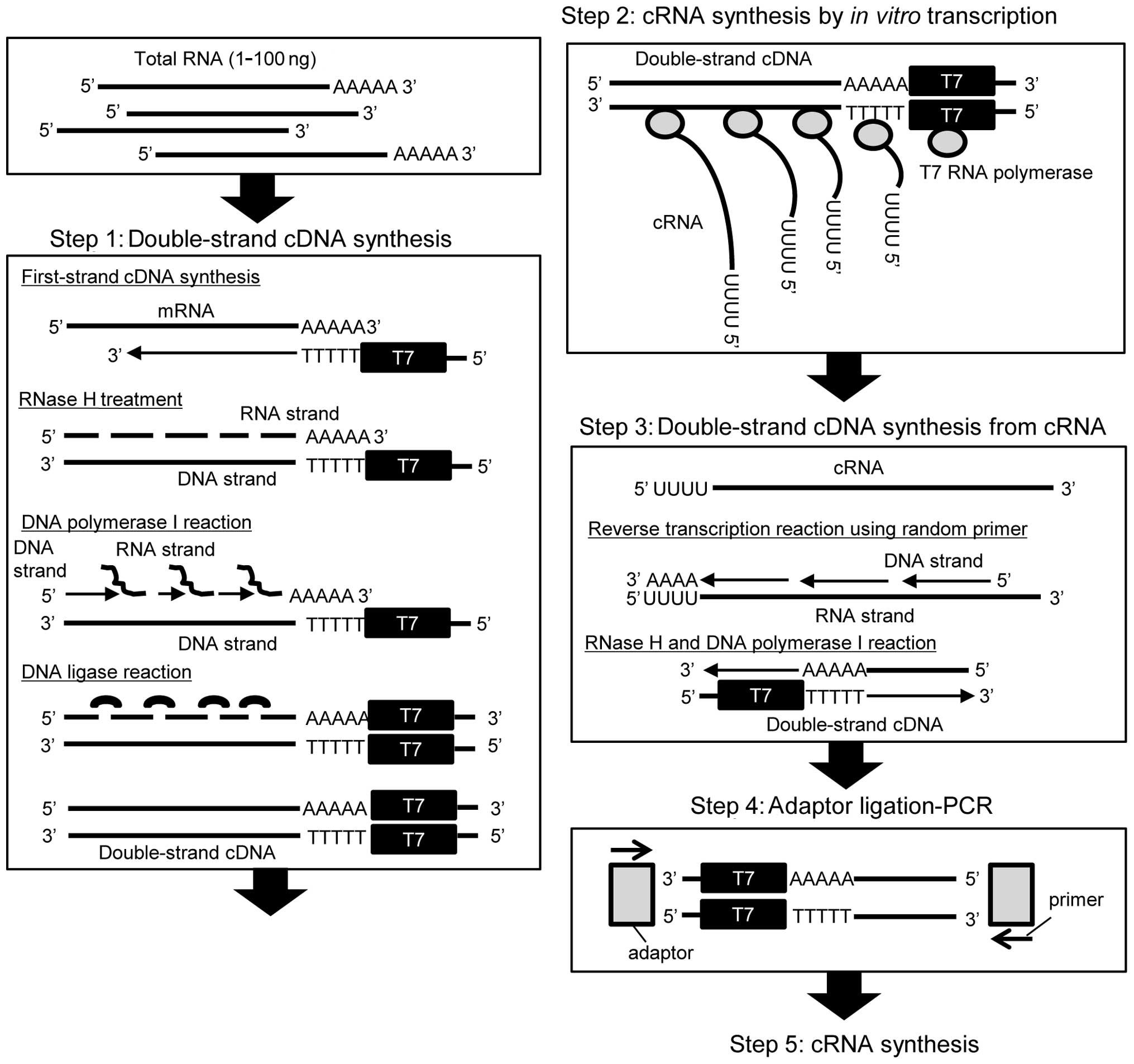
Introduction
Gene expression is a multistep process involving the
transcription, translation and turnover of messenger RNA (mRNA) and
proteins. Recently, analyses of gene expression have been used to
understand various biological phenomena. Reverse-transcription
polymerase chain reaction (RT-PCR) is a method used for analyzing
the mRNA expression of a single gene. During the 1990s, a
microarray method, with the potential to analyze the expression of
a large number of genes, was developed (1). In the 2000s, the word ‘transcriptome’
was coined, following the development of the microarray method and
sequencing of the entire human genome (2–4) as
well as the full-length cDNA (5,6). More
recently, transcriptome analyses have been accelerated by the
development of deep sequencers.
Microarray analysis is a method that employs Cyanine
3 (Cy3)- and/or Cyanine 5 (Cy5)-labeled complementary RNA (cRNA) or
cDNA, and requires a large amount of total RNA. Therefore, it is
difficult to obtain comprehensive information of gene expression
from a small amount of samples, such as clinical samples, tissue
sections and cells sorted by flow cytometry. Aoyagi et al
developed the T7 RNA polymerase-mediated transcription,
adaptor
ligation and PCR
amplification followed by the T7-transcription (TALPAT) method,
which markedly improved amplification efficiency, to resolve this
issue (7,8). The TALPAT method combines the T7 in
vitro transcription (9) with
the adaptor ligation PCR method (10,11)
and is able to amplify a small amount of mRNA to 5–10 mg cRNA
(7,8). Comprehensive analyses of gene
expression from a small amount of total RNA were enabled by the
development of the TALPAT method. However, this method is not
commonly known as the amplification method for a small amount of
total RNA.
In the present study, we investigated the
reproducibility and application of the TALPAT method using a small
amount of diluted total RNA.
Materials and methods
Cell line and cell culture
The human A549 lung cancer cell line (JCRB0076) was
purchased from the Human Science Research Resources Bank (Osaka,
Japan). The cells were cultured in Dulbecco’s minimum essential
medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100
U/ml penicillin and 100 μg/ml streptomycin. The cells were cultured
at 37°C in an atmosphere of 5% CO2. Cell counts were
performed by trypan blue staining.
Total RNA extraction
Total RNA from the A549 cells was extracted using
ISOGEN (Nippon Gene, Tokyo, Japan), according to the manufacturer’s
instructions. The concentration of total RNA extracted was examined
using the NanoDrop Spectrophotometer (ThermoFisher Scientific,
Wilmington, DE, USA), according to the manufacturer’s
instructions.
TALPAT-step 1
To synthesize first-strand cDNA, 1 μl of 100 μM
T7-oligo dT24 primer
(5′-pGGCCAGTGAATTGTAATACGACTCACTATAGGGAGGCGGTTTTTTTTTTTTTTTTTTTTTTTT-3′)
was added to 10 μl of a solution containing total RNA. The solution
was incubated at 65°C for 10 min and then chilled on ice. Four
microliters of 5X first-strand buffer (Life Technologies, Carlsbad,
CA, USA), 2 μl of 0.1 M dithiothreitol (DTT), 1 μl of 10 mM dNTP
and 1 μl of 40 U/μl RNasin Plus RNase inhibitor (Promega, Madison,
WI, USA) were added to the RNA solution. The solution was incubated
at 37°C for 2 min, and 1 μl of 200 U/μl SuperScript II RT (Life
Technologies) was then added to it. The solution was incubated at
37°C for 60 min. For the synthesis of second-strand cDNA, 91 μl of
RNase-free water, 30 μl of 5X second-strand buffer (Life
Technologies), 3 μl of 10 mM dNTPs, 4 μl of 10 U/μl E. coli
DNA polymerase I (Life Technologies), 1 μl of 10 U/μl E.
coli DNA ligase (Life Technologies) and 1 μl of 2 U/μl E.
coli RNase H (Life Technologies) were added to the first-strand
cDNA solution. The solution was incubated at 16°C for 2 h. For the
end smoothing of double-stranded cDNA, 2 μl of 5 U/μl T4 DNA
polymerase (Life Technologies) was added. The solution was
incubated at 16°C for 5 min. The synthesized double-strand cDNA was
purified using phenol/chloroform, isopropanol and ethachinmate
(Nippon Gene). The obtained pellets were washed twice with 70%
ethanol and resolved in 6.3 μl RNase-free water.
TALPAT-step 2
To synthesize cRNA by T7 in vitro
transcription, the AmpliScribe T7-Flash Transcription kit
(Epicentre Biotechnologies, Madison, WI, USA) was used. Two
microliters of 10X reaction buffer; 2.0 μl of 100 mM DTT as well as
1.8 μl of each of 100 mM dATP, dCTP, dGTP and dUTP; 0.5 μl of
RiboGuard RNase Inhibitor and 2 μl of AmpliScribe T7-Flash Enzyme
Solution were added to 6.3 μl of the double-strand cDNA solution
described above. The solution was then incubated at 37°C for 16 h.
To degrade double-strand cDNA, 1 μl of RNase-free DNase I solution
was added to the solution. The solution was incubated at 37°C for
15 min, and 400 μl of ISOGEN reagent was then added to it. The
synthesized cRNA was extracted using ISOGEN reagent, according to
the manufacturer’s instructions. The cRNA pellets were resuspended
in 10 μl of RNase-free water.
TALPAT-step 3
Random hexamer primer (1 μl) was added to the cRNA
solution obtained from step 2. Each solution was incubated at 68°C
for 10 min and then chilled on ice. To synthesize first-strand cDNA
from cRNA, 4 μl of 5X first-strand buffer (Life Technologies), 2 μl
of 0.1 M DTT, 1 μl of 10 mM dNTP and 1 μl of 40 U/μl RNasin Plus
RNase inhibitor (Promega) were added. The solution was incubated at
37°C for 2 min. Then, 1 μl of 200 U/μl SuperScript II RT (Life
Technologies) was added, and the solution was incubated at 37°C for
60 min. Following reverse transcription, 1 μl of 2 U/μl E.
coli RNase H (Life Technologies) was added to the first-strand
cDNA solution. The solution was incubated at 37°C for 20 min.
To anneal the primers in the synthesis of
second-strand cDNA, 1 μl of 100 μM T7-oligo dT24 primer
was added and incubated at 65°C for 5 min, and at 42°C for 10 min.
To synthesize second-strand cDNA, 90 μl of RNase-free water, 30 μl
of 5X second-strand buffer (Life Technologies), 3 μl of 10 mM
dNTPs, 4 μl of 10 U/μl E. coli DNA polymerase I (Life
Technologies) and 1 μl of 2 U/μl E. coli RNase H (Life
Technologies) were added to the first-strand cDNA solution. The
solution was incubated at 16°C for 2 h. For the end smoothing of
double-strand cDNA, 2 μl of 5 U/μl T4 DNA polymerase (Life
Technologies) was added, and the solution was incubated at 37°C for
5 min. The synthesized double-strand cDNA was purified using
phenol/chloroform, isopropanol and ethachinmate (Nippon Gene). The
obtained pellets were washed twice with 70% ethanol and resolved in
14 μl of RNase-free water.
TALPAT-step 4
To combine adaptor sequences of double-stranded
cDNA, 2 μl of 50 μM EcoRI-NotI-BamHI adaptor (Takara Bio, Shiga,
Japan), 2 μl of 10X T4 DNA ligase reaction buffer, 1 μl of 10 mM
ATP and 1 μl of 350 U/μl T4 DNA ligase (Takara Bio) were added to
the double-strand solution. The solution was incubated at 16°C for
16 h.
To amplify adaptor-ligated double-strand cDNA by
PCR, 63 μl of DNase-free water, 10 μl of 10X Ex Taq buffer, 12 μl
of 25 mM MgCl2, 10 μl of 2.5 mM dNTP, 3 μl of 100 μM
adaptor primer (5′-GGAATTCGGCGGCCGCGGATCC-3′), 1 μl of
adaptor-ligated double-strand cDNA, and 1 μl of 5 U/μl Ex Taq
polymerase (Takara Bio) were mixed. PCR was performed using the
Veriti 96 Well Thermal Cycler (Life Technologies) under the
following conditions: denaturation at 95°C for 5 min, 30 cycles
each of denaturation at 95°C for 1 min and annealing and extension
at 72°C for 3 min, with a final extension step at 72°C for 10 min.
PCR products were purified using phenol/chloroform, isopropanol and
ethachin-mate (Nippon Gene). The obtained pellets were washed twice
with 70% ethanol, resolved in Tris-HCl (TE) buffer (10 mM Tris-HCl
and 1 mM EDTA, pH 8.0), and produced at a concentration of 0.5
μg/μl.
TALPAT-step 5
To synthesize cRNA by T7 in vitro
transcription, the AmpliScribe T7-Flash Transcription kit
(Epicentre Biotechnologies) was used. Two microliters of 10X
reaction buffer; 2.0 μl of 100 mM DTT; 1.8 μl of each of 100 mM
dATP, dCTP, dGTP and dUTP; 0.5 μl of RiboGuard RNase Inhibitor; 2
μl of AmpliScribe T7-Flash Enzyme Solution and 1.0 μl of 0.5 μg/μl
PCR product solution from step 4 were added to 5.3 μl of RNase-free
water. The solution was then incubated at 37°C for 16 h. To degrade
double-strand cDNA, 1 μl of RNase-free DNase I solution was added
to this solution. The solution was incubated at 37°C for 15 min,
and 400 μl of ISOGEN reagent was then added to it. The synthesized
cRNA was extracted using the ISOGEN reagent, according to the
manufacturer’s instructions, and cRNA pellets were resuspended in
10 μl of RNase-free water.
Electrophoresis of cRNA synthesized by
the TALPAT method
To confirm the size of synthesized cRNA,
electrophoresis on 1% denaturing agarose gel containing
formaldehyde was performed using MOPS buffer (20 mM MOPS, 2 mM
sodium acetate and 1 mM EDTA, pH 7.0) at 100 V for 20 min.
Subsequent to electrophoresis, the gel was stained with ethidium
bromide and washed twice with RNase-free water. Bands were then
detected by UV irradiation.
Real-time PCR
To confirm reproducibility and relative expression
ratios of cRNA synthesized in TALPAT-step 2 and step 5, gene
expression of the seven housekeeping genes,
glyceraldehyde-3-phosphate dehydrogenase (GAPDH),
hydroxymethylbilane synthase (HMBS), hypoxanthine
phosphoribosyltransferase (Lesch-Nyhan syndrome) (HPRT1),
ribosomal protein L13a (RPL13A), succinate dehydrogenase
complex subunit A flavoprotein (Fp) (SDHA), TATA box binding
protein (TBP) and ubiquitin C (UBC) were examined by
real-time PCR using the primer pairs shown in Table I.
 | Table I.Primer sequences for real-time
PCR. |
Table I.
Primer sequences for real-time
PCR.
| Primer | Sequence (5′→3′) | Size (mer) | PCR products
(bp) |
|---|
| hsGAPDH F |
CCATGTAGACCCCTTGAAG | 19 | 83 |
| hsGAPDH R |
GGTTGAGCACAGGGTACTTT | 20 | |
| hsHMBS F |
GAGAAGTCCAAGCAACAGC | 19 | 61 |
| hsHMBS R |
CCTTCAGAACTGGTTTATTAGTAGG | 25 | |
| hsHPRT1 F |
GTAGTGTTTCAGTAATGTTGACTG | 24 | 73 |
| hsHPRT1 R |
AACTGCTGACAAAGATTCACTG | 22 | |
| hsRPL13A F |
GCATGAGCTTGCTGTTGTACAC | 22 | 90 |
| hsRPL13A R |
CATGGGCGATGCCTGTAAC | 19 | |
| hsSDHA F |
GAGATTGGCACCTAGTGGC | 19 | 94 |
| hsSDHA R |
CATCTCACAAGAATGAAGCAAGGG | 24 | |
| hsTBP F |
CAGTATTGCAGGACAGAATATATG | 24 | 83 |
| hsTBP R |
TTGTACAGAGTACTCTGAAGAAAG | 24 | |
| hsUBC F |
AAAGAGTCCACTCTGCAC | 18 | 101 |
| hsUBC R |
CTTTATTGAAAGGAAAGTGCAATG | 24 | |
To obtain cDNA derived from cRNA, 500 ng of cRNA was
used for the reverse transcription reaction. These reactions were
performed using the High Capacity cDNA Reverse Transcriptase kit
(Life Technologies), according to the manufacturer’s instructions.
Real-time PCR was performed using cDNA derived from cRNA, Power
SYBR-Green Master mix (Life Technologies), primer pairs shown in
Table I and the StepOne Plus
Real-Time PCR system (Life Technologies), under the following
conditions: 10 min at 95°C, followed by 40 cycles each of 95°C for
15 sec and 60°C for 60 sec. Relative expression ratios were
compared using the comparative Ct (ΔΔCt) method.
Results and Discussion
Correlation between the quantity of
starting samples and reaction time of T7 in vitro
transcription
Total RNA from the A549 cells was extracted using
the ISOGEN reagent, according to the manufacturer’s instructions.
To estimate the quality of total RNA, the measurement of absorbance
and electrophoresis of total RNA obtained were carried out. The
260/280 nm absorbance ratio of total RNA was 2.0 (data not shown).
In addition, the bands of 18S and 28S ribosomal RNA were detected
at a ratio of ∼1:2, after electrophoresis of total RNA on 1%
denaturing agarose gel (data not shown), indicating that total RNA
obtained was suitable for mRNA amplification.
To examine the correlation between the quantity of
the starting sample and the reaction time of T7 in vitro
transcription, TALPAT-step 1 was performed using 1, 10 and 100 ng
of diluted total RNA as the starting sample. In addition, the T7
in vitro transcription reaction in TALPAT-step 2 was
performed for 30 min, 1, 2, 4, 8 and 16 h. As shown in Table II, cRNA was amplified >100-fold,
when the T7 in vitro transcription reaction was performed
for 30 min in TALPAT-step 2, using double-strand cDNA derived from
1 ng of total RNA. In addition, the quantity of synthesized cRNA in
TALPAT step 2 increased depending on the quantity of the starting
sample and reaction time. This finding indicates that the most
efficient cRNA amplification is obtained when the T7 in
vitro transcription reaction in TALPAT-step 2 is performed for
30 min and, that the quantity of synthesized cRNA is dependent on
the quantity of the starting sample and reaction time.
| Table II.Quantity of cRNA synthesized in
TALPAT-step 2. |
Table II.
Quantity of cRNA synthesized in
TALPAT-step 2.
| Reaction time of T7
in vitro transcription in TALPAT-step 2
|
|---|
| Quantity of cRNA
synthesized (ng) | 30 min | 1 h | 2 h | 4 h | 8 h | 16 h |
|---|
| Quantity of starting
samples in TALPAT-step 1 (ng) | | | | | | |
| 100 | 694 | 1,382 | 2,034 | 2,399 | 4,208 | 5,748 |
| 10 | 133 | 185 | 248 | 446 | 757 | 1,720 |
| 1 | 105 | 144 | 203 | 314 | 587 | 1,161 |
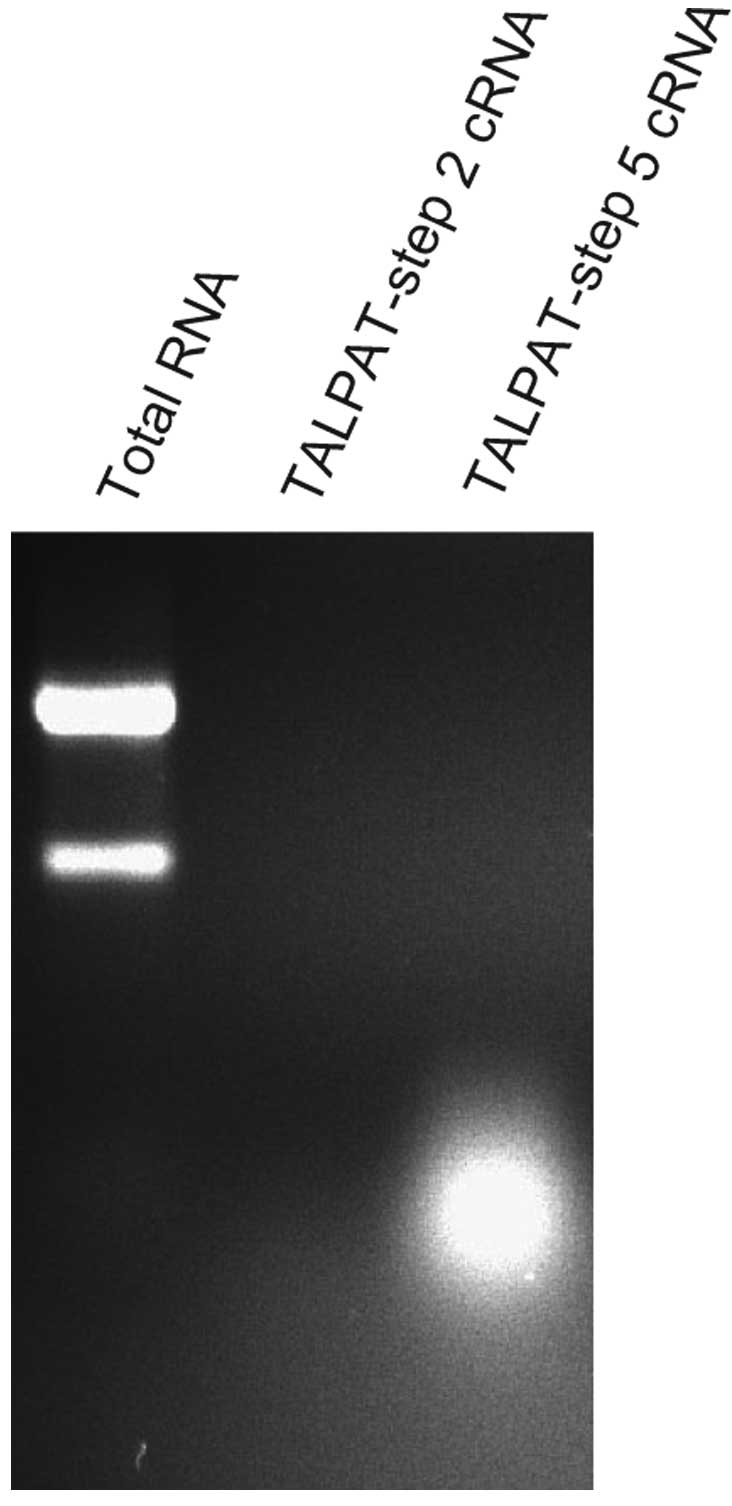
Detection of cRNA fragments synthesized
in TALPAT-step 2 and -step 5
To amplify the additional quantity of cRNA, TALPAT
step 3-step 5 were performed using cRNA fragments synthesized in
step 2. It is assumed that the mean length of the first-strand cDNA
is reduced in step 3, since random hexamer primers are used for the
synthesis of first-strand cDNA from cRNA obtained from step 2
(Fig. 1). Therefore, we examined
the length of cRNA synthesized in step 2 and step 5 by
electrophoresis on 1% denaturing agarose gel and detected the
fragments of cRNA using ethidium bromide staining. In this
experiment, 10 ng of total RNA obtained from TALPAT-step 1 was
used. T7 in vitro transcription in TALPAT-step 2 was
performed for 16 h. As shown in Fig.
2, the size of cRNA amplified in TALPAT-step 2 was ∼0.2–3.0 kb
and the size of cRNA amplified in TALPAT-step 5 was ∼0.2–0.5 kb.
This observation shows that the region up to 0.2–0.5 kb from the 3′
end of mRNA is primarily amplified in TALPAT-step 5 and, that the
size of amplified cRNA decreases depending on the processing step
of the TALPAT method. cRNA amplified by the TALPAT method may be
able to be used in microarray systems, such as Whole Human Genome
DNA microarray provided by Agilent Technologies, since the sequence
regions for mRNA detection are generically designed in the region
at the 3′ end of mRNA.
As shown in Fig. 1,
the TALPAT method is suitable for amplification of poly
(A)-positive RNA, such as mRNA, since T7-oligo dT24
primer is used for the synthesis of the first-strand cDNA.
Previously, natural antisense transcripts (NATs), which are
transcribed from the DNA strand as opposed to the sense strand,
have been identified by full-length cDNA analyses in humans and
mice (12,13). Several NATs, such as HIF-1α NATs
have poly(A)-tails at the 3′ end (14). In addition, large intervening
non-coding RNA (linc RNA), a long non-coding RNA with a
poly(A)-tail at the 3′ end, has been identified (15). The TALPAT method may be suitable for
amplifying poly (A)-positive non-coding RNA, such as several NATs
and linc RNA.
Reproducibility of the TALPAT method
To confirm the reproducibility of the TALPAT method,
real-time PCR analysis was performed using cRNA obtained from two
samples by the procedures described in Materials and methods. Ct
values of seven housekeeping genes, GAPDH, HMBS,
HPRT1, RPL13A, SDHA, TBP and
UBC, were determined by real-time PCR using cRNA obtained
from TALPAT-step 2 and -step 5. Scatter plot analysis was performed
using Ct values obtained by real-time PCR. The square of the
correlation coefficient (R2) of the seven housekeeping
genes was calculated to estimate reproducibility of the TALPAT
method. The linearity of the scatter plots was highly correlated
between the two samples in step 2 (R2=0.9885) as well as
step 5 (R2=0.9954) (Fig.
3), indicating the high reproducibility of this method.
Relative changes in gene expression by
the TALPAT method
To confirm relative changes in gene expression by
the TALPAT method, real-time PCR of the above-mentioned seven
housekeeping genes was performed using cRNA synthesized in
TALPAT-step 2 and -step 5. Analysis of gene expression from total
RNA without amplification by the TALPAT method was performed using
the oligo-dT priming method. To compare real-time PCR results,
expression values of the housekeeping genes were normalized based
on expression values of TBP in the amplified samples. As
shown in Fig. 4, few changes were
observed among the relative expression ratios of the examined
housekeeping genes. This indicates that the TALPAT method can
synthesize cRNA with a constant ratio among the seven housekeeping
genes.
Amplification from total RNA of a single
cell equivalent amount
To determine applications of the TALPAT method, we
examined whether or not amplification from total RNA of a single
cell equivalent amount is possible. Twenty picograms of total RNA
from the A549 cells were used in this experiment. Amplified cRNA
from TALPAT-step 2 and -step 5 was electrophoresed on 1% denaturing
agarose gel and detected by ethidium bromide staining. As shown in
Fig. 5, cRNA amplified in
TALPAT-step 2 is difficult to detect, due to the extremely low
quantities of the starting sample. However, cRNA of 0.2–0.5 kb was
detected in TALPAT-step 5, indicating that mRNA amplification from
a single cell may be possible using this method.
Aoyagi et al demonstrated that high-fidelity
mRNA amplification from a small amount of total RNA obtained by
laser-captured microdissection was possible using the TALPAT method
(7). Recently, a small amount of
RNA has been detected from exosomes of various body fluids, such as
serum/plasma, urine, as well as amniotic and ascites fluid
(16–20). The TALPAT method may be suitable for
mRNA and poly(A)-positive non-coding RNA amplification using a
small amount of total RNA from body fluids, and thus be useful in
the identification of biomarkers.
In conclusion, cRNA amplification by the TALPAT
method was confirmed to be highly reproducible. Relative expression
ratios among the housekeeping genes examined were constant. In
addition, cRNA amplification from 20 pg of total RNA was possible.
This method may be suitable for mRNA and poly (A)-positive
non-coding RNA amplification using a small amount of RNA from
single, laser-captured or sorted cells, as well as exosomes from
serum, urine or other body fluids.
Acknowledgements
This study was supported in part by a
grant from KAKENHI (no. 23790613), Grant-in-Aid for Young
Scientists (B). This study was also supported by the Takeda Science
Foundation and the Hirosaki University Grant for Exploratory
Research by Young Scientists.
References
|
1.
|
Schena M, Shalon D, Davis RW and Brown PO:
Quantitative monitoring of gene expression patterns with a
complementary DNA microarray. Science. 270:467–470. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Lander ES, Linton LM, Birren B, et al:
Initial sequencing and analysis of the human genome. Nature.
409:860–921. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Venter JC, Adams MD, Myers EW, et al: The
sequence of the human genome. Science. 291:1304–1351. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
International Human Genome Sequencing
Consortium: Finishing the euchromatic sequence of the human genome.
Nature. 431:931–945. 2004. View Article : Google Scholar
|
|
5.
|
Okazaki Y, Furuno M, Kasukawa T, et al:
Analysis of the mouse transcriptome based on functional annotation
of 60,770 full-length cDNAs. Nature. 420:563–573. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Carninci P, Kasukawa T, Katayama S, et al:
The transcriptional landscape of the mammalian genome. Science.
309:1559–1563. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Aoyagi K, Tatsuta T, Nishigaki M, et al: A
faithful method for PCR-mediated global mRNA amplification and its
integration into microarray analysis on laser-captured cells.
Biochem Biophys Res Commun. 300:915–920. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Isohata N, Aoyagi K, Mabuchi T, et al:
Hedgehog and epithelial-mesenchymal transition signaling in normal
and malignant epithelial cells of the esophagus. Int J Cancer.
125:1212–1221. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Wang E, Miller LD, Ohnmacht GA, Liu ET and
Marincola FM: High-fidelity mRNA amplification for gene profiling.
Nat Biotechnol. 18:457–459. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Ko MS, Ko SB, Takahashi N, Nishiguchi K
and Abe K: Unbiased amplification of a highly complex mixture of
DNA fragments by ‘lone linker’-tagged PCR. Nucleic Acids Res.
18:4293–4294. 1990.PubMed/NCBI
|
|
11.
|
Lucito R, Nakimura M, West JA, et al:
Genetic analysis using genomic representations. Proc Natl Acad Sci
USA. 95:4487–4492. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Rosok O and Sioud M: Systematic
identification of sense-antisense transcripts in mammalian cells.
Nat Biotechnol. 22:104–108. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Katayama S, Tomaru Y, Kasukawa T, et al:
Antisense transcription in the mammalian transcriptome. Science.
309:1564–1566. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Bertozzi D, Iurlaro R, Sordet O, et al:
Characterization of novel antisense HIF-1α transcripts in human
cancers. Cell Cycle. 10:3189–3197. 2011.PubMed/NCBI
|
|
15.
|
Guttman M, Amit I, Garber M, et al:
Chromatin signature reveals over a thousand highly conserved large
non-coding RNAs in mammals. Nature. 458:223–227. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Mathivanan S, Ji H and Simpson RJ:
Exosomes: extra-cellular organelles important in intercellular
communication. J Proteomics. 73:1907–1920. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Michael A, Bajracharya SD, Yuen PS, et al:
Exosomes from human saliva as a source of microRNA biomarkers. Oral
Dis. 16:34–38. 2010.PubMed/NCBI
|
|
18.
|
Moon PG, You S, Lee JE, Hwang D and Baek
MC: Urinary exosomes and proteomics. Mass Spectrom Rev.
30:1185–1202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Brase JC, Wuttig D, Kuner R and Sultmann
H: Serum microRNAs as non-invasive biomarkers for cancer. Mol
Cancer. 9:3062010. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Lasser C, Alikhani VS, Ekstrom K, et al:
Human saliva, plasma and breast milk exosomes contain RNA: uptake
by macrophages. J Transl Med. 9:92011. View Article : Google Scholar : PubMed/NCBI
|