Introduction
Receptor tyrosine kinases (RTKs) are cell surface
receptors with a high-affinity for growth hormones/factors and
which bind the majority of identified tyrosine kinases of cells.
RTKs commonly consist of a single subunit with three domains: an
extracellular N-terminal, a single hydrophobic
transmembrane-spanning and an intracellular C-terminal region. The
intracellular C-terminal region contains a tyrosine kinase domain
and a segment with multiple tyrosine sites for
auto-phosphorylation. This component of RTKs exhibits the highest
level of conservation and comprises catalytic domains responsible
for the kinase activity of these receptors, which catalyses the
tyrosine phosphorylation of RTKs as well as of their substrates.
Ligand binding to the extracellular domain of RTKs induces a series
of events including the formation of receptor dimers, activation of
the tyrosine kinase, tyrosine phosphorylation of the receptor or
the substrate of the kinase and consequent signaling (1–4).
RTKs have been shown to be key regulators of normal
cell processes and to play a crucial role in the development and
progression of various types of cancer. There are >10 classes of
RTKs, of which the most intensively studied are: i) the epidermal
growth factor receptor (EGFR) class: EGFR class has four
structurally related RTKs designated as ErbB-1 through ErbB-4.
Among these, ErbB-1 (EGFR) and ErbB-2 are found in numerous human
cancers and their excessive signaling may be critical in tumor
development and their malignant potential (5); ii) the vascular endothelial growth
factor receptor (VEGFR) class: this RTK class is one of the main
inducers of endothelial cell proliferation and permeability of
blood vessels. VEGFR-1 and VEGFR-2 are the main members of this
class (6). VEGFR-2 appears to
mediate almost all the known cell responses to VEGF and
accumulating data have shown that this kinase was closely
associated with the metastasis of tumor cells by accelerating
neovascularization (7); iii) the
platelet-derived growth factor receptor (PDGFR) class: PDGFs and
their cognate tyrosine kinase receptors are involved in multiple
tumor-associated processes, including the autocrine growth
stimulation of tumor cells, stimulation of tumor angiogenesis and
recruitment and regulation of tumor fibroblasts (8); iv) the insulin-like growth factor-1
receptor class (IGF-1R): RTKs in this class stimulate growth in
several different cell types, inhibit apoptosis, act as an
intermediate in numerous growth hormone responses and stimulate the
growth of certain types of cancer, including breast, prostate and
lung cancer (9,10). Since RTKs are closely associated
with the biological activities of tumor cells, a wide range of
inhibitory effects of RTK activation is likely to be of
significance in tumor suppression.
Type II cGMP-dependent protein kinase (PKG II) is a
serine-threonine kinase that is important in the regulation of cell
proliferation and apoptosis (11–14).
Of note, accumulating evidence has demonstrated that PKG II is a
potential tumor suppressor. Swartling et al(15) reported that PKG II inhibited the
proliferation of human neuroglioma cells and this inhibition was
associated with a decrease of the expression of transcription
factor Sox9 and the phosphorylation of Akt. Wang et
al(16) reported that PKG II
regulated cell proliferation and differentiation in the colonic
mucosa and suppressed the growth of colon cancer cells. Research
data from our laboratory demonstrated that the expression and
activity of PKG II in human gastric cancer cell lines was
significantly lower compared to that of normal cells (17) and the increase of expression and
activity of PKG II inhibited the proliferation of gastric cancer
cell lines (18). Furthermore, we
observed that PKG II inhibited EGF-induced signal transduction of
several pathways by inhibiting the activation of EGFR (19,20).
Since RTKs share a high conservation in their structure and the
kinase phosphorylating site-analyzing software predicted potential
serine/threonine phosphorylation sites on certain RTKs, we
hypothesized that PKG II may exert a similar inhibitory effect on
the activation of other members of the RTK family. This study was
designed to investigate the inhibitory effect of PKG II on the
phosphorylation/activation of the VEGFR, PDGFR and IGF-1R classes
of RTKs.
Materials and methods
Cell lines and reagents
The AGS human gastric cancer cell line was purchased
from ATCC (ATCC® no: CRL-1793TM). Adenoviral vectors
encoding β-galactosidase (Ad-LacZ) and PKG II (Ad-PKG II) were kind
gifts from Dr Gerry Boss and Dr Renate Pilz, University of
California, San Diego, USA. Dulbecco's Modified Eagle's Media
(DMEM) and fetal calf serum (FCS) were purchased from Gibco (Grand
Island, NY, USA). The antibody anti-PKG II was purchased from
Abgent Biotechnology (San Diego, CA, USA). Goat anti-β-actin and
mouse anti-p-ERK antibodies were obtained from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Rabbit anti-ERK antibody
was obtained from Cell Signaling Technology, Inc. (Danvers, MA,
USA). Rabbit anti-p-VEGFR2, rabbit anti-VEGFR2, rabbit
anti-p-PDGFRβ, rabbit anti-PDGFRβ, rabbit anti-p-IGF-1R, rabbit
anti-IGF-1R, rabbit anti-p-PI3K p85 and rabbit anti-PI3K p85
antibodies were purchased from Bioworld Technology (St. Louis Park,
MN, USA). The horseradish peroxidase-conjugated secondary
antibodies were purchased from Jackson ImmunoResearch Laboratories
(West Grove, PA, USA). The cellular permeable cGMP analog
8-pCPT-cGMP was obtained from Calbiochem (San Diego, CA, USA).
Recombinant human VEGF-C, PDGF-BB and IGF-1 were obtained from
Peprotech (Princeton, NJ, USA). Electrochemiluminescence (ECL)
reagents were obtained from Millipore (Billerica, MA, USA). Other
reagents used were of analytical grade.
Preparation of adenoviral vectors
293A cells were transfected with adenoviral vector
encoding cDNA of LacZ and PKG II and cultured for up to 10 days
until CPE was detected. The cells and culture medium were collected
and underwent three freezing-thawing cycles. The supernatants
containing the adenoviruses (Ad-LacZ and Ad-PKG II) were used to
infect new 293A cells to amplify adenoviruses. Amplified adenoviral
preparations were titrated and the pfu/ml was determined and
maintained at −80°C until use.
Cell culture and infection with
adenoviral vectors
AGS cells were cultured in DMEM supplemented with
10% FCS and maintained at 37°C in a humidified incubator with 95%
air and 5% CO2. The medium was changed every two days
and the cells were sub-cultured at confluence. On the day prior to
infection, cells were freshly planted at 70–80% confluence and
transinfection was performed with a multiplicity of infection (MOI)
of 100%.
Western blot analysis
Proteins extracted from whole-cell lysates were
separated by 10% SDS-PAGE and were transferred onto the PVDF
membrane. The primary antibodies were incubated overnight at 4°C
and incubated with the corresponding secondary antibodies for 1 h
at RT, with three washes following each incubation. ECL reagents
were used to demonstrate the positive bands on the membrane.
Prediction of PKG II-specific
serine/threonine phosphorylation sites on RTKs
Protein phosphorylation site program GPS2.0 was used
to indicate the potential PKG II-specific Ser/Thr phosphorylation
site on the RTKs. The results demonstrated that the RTKs
investigated in this study possessed potential serine/threonine
phosphorylation sites. This observation suggested that PKG II
affects the activation of these RTKs by phosphorylating specific
Ser/Thr site on the receptors (Table
I).
 | Table IPotential serine/threonine
phosphorylation sites of selected RTKs. |
Table I
Potential serine/threonine
phosphorylation sites of selected RTKs.
| RTK | Phosphorylation
site | Kinase | Related AA
sequence |
|---|
| PDGFRβ | | | |
| 180 S | AGC/PKG/PKG2 | YDHQRGFSGIFEDRS |
| 644 S | AGC/PKG/PKG2 | LKSTARSSEKQALMS |
| 651 S | AGC/PKG/PKG2 | SEKQALMSELKIMSH |
| 713 S | AGC/PKG/PKG2 | SDKRRPPSAELYSNA |
| 749 S | AGC/PKG/PKG2 | MDMSKDESVDYVPML |
| 856 S | AGC/PKG/PKG2 | ARDIMRDSNYISKGS |
| 863 S | AGC/PKG/PKG2 | SNYISKGSTFLPLKW |
| VEGFR2 | | | |
| 393 T | AGC/PKG/PKG2 | MEVSERDTGNYTVIL |
| 439 T | AGC/PKG/PKG2 | VDSYQYGTTQTLTCT |
| 1231 S | AGC/PKG/PKG2 | LQNSKRKSRPVSVKT |
| IGF-1R | | | |
| 5 S | AGC/PKG/PKG2 | ***MKSGSGGGSPTS |
| 293 S | AGC/PKG/PKG2 | LSAESSDSEGFVIHD |
| 316 S | AGC/PKG/PKG2 | SGFIRNGSQSMYCIP |
| 398 S | AGC/PKG/PKG2 | RHSHALVSLSFLKNL |
| 473 S | AGC/PKG/PKG2 | TGTKGRQSKGDINTR |
| 871 S | AGC/PKG/PKG2 | MYEIKYGSQVEDQRE |
Results
PKG II inhibits the activation of
VEGFR
Two membrane receptors exhibited a high affinity for
VEGF: VEGF-R1 and VEGF-R2. In response to VEGF binding, VEGF-R2
underwent auto-phosphorylation on multiple tyrosine residues
including Tyr 951, 996, 1054 and 1059. Since the phosphorylated
sites were involved in the regulation of kinase activity, the
phosphorylation of these sites represented the activation of
VEGF-R2. In this experiment, western blot analysis with
anti-phospho-VEGFR2 (Tyr 951) antibody was used to detect the
phosphorylation/activation of VEGF-R2. AGS cells were infected with
adenoviral vector encoding LacZ or PKG II overnight, treated with
8-pCPT-cGMP for 1 h and then with 100 ng/ml VEGF-C for 5 min. The
cell lysate was subjected to western blot analysis. The result
demonstrated that VEGF treatment led to a clear increase of Tyr 951
phosphorylation on VEGFR2, while PKG II effectively prevented the
phosphorylation (Fig. 1).
PKG II inhibits the activation of
PDGFR
The PDGF molecule consists of two amino acid chains
that dimerize to form PDGF-AA, PDGF-AB and PDGF-BB. These
functionally distinct isoforms may bind to the receptors PDGFRα and
PDGFRβ, leading to their phosphorylation. We performed western blot
analysis with antibody against p-PDGFRβ (Tyr 751) to detect the
activating effect of PDGF-BB on PDGFRβ and the inhibitory effect of
PKG II on this action. AGS cells were treated as described above
and western blot analysis with anti p-PDGFRβ (Tyr 751) was
performed. The results demonstrated that stimulating with PDGF-BB
(100 ng/ml) for 5 min led to a significant increase of Tyr 751
phosphorylation on PDGFRβ. Pre-infection with Ad-PKG II and
treatment with 8-pCPT-cGMP effectively inhibited the
phosphorylation/activation of PDGFRβ induced by PDGF-BB (Fig. 2).
PKG II inhibits the activation of
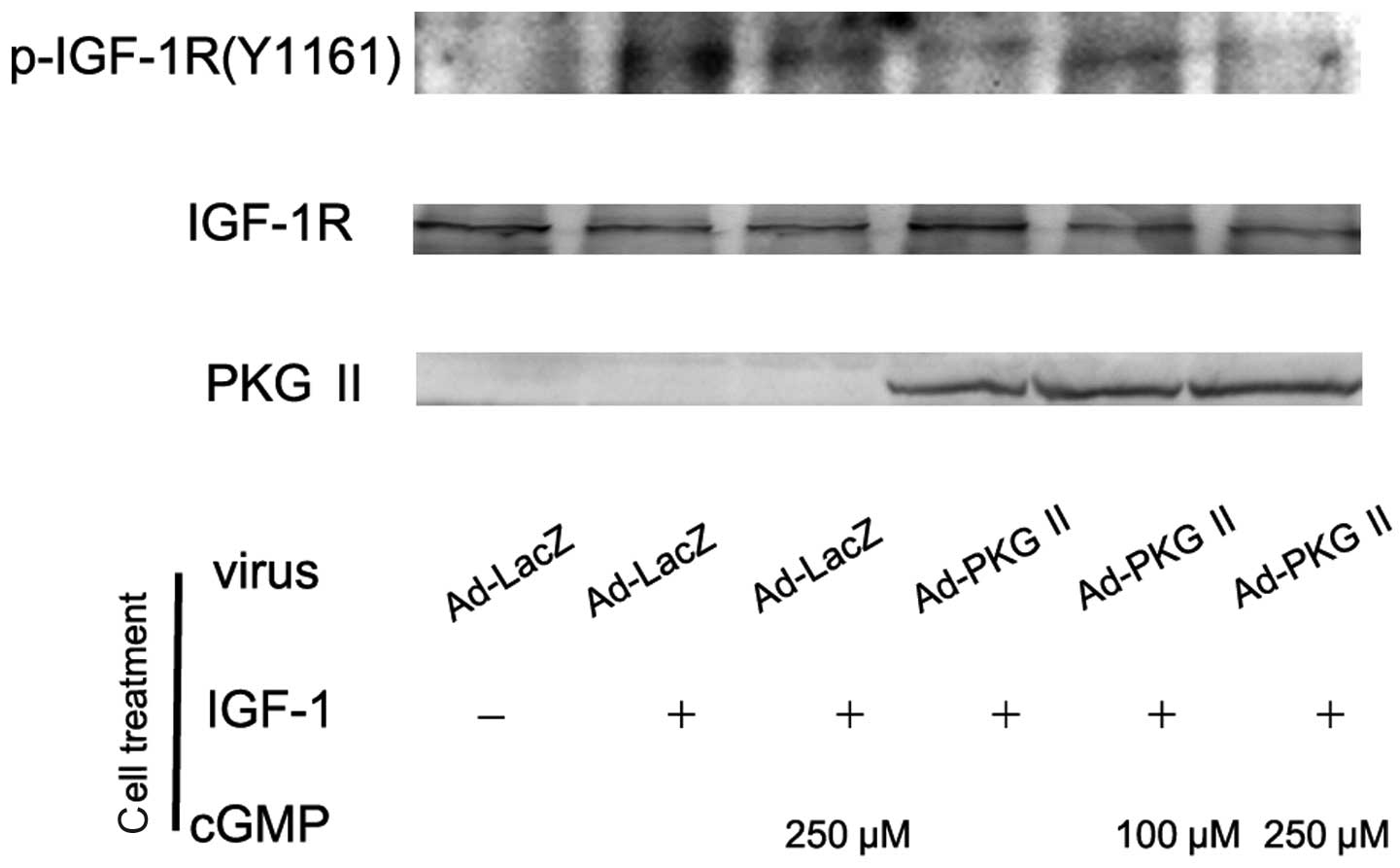
IGF-1R
There are two types of IGF receptors based on their
relative affinities, IGF-1 and IGF-2. IGF-1 receptor (IGF-1R)
consists of two α subunits and two β subunits and is therefore
preferred over IGF-2. In response to ligand binding, the α chain
induces tyrosine autophosphorylation of the β chain. This event
triggers a cascade of intracellular signaling. Thus, the tyrosine
phosphorylation of the β subunit represents the activation of
IGF-1R. In this experiment, AGS cells were infected with Ad-PKG II,
treated with 8-pCPT-cGMP and stimulated with IGF-1 (100 ng/ml)
respectively as described above. Western blot analysis with
antibody against Tyr 1161 phosphorylated IGF-1R was employed to
investigate the inhibitory effect of PKG II on the IGF-1-induced
activation of IGF-1R. The results showed that IGF-1 treatment
caused Tyr 1161 phosphorylation on IGF-1R and PKG II prevented the
process (Fig. 3).
PKG II inhibits the ERK activation
induced by VEGF, PDGF and IGF-1
Activation of RTK initiates several signal
transduction pathways. Of these the MAPK/ERK-mediated pathway is
the one closely associated with cell proliferation and
differentiation. Tyrosine phosphorylated RTKs are able to recruit
the adaptor protein to phosphorylated sites and trigger a
serine/threonine kinase cascade to phosphorylate ERK.
Phosphorylation at Thr185 and 204 is required for the activation of
ERK. Studies have shown that VEGF, PDGF and IGF are able to induce
the activation of MAPK/ERK (21,22).
In this experiment, western blot analysis with anti phospho-ERK
antibody was employed to detect the activation of this signal
component induced by VEGF, PDGF and IGF-1 and the inhibition of PKG
II on this activation. The results showed that 100 ng/ml of VEGF-C,
PDGF-BB and IGF-1 caused phosphorylation/activation of ERK and PKG
II prevented their phosphorylation/activation (Fig. 4).
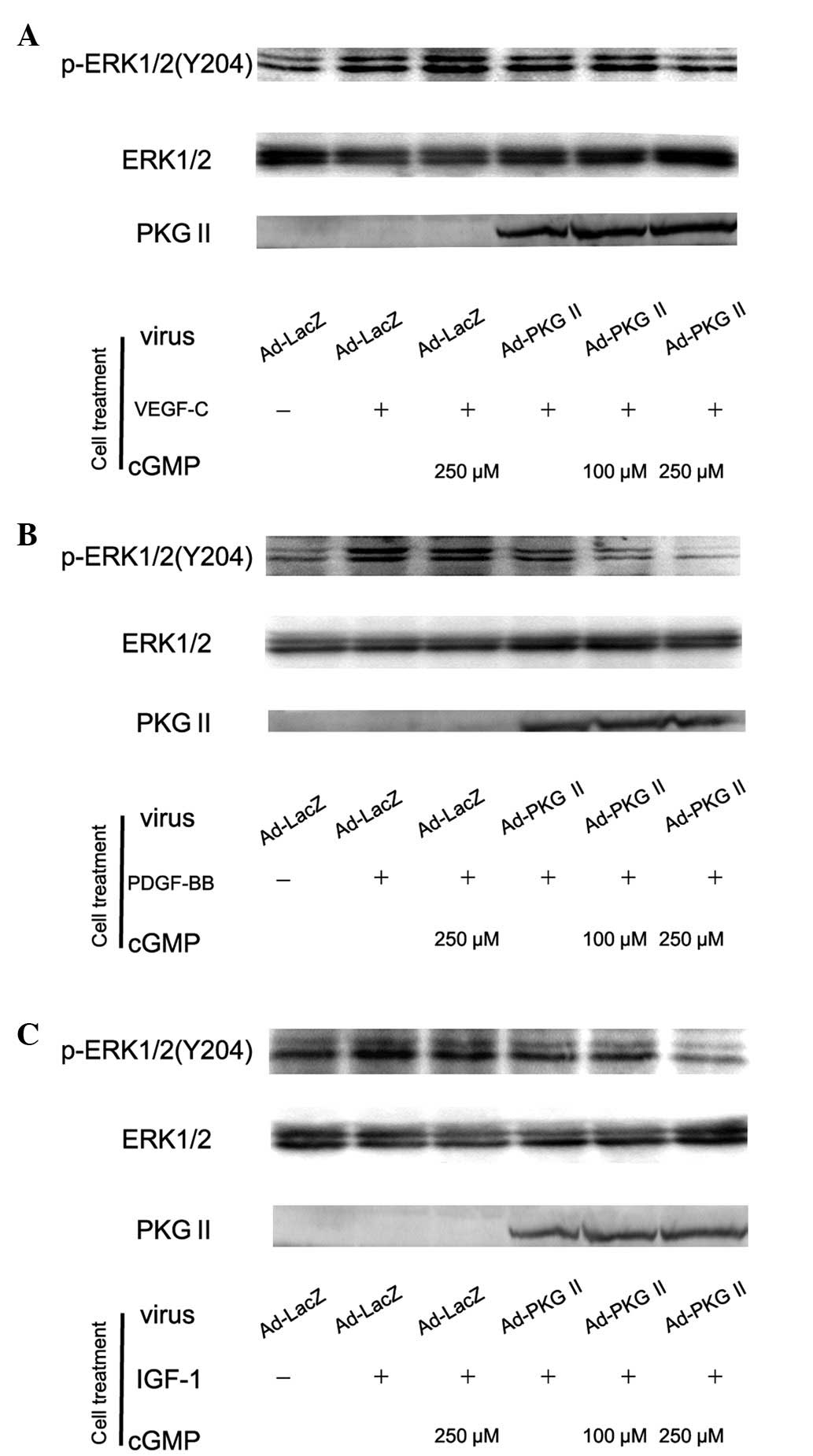 | Figure 4Type II cGMP-dependent protein kinase
(PKG II) blocks Tyr 204 phosphorylation of ERK1/2 induced by
vascular endothelial growth factor (VEGF)-C, platelet-derived
growth factor (PDGF)-BB and insulin-like growth factor (IGF)-1
respectively. AGS cells were infected with Ad-PKG II for 48 h,
serum starved overnight for 14 h, incubated with 8-pCPT-cGMP for 1
h and then stimulated with VEGF-C (100 ng/ml), PDGF-BB (100 ng/ml)
and IGF-1 (100 ng/ml) respectively for 5 min. Western blot analysis
was used to detect the phosphorylation of ERK1/2. Within 5 min
after adding the growth factors to culture medium, phosphorylation
of ERK1/2 (Tyr 204) increased significantly (panel A, Ad-LacZ +
VEGF-C group; panel B, Ad-LacZ + PDGF-BB group; panel C, Ad-LacZ +
IGF-1 group) and the phosphorylation was inhibited by pre-infecting
the cells with Ad-PKG II and stimulating the enzyme with
8-pCPT-cGMP (panel A, Ad-PKG II + cGMP + VEGF-C groups; panel B,
Ad-PKG II + cGMP + PDGF-BB groups; panel C, Ad-PKG II + cGMP +
IGF-1 groups). The results shown are representative of three
separate experiments. |
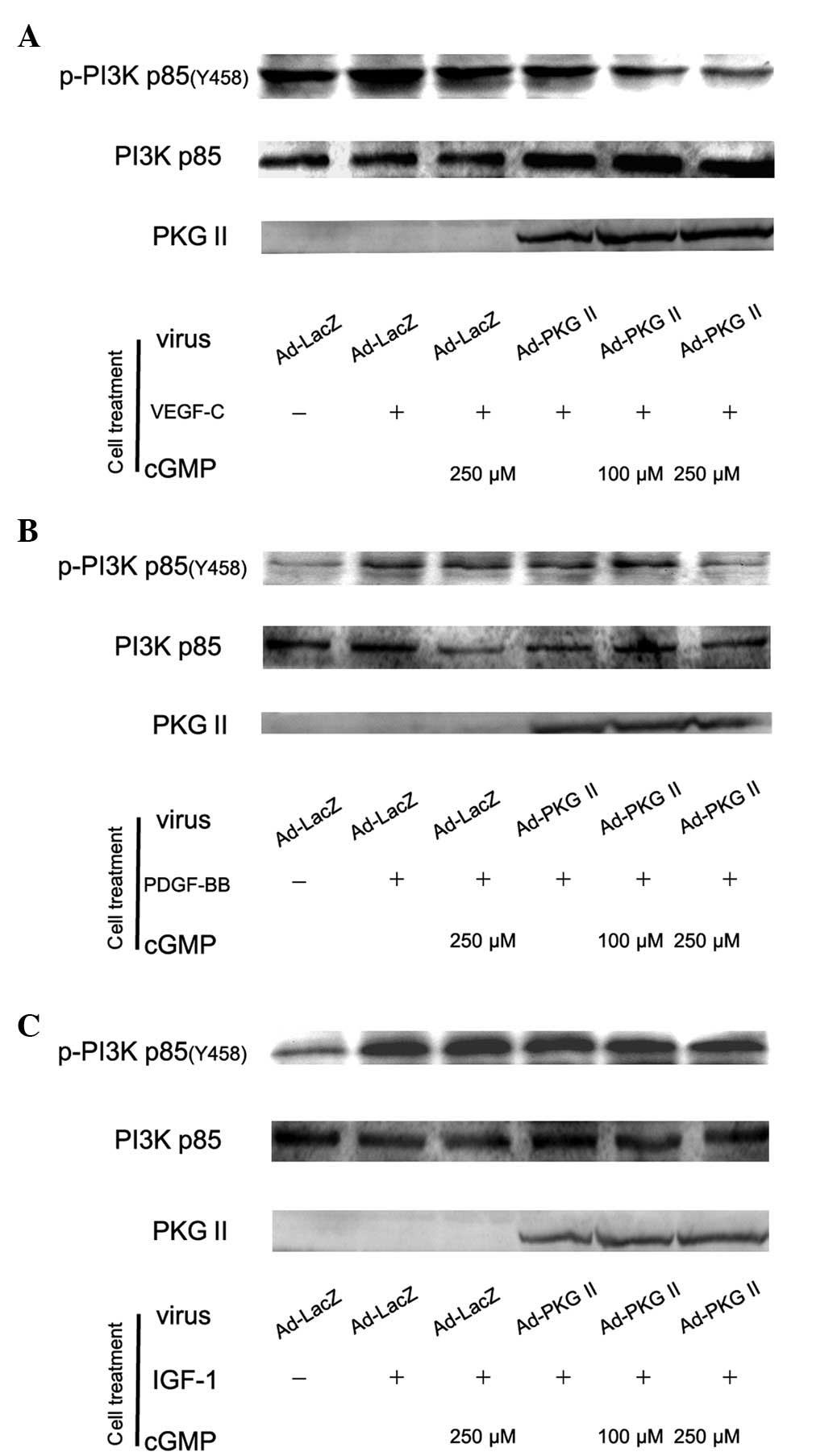
PKG II inhibits the activation of PI3K
induced by VEGF, PDGF and IGF-1
The PI3K-mediated pathway is another signal
transduction pathway that can be activated by tyrosine kinase
receptors and is responsible for the production of lipids with
biological activity. PI3Ks are composed of a catalytic (p110) and a
regulatory subunit. Various isoforms of the catalytic subunit
(p110α, p110β, p110γ and p110δ) have been isolated. p85α and p85β
are the regulatory subunits that associate with p110α, p110β and
p110δ. Upstream signaling causes the phosphorylation of p85 subunit
of PI3K. In this experiment, western blot analysis with anti p85
(Tyr 458) antibody was applied to detect the phosphorylation of p85
induced by VEGF, PDGF and IGF-1 and the inhibition of PKG II on
this process. The results showed that treatment with 100 ng/ml of
VEGF-C, PDGF-BB and IGF-1 caused an increase of Tyr 458
phosphorylation of p85 and PKG II inhibited the phosphorylation
processes (Fig. 5).
Discussion
RTKs are crucial in the regulation of critical cell
processes, including proliferation and differentiation, cell
survival and metabolism, cell migration and cell-cycle control.
Numerous diseases result from genetic changes or abnormalities that
alter the activity, abundance, cell distribution or regulation of
RTKs. Mutations in RTKs and aberrant activation of their
intracellular signaling pathways have been associated with various
diseases such as diabetes, inflammation, severe bone disorders,
arteriosclerosis and angiogenesis, particularly with regard to
cancer (3). Therefore, inhibiting
the activation/activity of these RTKs is crucial in tumor
suppression.
EGFR exists on the cell surface. Overexpression and
mutation of EGFR often occurs in most cancers (23) and patients with overexpression of
EGFR usually have a poor prognosis (24). The activating process of EGFR
includes the phosphorylation of its intracellular domain and
different phosphorylation sites are associated with different
signal pathways. For example, phosphorylation of Tyr 1068 and 1086
on EGFR is associated with the MAPK/ERK-mediated pathway (25). Previously, we showed that PKG II
inhibits the EGF-induced activation of the key signal transduction
components in the MAPK/ERK-mediated pathway and inhibits the
phosphorylation/activation of EGFR induced by EGF (19,20),
revealing that PKG II exerts an inhibitory effect on TK activity of
EGFR and the consequent inhibition of EGF/EGFR-mediated signal
transductions.
The VEGF family comprises the members VEGF-A,
VEGF-B, VEGF-C, VEGF-D, VEGF-E, PLGF-1 and PLGF-2. These members
are important signaling proteins involved in tumor growth and
angiogenesis. The main receptors, VEGFR1 and VEGFR2, are responders
of VEGFs. VEGF binds mainly with VEGFR2 and the receptor exhibits
robust protein-tyrosine kinase activity in response to its ligands
and is the predominant mediator of VEGF-stimulated endothelial cell
migration, proliferation, survival and enhanced vascular
permeability (26). A previous
study has shown that VEGF expression in gastric cancer was
increased and was associated with an increase of blood vessel
density and invasion as well as metastasis of lymph node, bone
marrow and other tissues (27).
When VEGF binds with VEGFR2, it induces the dimerization of VEGFR2
that leads to autophosphorylation and activation of the receptor.
Major phosphorylation sites on VEGFR2 include Tyr 951, 1054, 1059,
1175 and 1214 (28). Our results
showed that in AGS cells, VEGF-C induced the phosphorylation of Tyr
951 on VEGFR2 and PKG II inhibited the phosphorylation process.
This suggests that PKG II is able to inhibit the activation of
other RTKs, with the exception of EGFR.
PDGFs consist of the amino acid chains, A and B,
which dimerize to form the isoforms PDGF-AA, PDGF-AB and PDGF-BB.
Of these, PDGF-BB stimulates cells to enter the G1-S phase and
proliferate (29). The PDGFR family
consists of PDGFRα and PDGFRβ and previous studies (30–33)
have shown that PDGFR expression increased in several cancers
including liver, pancreatic, colon and gastric cancer. Similar to
the activation of EGFR and VEGFR, the activation of PDGFR by PDGFs
includes the binding of PDGFs with the extracellular domain of the
receptor, with the consequent phosphorylation of intracellular
domains of the receptor and Tyr 751 being one of the
phosphorylation sites (34). Our
results have demonstrated that in the AGS gastric cancer cell line,
PDGF-BB induced the phosphorylation of Tyr 751 on PDGFRβ and PKG II
inhibited the phosphorylation process, providing further evidence
to identify PKG II as a cancer inhibitor by suppressing the
activation of RTKs.
IGFR is a tetramer comprising α and two β chains.
The intracellular part of β chains has tyrosine kinase activity.
IGF-1 and -2 are capable of binding with α chains and cause
activation of the kinase. The phosphorylation of Tyr 1161 on the
receptor is an important event for the activation (35). We assessed anti-phospho-IGF-1R (Tyr
1161) to detect the phosphorylation/activation of IGF-1R and
observed that in AGS cells, PKG II inhibited the activating effect
of IGF-1R on its receptor. Findings of a previous study showed that
IGF-1R is widely expressed in cancer tissues, including renal,
colon and gastric cancer (11). In
cancer cells, IGF-1R is able to inhibit apoptosis, stimulate
vasculogenesis and angiogenesis, regulate migration and promote
cell adherence (36). IGF-1R has
become a crucial target gene in cancer therapy. Our results showed
that PKG II inhibited the activation of IGF-1R, confirming the
potential cancer inhibitory effect of this protein kinase by
inhibiting RTK activity as well as indicating that PKG II inhibits
the activation of RTK with structure difference (a tetramer but not
dimer).
In conclusion, our results have demonstrated that
PKG II exerts an inhibitory effect on the activation of key RTK
members. This suggests that PKG II exerts wide range inhibition on
tumor cells. However, more studies are required to confirm this
kinase as a potential tumor suppressor.
Acknowledgements
This study was supported by the
National Natural Science Foundation of China (nos. 81272755,
31040002 and 81201959); The Innovation Grant of Jiangsu
University.
References
|
1
|
Robinson DR, Wu YM and Lin SF: The protein
tyrosine kinase family of the human genome. Oncogene. 19:5548–5557.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zwick E, Bange J and Ullrich A: Receptor
tyrosine kinase signalling as a target for cancer intervention
strategies. Endocr Relat Cancer. 8:161–173. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lemmon MA and Schlessinger J: Cell
signaling by receptor-tyrosine kinases. Cell. 141:1117–1134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Choura M and Rebaï A: Receptor tyrosine
kinases: from biology to pathology. J Recept Signal Transduct Res.
31:387–394. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang H, Berezov A, Wang Q, et al: ErbB
receptors: from oncogenes to targeted cancer therapies. J Clin
Invest. 117:2051–2058. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stuttfeld E and Ballmer-Hofer K: Structure
and function of VEGF receptors. IUBMB Life. 61:915–922. 2009.
View Article : Google Scholar
|
|
7
|
Holmes K, Roberts OL, Thomas AM and Cross
MJ: Vascular endothelial growth factor receptor-2: structure,
function, intra-cellular signalling and therapeutic inhibition.
Cell Signal. 19:2003–2012. 2007. View Article : Google Scholar
|
|
8
|
Suzuki S, Dobashi Y, Hatakeyama Y, et al:
Clinicopathological significance of platelet-derived growth factor
(PDGF)-B and vascular endothelial growth factor-A expression, PDGF
receptor-β phosphorylation, and microvessel density in gastric
cancer. BMC Cancer. 30:659–668. 2010.PubMed/NCBI
|
|
9
|
Hartog H, Wesseling J, Boezen HM and van
der Graaf WT: The insulin-like growth factor 1 receptor in cancer:
old focus, new future. Eur J Cancer. 43:1895–1904. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ouban A, Muraca P, Yeatman T and Coppola
D: Expression and distribution of insulin-like growth factor-1
receptor in human carcinomas. Hum Pathol. 34:803–808. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cook AL and Haynes JM: Protein kinase G
II-mediated proliferative effects in human cultured prostatic
stromal cells. Cell Signal. 16:253–261. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cook AL and Haynes JM: Phosphorylation of
the PKG substrate, vasodilator-stimulated phosphoprotein (VASP), in
human cultured prostatic stromal cells. Nitric Oxide. 16:10–17.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chiche JD, Schlutsmeyer SM, Bloch DB, et
al: Adenovirus-mediated gene transfer of cGMP-dependent protein
kinase increases the sensitivity of cultured vascular smooth muscle
cells to the antiproliferative and pro-apoptotic effects of nitric
oxide/cGMP. J Biol Chem. 273:34263–34271. 1998. View Article : Google Scholar
|
|
14
|
Hood J and Granger HJ: Protein kinase G
mediates vascular endothelial growth factor-induced Raf-1
activation and proliferation in human endothelial cells. J Biol
Chem. 273:23504–23508. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Swartling FJ, Ferletta M, Kastemar M, et
al: Cyclic GMP-dependent protein kinase II inhibits cell
proliferation, Sox9 expression and Akt phosphorylation in human
glioma cell lines. Oncogene. 28:3121–3131. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang R, Kwon IK, Thangaraju M, et al: Type
2 cGMP-dependent protein kinase regulates proliferation and
differentiation in the colonic mucosa. Am J Physiol Gastrointest
Liver Physiol. 303:G209–G219. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang SQ, Chen YC, Wang Y, et al:
Expression of cGMP dependent protein kinase II in cancer cell lines
was obviously decreased. J Jiangsu Univ (Medicine edition). 18:1–5.
2008.
|
|
18
|
Chen YC, Ren F, Sang JR, et al: Type II
cGMP-dependent protein kinase inhibits proliferation of the gastric
cancer cell line BGC-823. Mol Med Rep. 3:361–366. 2010.PubMed/NCBI
|
|
19
|
Wu Y, Chen Y, Qu R, et al: Type II
cGMP-dependent protein kinase inhibits EGF-triggered signal
transduction of the MAPK/ERK-mediated pathway in gastric cancer
cells. Oncol Rep. 27:553–558. 2010.PubMed/NCBI
|
|
20
|
Lan T, Chen Y, Sang J, et al: Type II
cGMP-dependent protein kinase inhibits EGF-induced MAPK/JNK signal
transduction in breast cancer cells. Oncol Rep. 27:2039–2044.
2012.PubMed/NCBI
|
|
21
|
Fournier NM, Lee B, Banasr M, et al:
Vascular endothelial growth factor regulates adult hippocampal cell
proliferation through MEK/ERK- and PI3K/Akt-dependent signaling.
Neuropharmacology. 63:642–652. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pratsinis H and Kletsas D: PDGF, bFGF and
IGF-I stimulate the proliferation of intervertebral disc cells in
vitro via the activation of the ERK and Akt signaling pathways. Eur
Spine J. 16:1858–1866. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Quatrale AE, Porcelli L, Silvestris N, et
al: EGFR tyrosine kinases inhibitors in cancer treatment: in vitro
and in vivo evidence. Front Biosci. 16:1962–1972. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Normanno N, Bianco C, De Luca A, et al:
Target-based agents against ErbB receptors and their ligands: a
novel approach to cancer treatment. Endocr Relat Cancer. 10:1–21.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Oda K, Matsuoka Y, Funahashi A and Kitano
H: A comprehensive pathway map of epidermal growth factor receptor
signaling. Mol Syst Biol. 1:2005.00102005.PubMed/NCBI
|
|
26
|
Hicklin DJ and Ellis LM: Role of the
vascular endothelial growth factor pathway in tumor growth and
angiogenesis. J Clin Oncol. 23:1011–1127. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gretschel S, Astrosini C, Vieth M, et al:
Markers of tumor angiogenesis and tumor cells in bone marrow in
gastric cancer patients. Eur J Surg Oncol. 34:642–647. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Koch S and Claesson-Welsh L: Signal
transduction by vascular endothelial growth factor receptors. Cold
Spring Harb Perspect Med. 2:a0065022012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Heldin CH: Structural and functional
studies on platelet-derived growth factor. EMBO J. 11:4251–4259.
1992.PubMed/NCBI
|
|
30
|
Li ZF, Su YF and Chen XP: Expression of
platelet-derived growth factor receptor in hepatocellular carcinoma
and portal vein tumor thrombus. Acta Academiae Medicinae Qingdao
Universitatis. 44:307–311. 2008.(In Chinese).
|
|
31
|
Luo HG, Ding YW, Ma TX, et al: Study on
the correlation between flt-1, bFGFR, PDGFR and tumor biology in
pancreatic carcinoma. Zhonghua Xiao Hua Wai Ke Za Zhi. 1:253–255.
2002.(In Chinese).
|
|
32
|
Zhu HT, Han J and Ma L: Expression and
clinical significance of PDGFRalpha and PDGFRbeta in colorectal
cancer. Ai Zheng. 27:654–660. 2008.(In Chinese).
|
|
33
|
Liu DC, Li RG, Zhou JP, et al:
Relationship between the expression of PDGF and PDGFR and MVC in
the gastric carcinoma. Zhongguo Lin Chuang Yi Sheng. 5:1061–1062.
2003.(In Chinese).
|
|
34
|
Board R and Jayson GC: Platelet-derived
growth factor receptor (PDGFR): a target for anticancer
therapeutics. Drug Resist Updat. 8:75–83. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jones JI and Clemmons DR: Insulin-like
growth factors and their binding proteins: biological actions.
Endocr Rev. 16:3–34. 1995.PubMed/NCBI
|
|
36
|
Neid M, Datta K, Stephan S, et al: Role of
insulin receptor substrates and protein kinase C-zeta in vascular
permeability factor/vascular endothelial growth factor expression
in pancreatic cancer cells. J Biol Chem. 279:3941–3948. 2004.
View Article : Google Scholar
|