Introduction
Free heme is released through various pathological
processes and may further damage tissues by generating reactive
oxygen species, such as the hydroxyl radical (1). Heme oxygenase 1 (HO-1) is the
rate-limiting enzyme in heme catabolism, thus protecting against
heme toxicity. The HO-1 gene (ho-1) is induced by free heme
and heme-independent oxidative stress and is suppressed by the
transcription factor Bach1 (2,3). Under
baseline conditions, Bach1 binds to small Maf proteins to form a
heterodimer that, in turn, binds to the Maf recognition element
(MARE) in the promoter region of ho-1 to repress
transcription (2,3). During oxidative stress and in the
presence of excess free heme, Bach1-Maf is released from MARE,
allowing transcriptional activation of ho-1 by nuclear
factor (erythroid-derived 2)-like 2 (Nrf2)-Maf heterodimers
(2,3).
Carbon tetrachloride (CCl4) was shown to
cause severe hepatic injury in animals (4,5).
CCl4 is reductively metabolized by hepatic cytochrome
P450 (CYP), producing a reactive intermediate that catalyzes the
production of lipid peroxides. This early lipid peroxidation
initiates an oxidation cycle that eventually results in the
breakdown of cell membranes (4). We
previously demonstrated that the treatment of rats with
CCl4 led to a rapid increase in microsomal heme
concentration, which was likely due to the destruction of hepatic
CYP and the significant HO-1 induction in hepatocytes (5). The concurrent inhibition of HO-1
activity resulted in a sustained increase in the microsomal heme
concentration, aggravation of hepatic injury and exacerbation of
the inflammatory response, suggesting that increased free heme
plays a significant role in CCl4-induced oxidative
injury (5).
Considering that Bach1 inactivation and
CCl4-induced tissue injury are caused by heme-dependent
oxidative stress, CCl4 may also regulate Bach1
expression. To test this hypothesis, rats were treated with
CCl4 and the mRNA expression levels of Bach1, HO-1 and
δ-aminolevulinate synthase (ALAS1; a heme biosynthesis enzyme),
were measured to assess hepatic oxidative injury.
Materials and methods
Animals and treatments
A total of 64 male Sprague-Dawley rats weighing
200–260 g were purchased from Clea Japan, Inc. (Tokyo, Japan) and
housed in a temperature-controlled room (25°C) with alternating
12-h light/dark cycles and were allowed free access to water and
chow until the start of the experiments.
The rats were randomly divided into two groups, the
CCl4-treated (n=44) and vehicle-treated (control) (n=20)
groups. The rats in the CCl4 group were
intraperitoneally (i.p.) injected with CCl4
(Sigma-Aldrich Japan Co., Tokyo, Japan) at doses of 0.1, 1.0 and
2.0 ml/kg body weight, dissolved in an equal volume of corn oil.
The control rats were i.p. injected with the same volume of corn
oil. Under light ether anesthesia, the rats were sacrificed at each
predefined time point (0–24 h) by exsanguinations from the
abdominal aorta. Briefly, the abdominal cavity was opened, blood
was collected through a catheter inserted into the aorta and the
liver was excised. The livers were immediately frozen in liquid
nitrogen and stored at −80°C until RNA extraction. For
determination of hepatic malondialdehyde (MDA) and glutathione
(GSH) content, the livers were perfused in situ via the
abdominal aorta with ice-cold 0.9% NaCl solution until the venous
effluent became clear. The livers were then removed, frozen and
stored as described above.
The animal experiments were approved by the Animal
Use and Care Committee of Okayama University Medical School
(Okayama, Japan). The care and handling of the animals were in
accordance with the National Institutes of Health Guidelines for
Animal Research.
cDNA probes
The template cDNAs used to generate probes for
northern blot analysis included rat pRHO-1 (6), provided by Dr S. Shibahara, rat
pKRA2cA (7) and ALAS1, provided by
Dr M. Yamamoto, and rat Bach1 cDNA, corresponding to base pairs
785–1382, provided by Dr K. Igarashi (Tohoku University, Sendai,
Japan). The rat Bach1 cDNA was prepared from C6 glioma RNA by
reverse transcription and polymerase chain reaction and constructed
in a pCR®-Blunt II-TOPO® Vector (Invitrogen
Life Technologies, Carlsbad, CA, USA) (8). All the cDNA probes used for northern
blot analysis were labeled with [α-32P]dCTP
(PerkinElmer, Inc., Yokohama, Japan) using the Amersham Rediprime
II DNA labeling system (GE Healthcare Japan Co., Tokyo, Japan)
according to the manufacturer’s instructions.
RNA isolation and northern blot
analysis
Total RNA was isolated from the rat tissues using
TRI-Reagent® (Sigma-Aldrich Japan Co.) according to the
manufacturer’s instructions. Northern blot analysis was performed
as previously described (9).
Briefly, total RNA (20 μg) was separated by electrophoresis on 1.2%
(w/v) agarose gel containing 6.5% (v/v) formaldehyde. After
blotting on a sheet of Bio-Rad Zeta-Probe GT blotting membrane
(Bio-Rad Laboratories, Richmond, CA, USA), RNA samples were
hybridized with [α-32P]dCTP-labeled cDNA probes,
followed by washing under stringent conditions. Each blotted
membrane was exposed to a sheet of Fuji Medical X-ray film
(Fujifilm Corp., Tokyo, Japan) with an intensifying screen at
−80°C. Target bands, as well as the 18S ribosomal RNA band on
autoradiographs, were quantified using a Gel Print™ 2000i image
scanner and Basic Quantifier™ version 3.0 image analysis software
(Genomic Solutions, Inc., Ann Arbor, MI, USA). The relative amounts
of hybridized radiolabeled cDNAs were normalized to 18S ribosomal
RNA levels to correct for differences in gel loading.
Assay of serum ALT activity
Serum was separated from whole blood by
centrifugation at 1,600 × g for 10 min at room temperature and
serum ALT activity was measured using an automatic biochemical
analyzer calibrated with quality control standards (Dade
Dimension® AR® Clinical Chemistry system;
Global Medical Instrumentation, Inc., Ramsey, MN, USA).
Measurement of hepatic MDA
concentration
Hepatic tissues were homogenized in 9 volumes of 0.1
M phosphate buffer (pH 7.4) (w/v) containing 5 mM butylated
hydroxytoluene (BHT) using a Potter-Elvehjem type glass-Teflin
homogenizer (AGC Techno Glass Co., Ltd., Shizuoka, Japan). BHT was
provided as a 500-mM solution in acetonitrile. Homogenized liver
samples were filtered through mesh gauze and the MDA concentration
was measured using the Bioxytech® MDA-586™ kit (Oxis
International, Inc., Foster City, CA, USA) according to the
manufacturer’s instructions. The results are expressed as μmol
MDA/mg protein. The protein concentration in the homogenized liver
samples was measured using the DC™ protein assay (Bio-Rad
Laboratories, Hercules, CA, USA).
Measurements of hepatic GSH content
Hepatic tissues were minced in 10 volumes of
ice-cold aqueous 5% metaphosphoric acid. The homogenized samples
were centrifuged at 3,000 × g for 10 min at 4°C and the upper clear
aqueous layer was collected for analysis. The GSH assays were
performed using the Bioxytech® GSH-400™ kit (Oxis
International, Inc.) according to the manufacturer’s instructions.
The GSH content is expressed as μmol/g fresh tissue weight.
Statistical analysis
Data are presented as the means ± standard
deviation. Continuous variables were compared by Student’s t-tests
or analysis of variance followed by Tukey-Kramer’s honestly
significant difference post hoc tests, as appropriate. The
correlation between Bach1 mRNA and serum ALT levels was assessed by
Pearson’s correlation coefficient and expressed in r2
and P-values. The JMP 10™ package (SAS Institute, Inc., Cary, NC,
USA) was used for all the statistical calculations. P<0.05 was
considered to indicate a statistically significant difference.
Results and Discussion
Effects of CCl4 treatment on
serum ALT, MDA and GSH levels
Serum ALT activity was assessed 24 h after i.p.
injection of CCl4 (1 ml/kg) as a measure of hepatic
dysfunction. The serum ALT levels in the CCl4-treated
rats were significantly increased compared to those in the control
rats (Table I). In this model,
hepatic injury is considered to be caused by
CCl4-mediated free radical production and ensuing lipid
peroxidation (4); thus, we
determined hepatic MDA levels 24 h after CCl4 treatment.
Hepatic tissue samples from the CCl4-treated rats
exhibited significantly elevated MDA levels compared to the samples
from the vehicle-treated controls (Table I) (5). Consistent with CCl4-induced
oxidative stress (10), hepatic
tissue homogenates from the CCl4-treated rats exhibited
a significantly lower GSH content compared to samples from the
vehicle-treated controls, reaching a nadir of ~75% of the baseline
at 3 h after injection (Table
I).
 | Table IEffects of CCl4 treatment
on serum ALT, hepatic MDA and GSH concentrations. |
Table I
Effects of CCl4 treatment
on serum ALT, hepatic MDA and GSH concentrations.
| Experimental
groups | |
|---|
|
| |
|---|
| Measurements | Control | CCl4 | P-values |
|---|
| ALT (IU/l) (n=8) | 32.13±3.68 | 384.38±333.39 | 0.05 |
| MDA (μmol/mg protein)
(n=6) | 0.19±0.01 | 0.29±0.04 | 0.005 |
| GSH (μmol/g FW)
(n=6) | 6.43±0.27 | 4.82±0.57 | 0.0005 |
Effects of CCl4 treatment on
HO-1 and ALAS1 gene expression
It was previously demonstrated that CCl4
treatment increases the microsomal free heme concentration, which
may exert marked effects on the heme regulatory enzymes ALAS1
(biosynthesis) and HO-1 (catabolism) (11,12).
HO-1 mRNA expression was barely detectable in the vehicle-treated
control liver (Fig. 1A); however,
it started to increase 1–3 h after CCl4 injection,
reaching a maximum at 6 h prior to a rapid decrease and a gradual
return to near baseline levels by 12 h (Fig. 1A). Unlike HO-1 expression, the
levels of hepatic ALAS1 mRNA, which is the target of heme feedback
control (7), increased immediately
on CCl4 injection, but decreased below baseline by 6 h
after treatment (at the peak time of HO-1 mRNA expression). Hepatic
ALAS1 mRNA expression reached a minimum of ~10% of that in the
untreated control liver at ~9 h after treatment, followed by a
gradual increase and return to baseline by ~18 h after treatment
(Fig. 1B). These changes are
consistent with those reported by our previous study (5). HO-1 was found to be upregulated,
whereas ALAS1 was downregulated by heme (7,12).
Thus, the reciprocal responses of the HO-1 and ALAS1 genes strongly
suggest an increase in hepatic intracellular free heme (13) following CCl4 treatment.
We previously demonstrated that inhibition of HO-1 resulted in a
sustained increase in the hepatic free heme concentration, possibly
due to CCl4-mediated destruction of hepatic CYP and
exacerbation of CCl4-induced hepatic injury (5). Thus, during CCl4-induced
hepatic injury, it is reasonable to hypothesize an increase in
intracellular heme concentration, which may compound free radical
production and exacerbate cell oxidative injury.
 | Figure 1Changes in hepatic HO-1 and ALAS1 gene
expression levels following CCl4 treatment. The rats
were sacrificed at 0, 1, 3, 6, 9, 12, 18 and 24 h after injection
of CCl4 (1.0 ml/kg, intraperitoneally), their livers
were excised and total RNA (20 μg) was subjected to northern blot
analysis. Autoradiographic signals of RNA blots hybridized with (A)
[α-32P]dCTP-labeled HO-1 or (B) ALAS1 cDNA probes are
shown. Ethidium bromide staining of the same RNA is shown as the
loading control. Closed arrowhead, 28S ribosomal RNA; and open
arrowhead, 18S ribosomal RNA. Three independent experiments showed
similar results and a typical example is shown. (C) Concentrations
of HO-1 mRNA are expressed as relative values to the concentration
of an untreated control spleen, in which HO-1 is known to be
constitutively expressed; and (D) concentrations of ALAS1 mRNA are
expressed as relative values to the concentration of an untreated
control liver. HO-1, heme oxygenase-1; ALAS1, δ-aminolevulinate
synthase; CCl4, carbon tetrachloride. |
Effects of CCl4 treatment on
Bach1 gene expression
The Bach1 transcription factor acts as a repressor
of ho-1 activation (2,3). Under
physiological conditions, Bach1 forms heterodimers with the basic
leucine zipper subfamily of small Maf proteins that bind to the
MARE in the promoter region of ho-1 and repress
transcription (2,3). During oxidative stress, Bach1 is
released from MARE, allowing transcriptional activation of
ho-1 by Nrf2-Maf heterodimers (2,3). An
increase in the intracellular heme concentration appears to release
Bach1 from MARE and promote Bach1 nuclear export by directly
binding to heme-binding motifs (Bach1 CP motifs), which in turn
allows the transcriptional activation of ho-1 (2,3). As
previously mentioned, CCl4 treatment induces hepatic
oxidative damage that is dependent, at least in part, on free heme
accumulation. Thus, CCl4 treatment may also affect
hepatic Bach1 expression. While Bach1 mRNA was not detectable in
the livers of the vehicle-treated control rats, its expression was
significantly increased in the livers of the rats injected with
≥0.5 ml/kg CCl4 and the increase was dose-dependent
(≤2.0 ml/kg) (Fig. 2A). Following
treatment with 1 ml/kg CCl4, hepatic Bach1 mRNA
expression started to increase after 1–3 h, reaching a maximum at 6
h prior to returning to near baseline levels by 12 h (Fig. 2B).
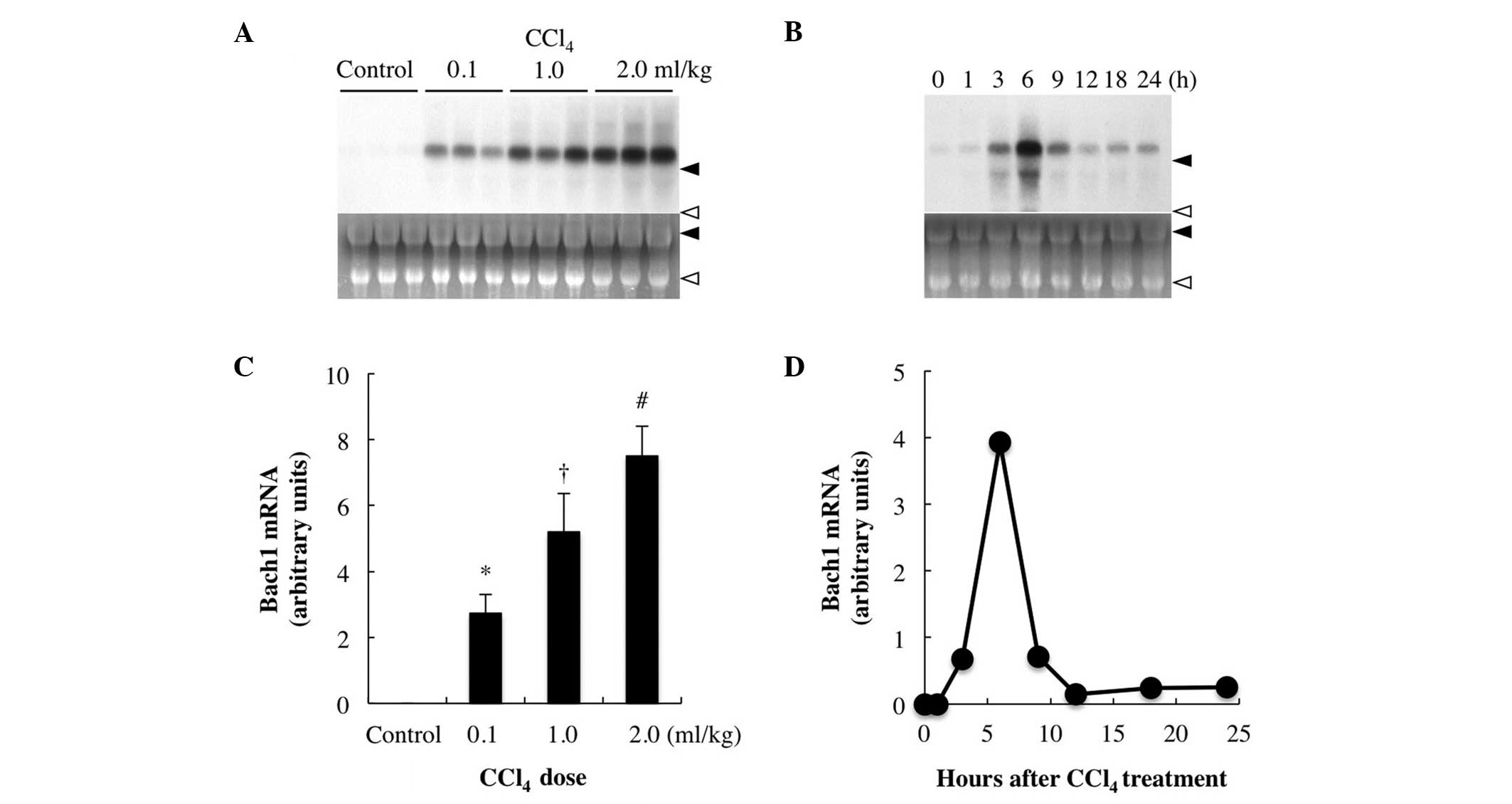 | Figure 2Effect of CCl4 treatment on
Bach1 gene expression. (A) Dose-response. The rats were injected
with either CCl4 [0.1, 1.0 or 2.0 ml/kg body weight,
intraperitoneally (i.p.)] or 2 ml of vehicle (corn oil), were
sacrificed 6 h after injection and their livers were excised for
northern blot analysis. (B) Timecourse of hepatic Bach1 gene
expression after CCl4 treatment. The rats were
sacrificed at 0, 1, 3, 6, 9, 12, 18 and 24 h after CCl4
injection (1.0 ml/kg, i.p.) and their livers were excised. Total
RNA (20 μg) was subjected to northern blot analysis.
Autoradiographic signals of RNA blots hybridized with a
[α-32P]dCTP-labeled Bach1 cDNA probe are shown. Ethidium
bromide staining of the same RNA is shown as the loading control.
Closed arrowhead, 28S ribosomal RNA; and open arrowhead, 18S
ribosomal RNA; control, vehicle (corn oil)-treated rats. Three
independent experiments yielded similar results and a typical
example is shown. (C and D) Bach1 gene expression levels expressed
as densitometric arbitrary units. Data are expressed as the means ±
standard deviation and were statistically evaluated using analysis
of variance followed by Tukey-Kramer’s honestly significant
difference test. *P<0.05 vs. control group;
†P<0.05 vs. 0.1 ml/kg CCl4; and
#P<0.05 vs. 1.0 ml/kg CCl4.
CCl4, carbon tetrachloride. |
To the best of our knowledge, this is the first
study to demonstrate the induction of Bach1 mRNA in vivo.
Consistent with our findings, a recent cell culture study reported
that Bach1 mRNA was upregulated by oxidative stress evoked by
ultraviolet A irradiation, a treatment that releases free heme from
microsomal heme-containing proteins (14,15).
Hypoxia, desferrioxamine and interferon-γ are among the other
treatments known to upregulate Bach1 in cultured cells (8). However, the elevated expression of
Bach1 repressed HO-1 expression in human vascular endothelial, T98G
glioblastoma and A549 lung cancer cells (8). Furthermore, interleukin-γ decreased
HO-1 expression through Bach1 induction in human retinal pigment
epithelial cells. By contrast, hypoxia induced HO-1 and Bach1 mRNA
expression (8). Thus, the
association between Bach1 and HO-1 mRNA expression appears to
differ according to the stimulus and may also be cell-specific.
Therefore, individual hepatic cell types may respond differently to
CCl4 treatment. In this case, the observed Bach1
expression response reflects all the cell types according to
response strength and cell fraction.
Although heme proteins are necessary for cell
viability, excess free heme is deleterious, as it acts as a potent
pro-oxidant (16). In fact, failure
to control the deleterious effects of free heme contributes to the
pathogenesis of a number of conditions, such as severe sepsis,
malaria and hemolysis associated with large-volume transfusion
(16), underscoring the utility of
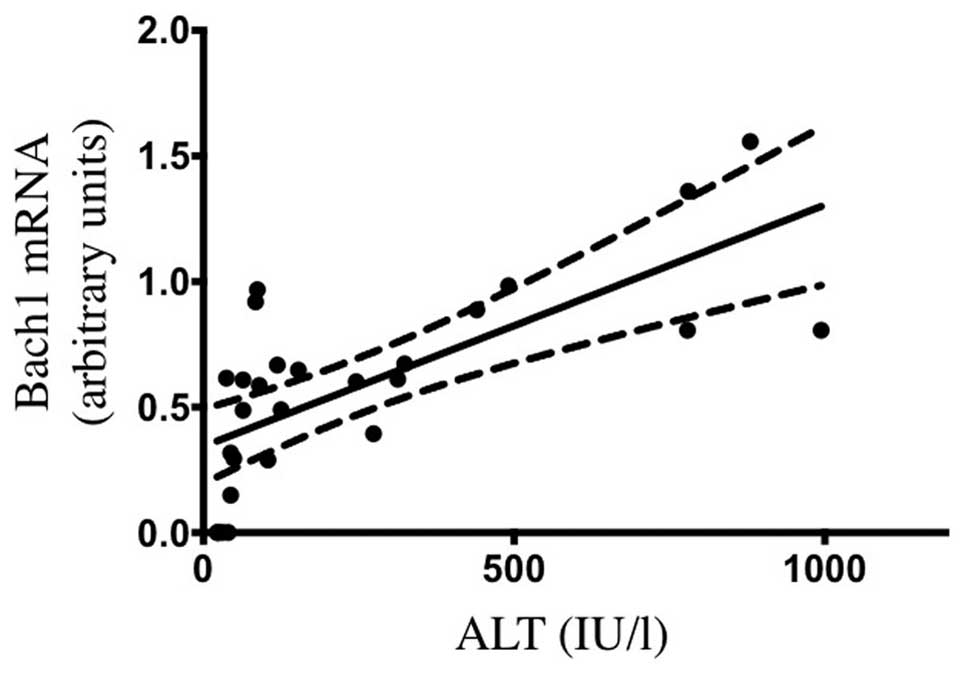
the CCl4-induced hepatic injury model (5). As illustrated in Fig. 3, the serum ALT levels at 6 h after
treatment were positively correlated with Bach1 mRNA levels
(r2=0.5033, P<0.001). Taken together, our findings
suggest that Bach1 mRNA expression may reflect the extent of
oxidative tissue injury aggravated by free heme.
The pathophysiological significance of Bach1 mRNA
induction by oxidative injury remains to be determined. The
activation of HO-1 by CCl4 is a compensatory stress
response that results in the clearance of excess heme (5). However, the overexpression of HO-1 is
likely to have deleterious consequences once this excess free heme
is catabolized, as HO-1 itself damages heme-containing proteins and
releases labile iron, which also catalyzes free radical formation
(17). Thus, the activation of
Bach1 expression by CCl4 may be a crucial compensatory
mechanism to accelerate the restoration of homeostasis under this
unique form of oxidative stress (15). Further studies measuring free heme,
Bach1 protein expression and subcellular localization and HO-1
expression in tandem are required to test this hypothesis.
In conclusion, to the best of our knowledge, this
study was the first to demonstrate that Bach1 mRNA is induced in
rat liver following i.p. administration of CCl4, a
compound that causes hepatic oxidative injury mediated partly
through increasing the concentration of free heme. A significant
positive correlation between hepatic Bach1 gene expression and
serum ALT levels following CCl4 treatment was also
demonstrated, suggesting that Bach1 mRNA expression may reflect the
extent of CCl4-induced oxidative tissue injury.
Acknowledgements
This study was supported by the Japan Society for
the Promotion of Science Grants-in-Aid for Scientific Research
(KAKENHI) (grant nos. 21592307 and 24592735). The authors would
like to thank Dr Shigeki Shibahara, Dr Masayuki Yamamoto and Dr
Kazuhiko Igarashi (Tohoku University, Sendai, Japan) for providing
cDNAs for HO-1, ALAS1 and Bach1, respectively, and Dr Reiko Akagi
(Yasuda University, Hiroshima, Japan) for her encouragement towards
this study.
References
|
1
|
Sassa S: Biological implications of heme
metabolism. J Clin Biochem Nutr. 38:138–155. 2006. View Article : Google Scholar
|
|
2
|
Igarashi K and Sun J: The heme-Bach1
pathway in the regulation of oxidative stress response and
erythroid differentiation. Antioxid Redox Signal. 8:107–118. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ozono R: New biotechnological methods to
reduce oxidative stress in the cardiovascular system: focusing on
the Bach1/heme oxygenase-1 pathway. Curr Pharm Biotechnol. 7:87–93.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
De Groot H and Sies H: Cytochrome P-450,
reductive metabolism, and cell injury. Drug Metab Rev. 20:275–284.
1989.PubMed/NCBI
|
|
5
|
Nakahira K, Takahashi T, Shimizu H, et al:
Protective role of heme oxygenase-1 induction in carbon
tetrachloride-induced hepatotoxicity. Biochem Pharmacol.
66:1091–1105. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shibahara S, Muller R, Taguchi H and
Yoshida T: Cloning and expression of cDNA for rat heme oxygenase.
Proc Natl Acad Sci USA. 82:7865–7869. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yamamoto M, Kure S, Engel JD and Hiraga K:
Structure, turnover, and heme-mediated suppression of the level of
mRNA encoding rat liver delta-aminolevulinate synthase. J Biol
Chem. 263:15973–15979. 1988.PubMed/NCBI
|
|
8
|
Kitamuro T, Takahashi K, Ogawa K, et al:
Bach1 functions as a hypoxia-inducible repressor for the heme
oxygenase-1 gene in human cells. J Biol Chem. 278:9125–9133. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maeshima K, Takahashi T, Nakahira K, et
al: A protective role of interleukin 11 on hepatic injury in acute
endotoxemia. Shock. 21:134–138. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Franco R and Cidlowski JA: Glutathione
efflux and cell death. Antioxid Redox Signal. 17:1694–1713. 2012.
View Article : Google Scholar
|
|
11
|
Granick S and Urata G: Increase in
activity of alpha-aminolevulinic acid synthetase in liver
mitochondria induced by feeding of
3,5-dicarbethoxy-1,4-dihydrocollidine. J Biol Chem. 238:821–827.
1963.PubMed/NCBI
|
|
12
|
Yoshinaga T, Sassa S and Kappas A: The
oxidative degradation of heme c by the microsomal heme oxygenase
system. J Biol Chem. 257:7803–7807. 1982.PubMed/NCBI
|
|
13
|
Jeney V, Balla J, Yachie A, Varga Z,
Vercellotti GM, Eaton JW and Balla G: Pro-oxidant and cytotoxic
effects of circulating heme. Blood. 100:879–887. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kvam E, Noel A, Basu-Modak S and Tyrrell
RM: Cyclooxygenase dependent release of heme from microsomal
hemeproteins correlates with induction of heme oxygenase 1
transcription in human fibroblasts. Free Radic Biol Med.
26:511–517. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Raval CM, Zhong JL, Mitchell SA and
Tyrrell RM: The role of Bach1 in ultraviolet A-mediated human heme
oxygenase 1 regulation in human skin fibroblasts. Free Radic Biol
Med. 52:227–236. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Larsen R, Gouveia Z, Soares MP and
Gozzelino R: Heme cytotoxicity and the pathogenesis of
immune-mediated inflammatory diseases. Front Pharmacol. 3:772012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ryter SW and Tyrrell RM: The heme
synthesis and degradation pathways: role in oxidant sensitivity.
Heme oxygenase has both pro- and antioxidant properties. Free Radic
Biol Med. 28:289–309. 2000. View Article : Google Scholar : PubMed/NCBI
|