Introduction
Rheumatoid arthritis (RA) is a common chronic
inflammatory disease that predominantly affects the small joints of
the hands and feet (1). The
propagation of inflammation is characterized by migration of
mononuclear cells into the local inflammatory sites, leading to the
hyperplasia of fibroblast-like synoviocytes (FLSs) primarily due to
resistance to apoptosis and damage to cartilage and bone (2). The chronic inflammation is attributed to
breakdown of immune homostasis, which results in activation of
immune cells and elevation of pro-inflammatory cytokines (3). The immune activation leads to deposition
of immune complexes and mononuclear cell infiltration into
susceptible organs, while the pro-inflammatory cytokines are
responsible for maintenance of the immune activation (4). The major mediators of chronic
inflammation in RA include tumor necrosis factor α (TNF-α),
interleukin (IL)-1β, IL-6, IL-8 and prostaglandin E2 (5). Induction of pro-inflammatory mediators
requires activation of the nuclear factor (NF)-κB inducing kinase-
or inhibitor of NF-κB (IκB) kinase-mediated NF-κB signal
transduction pathways (6,7). The transcription factor NF-κB has been
well recognized as a key regulator of inflammation in RA (8–10).
TNF-α-induced protein-3 (TNFAIP3) is an
important negative immunoregulatory gene. TNFAIP3 is a dual
ubiquitin-editing enzyme, whose expression is induced by a large
number of stimuli in a wide variety of cells (11). Evidence from animal models indicates
that TNFAIP3 is a plausible candidate gene in RA
susceptibility (11). In
TNFAIP3 knockout mice, its deficiency leads to death shortly
following birth by severe inflammation and tissue damage in
multiple organs (12,13). In immune cells, overexpression of
TNFAIP3 can terminate NF-κB signaling transduced from TNF
receptors, toll-like receptors, nucleotide-binding oligomerization
domain containing 2 receptors or T cell receptors (14,15). The
zinc-finger protein A20 is encoded by an immediate early response
gene and acts as a potent IκB signaling pathway (13). Of note, the expression of
TNFAIP3 itself is under the control of NF-κB, suggesting
that TNFAIP3 is involved in the negative-feedback regulation
of NF-κB activation (13).
TNFAIP3-deficient mice develop severe inflammation, which
includes inflammation of the joints (16).
Although there is an association between
single-nucleotide polymorphisms (SNPs) of the TNFAIP3 gene
and RA disease (17–19), the expression level of TNFAIP3
in immune cells from RA patients is not clear. Therefore, the
expression of TNFAIP3 mRNA was compared in peripheral blood
mononuclear cell (PBMC) between RA patients and healthy controls
and the association between TNFAIP3 expression level and
disease activity was analyzed in order to elucidate the role of
TNFAIP3 expression in the pathogenesis of RA.
Materials and methods
Human subjects
A total of 48 patients of Northern Han Chinese
descent with RA that was initially diagnosed according to the
criteria of the American College of Rheumatology (ACR) and European
League Against Rheumatism (EULAR) (2010) were enrolled. At the same
time, 41 healthy controls were recruited, who were ethnicity,
gender- and age-matched with the patients and did not have any
rheumatological conditions. RA score, including joint involvement,
serology, duration of symptoms and acute phase reactants for
diagnostic purposes, was assessed using the RA classification
criteria and scoring system revised by ACR/EULAR in 2010 (20). Peripheral bloods were sampled from all
the patients prior to the administration of any immunosuppressive
drug to exclude the influence of the drug on TNFAIP3
expression. All the blood samples from the patients and healthy
controls were used with informed consent and approval from the
Ethics Committee of Qingdao Municipal Hospital (Qingdao, Shandong,
China). The characteristics of the patients and healthy subjects
are shown in Table I.
 | Table I.Demographic characteristics, clinical
features and laboratory measurements of the studied subjects. |
Table I.
Demographic characteristics, clinical
features and laboratory measurements of the studied subjects.
|
Characteristics | RA patients
(n=48) | Healthy controls
(n=41) |
|---|
| Demographic
characteristics |
|
|
| Female,
no. (%) | 40 (83) | 33 (80) |
| Male,
no. (%) | 8 (17) | 8 (20) |
| Age,
years (range) | 35 (19–50) | 33 (20–51) |
| Clinical
features |
|
|
| RA
standard rating | 8 (6–10) | – |
| Laboratory
measurements |
|
|
| C3,
g/l | 1.1 (0.9–1.9) | – |
| C4,
g/l | 0.2 (0.1–0.5) | – |
| IgG,
g/l | 15.5
(7.0–35.2) | – |
| IgA,
g/l | 1.1 (0.7–4.0) | – |
| IgM,
g/l | 1.3 (0.4–2.3) | – |
| RF,
IU/ml | 352 (28–734) | – |
| CRP,
mg/l | 54 (5–198) | – |
| AKA,
no. (%) | 13 (72) | – |
|
Anti-CCP Ab, U/ml | 305 (58–1,892) | – |
| ESR,
mm/h | 58 (10–142) | 8 (3–20) |
Laboratory measurement
For all the RA patients, serum levels of C3, C4,
immunoglobulin G (IgG), IgA, IgM, rheumatoid factor (RF) and
C-reactive protein (CRP) were analyzed by an automatic
nephelometric immunoassay analyzer (Siemens, Munich, Germany).
Anti-keratin antibody (AKA) was analyzed by the indirect
immunofluorescent assay (Euroimmun AG, Lübeck, Germany).
Anti-cyclic citrullinated peptide (CCP) antibodies were detected
using the quantitative enzyme-linked immunoabsorbent assay
according to the instructions of the manufacturer (Euroimmun AG).
For all the subjects, including the patients and healthy controls,
erythrocyte sedimentation rates were determined by Westergren test
(Monitor-J+ analyzer; Electa Lab Srl, Forli, Italy).
Preparation of PBMCs and extraction of
RNA
Peripheral blood was sampled in sodium
citrate-containing cell preparation tubes. PBMCs were separated by
density gradient centrifugation from the peripheral blood
anticoagulated with sodium citrate. Total RNA was extracted from
PBMCs (5×105) using TRIzol (Takara, Dalian, China) and
treated with RNase-free DNase (Sangon Biotech Inc., Shanghai,
China) to remove genomic DNA contamination. RNA (1 µg) was
reversely transcribed to cDNA using a reverse transcription system
kit (Sangon Biotech Inc.) for each sample, and subsequently
quantified by photometrical measurement.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The expression of TNFAIP3 mRNA was evaluated
by qPCR in triplicate and the level of β-actin mRNA was also
detected as an internal control. qPCR was performed using the
SYBR-Green I qPCR kit in accordance with the manufacturer's
instructions (Takara) in an ABI PRISM® 7500 Sequence Detection
System (Perkin-Elmer, Norwalk, CT, USA). Amplification conditions
were as follows: 95°C for 10 sec, followed by 40 cycles of 95°C for
5 sec and 60°C for 40 sec. Primers were synthetized as described by
Li et al (3). The primers used
were as follows: TNFAIP3 forward,
5′-CGTCCAGGTTCCAGAACACCATTC-3′ and reverse,
5′-TGCGCTGGCTCGATCTCAGTTG-3′; and β-actin forward,
5′-GACTACCTCATGAAGATCCTCACC-3′ and reverse,
5′-TCTCCTTAATGTCACGCACGATT-3′. Each sample was run in triplicate.
The PCR products were run in an agarose gel and were in all cases
confined to a single band of the expected size. A melting-curve
analysis was also performed to ensure the specificity of the
products. The expression of the TNFAIP3 gene was normalized
to β-actin and the relative mRNA expression of TNFAIP3 was
determined using the comparative (2−ΔΔCt) method.
Statistical analysis
Statistical analysis was performed in the SPSS 13.0
software (SPSS, Inc., Chicago, IL, USA). Data are expressed as the
mean ± standard deviation. The difference in TNFAIP3 mRNA
level between subject groups was analyzed using the Student's
t-test independently. Correlations analysis was performed using the
Spearman's rank test. P<0.05 was considered to indicate a
statistically significant difference. Figs. 1–5 were
generated with the GraphPad Prism software, version 5.0 (San Diego,
CA, USA).
Results
Laboratory measurements of the
patients with RA
The demographic characteristics, clinical
manifestation and laboratory measurements in the RA patients are
presented in Table I. The positive
results of anti-CCP antibodies, RF and AKA in the RA patients were
found in 32, 34 and 13 patients, respectively. The mean value of
the RA standard rating score was 7.56 (range, 6–10). The mean value
of anti-CCP antibodies for the patients was 305 U/ml (range,
58–1,892 U/ml). The mean value of CRP for the patients was 54.4
mg/l (range, 5–198 mg/l).
Quantification of TNFAIP3 mRNA
expression in PBMC from RA patients and healthy controls by
RT-qPCR
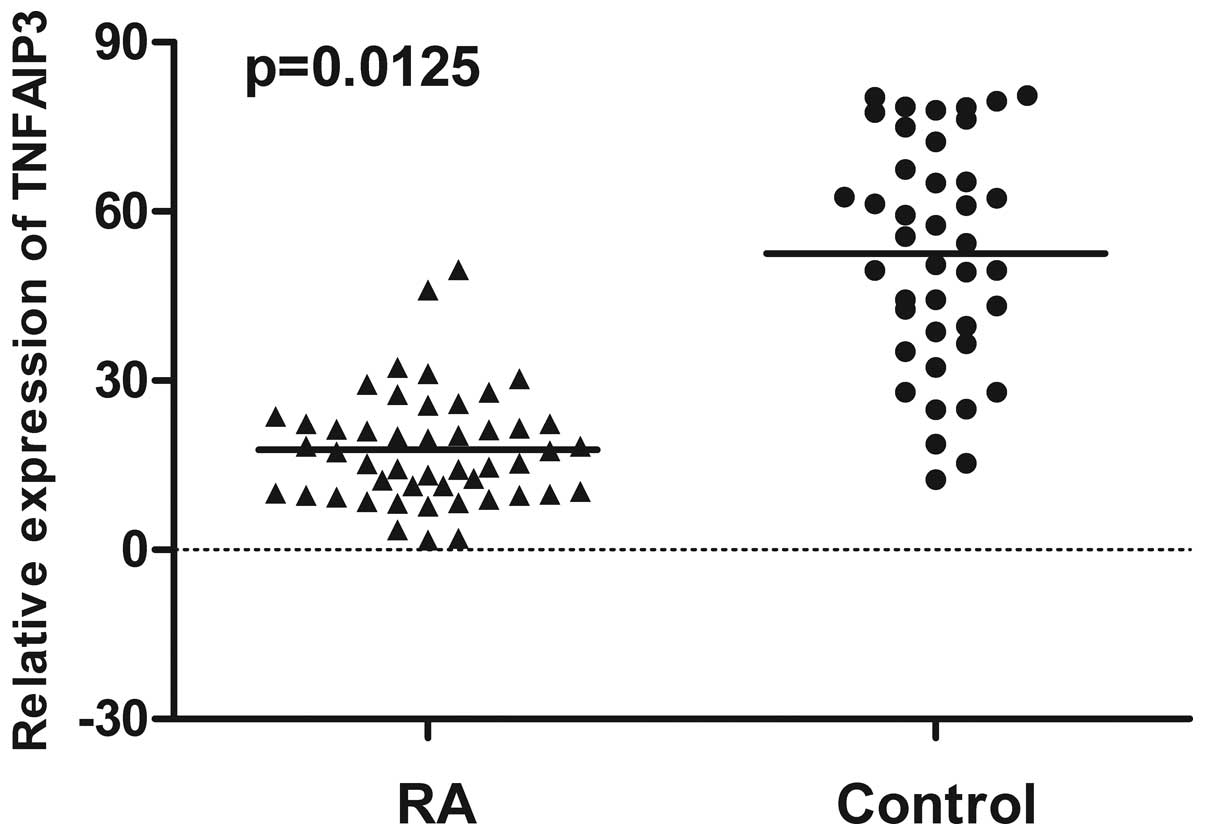
The expression of TNFAIP3 mRNA in PBMC from
48 RA patients and 41 gender- and age-matched healthy controls was
examined using RT-qPCR. The mean of TNFAIP3 mRNA expression
in PBMC from RA patients (21.32) was significantly decreased
compared to healthy controls (52.58) (P=0.0125) (Fig. 1). In addition, the expression of
TNFAIP3 mRNA in RA patients with the positive result of
anti-CCP antibodies (19.44) was significantly lower than in those
without a positive result of anti-CCP antibodies (27.67) (P=0.0134)
(Fig. 2).
Analysis of the associations between
TNFAIP3 mRNA expression and the characteristics or
laboratory parameters in the patients with RA
Association of TNFAIP3 with demographic
characteristics, clinical manifestations and laboratory parameters
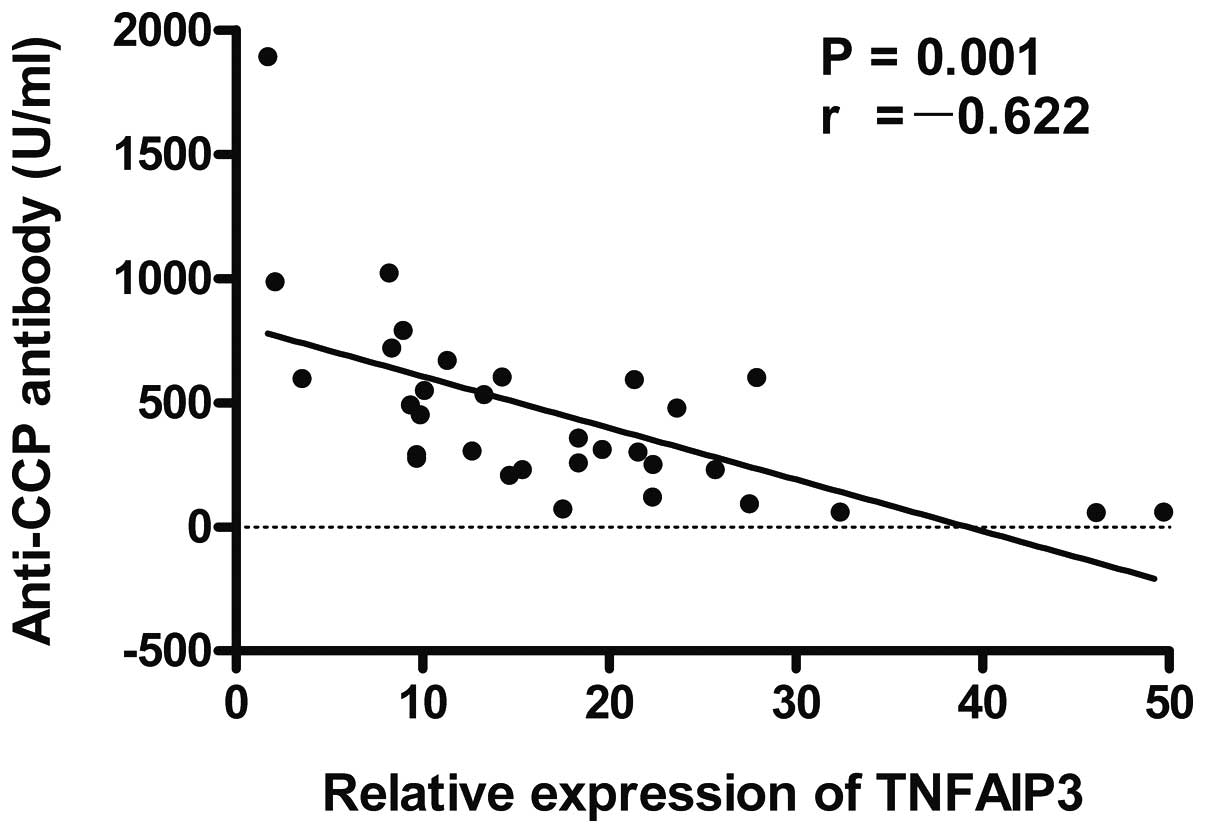
were analyzed. The results showed that the expression level of
TNFAIP3 mRNA was negatively correlated with the RA score
(r=-0.596, P=0.001; Fig. 3), anti-CCP
antibodies (r=-0.622, P=0.001; Fig. 4)
and CRP (r=-0.591, P=0.001; Fig. 5) in
RA patients. No statistically significant associations were
identified between TNFAIP3 mRNA expression levels and other
characteristics, clinical manifestations or laboratory parameters
in the patients with RA.
Discussion
RA is a chronic destructive disease of the joints
that is characterized by hyperplastic synovitis due to resistance
to apoptosis, infiltration of inflammatory cells into synovial
tissue and joint destruction (21). As
one of the NF-κB target genes, TNFAIP3 has been well
established for its negative-feedback mechanism to block NF-κB
activation through its ubiquitin-editing function in response to
various inflammatory signaling, including TNF, IL-1β and
lipopolysaccharides (12,13,22,23). Inactivation of TNFAIP3 by the
typical two mechanisms, deletions and inactivation mutations, has
been reported in marginal zone lymphomas, Hodgkin's lymphoma,
primary mediastinal B cell lymphoma and activated B cell-like
diffuse large B cell lymphoma, which may contribute to
lymphomagenesis (24,25).
The TNFAIP3 gene plays a critical role in the
negative regulation of immunity. To the best of our knowledge, this
is the first study to report the downregulation of TNFAIP3
expression in RA patients and the negative correlation between
TNFAIP3 expression and RA score, anti-CCP antibodies, CRP.
The results suggest that the decreased expression of TNFAIP3
may be involved in the diagnosis and pathogenesis of RA.
The deficiency of TNFAIP3 expression may
contribute to the pathogenesis of RA through several mechanisms.
One of the possible mechanisms is that the insufficient expression
of TNFAIP3 may cause hyperactivation of autoreactive T cell.
TNFAIP3 has ubiquitinating and deubiquitinating enzymatic
activity in the TNF receptor-signaling pathway (26). TNFAIP3 contains an N-terminal
domain that belongs to the ovarian tumor superfamily of
deubiquitinating cysteine proteases and deubiquitinates K63-linked
polyubiquitinated receptor-interacting protein, thereby blocking
TNF-α induced NF-κB signaling (15).
Since the TNFAIP3 gene has a function in limiting the
activation of T cell by deubiquitinating mucosa-associated lymphoid
tissue 1 to disrupt T cell receptor signaling to NF-κB (18), using TNFAIP3 to block the NF-κB
pathway in rheumatoid joints reduces the inflammatory response and
the tissue destruction (26).
TNFAIP3 decreased expression may induce T cell
hyperactivation and the subsequent tissue damage occurring in RA
patients. One of the major transcriptional circuits indicated in
joint inflammation is the NF-κB pathway. Within the joints,
activation of NF-κB leads to the expression of mediators of
inflammation that include cytokines, chemokines and adhesion
molecules (26). TNFAIP3
restricts B cell survival and prevents dendritic cell activation
(27,28). By contrast, TNFAIP3 deficiency
in B cells enhances B cell proliferation (29) and results in the development of the
immune complex. Therefore, the decreased expression of
TNFAIP3 may lead to hyperactivation of B cells and
accumulation of the immune complex.
In RA, NF-κB regulates production of
pro-inflammatory cytokines such as TNF-α, IL-1β and IL-6, cell
cycle progression, cell survival, adhesion and inhibition of
apoptosis of FLS (30–32). Constitutive activation of NF-κB in
certain autoimmune diseases, including RA, has been reported
(33). One possible mechanism in this
case is a defector insufficient activity of physiological IκB
pathway, such as TNFAIP3 involved in a negative-feedback
loop. As a result, the decreased expression of TNFAIP3 in RA
patients may also contribute to a high serum level of
proinflammatory cytokines, including TNF-α, IL-1β and IL-6, which
is another characteristic of RA. The major source of the
proinflammatory cytokines is monocytes and macrophages.
Myeloid-TNFAIP3-deficient mice have high levels of
inflammatory cytokines in their serum, consistent with a sustained
NF-κB activation and higher TNF-α production by the macrophages
(34). Thus, the increased production
of proinflammatory cytokines in RA may be partially ascribed to the
insufficient expression of TNFAIP3.
Multiple polymorphisms in TNFAIP3 are closely
associated with numerous pathological conditions, including
systemic lupus erythematosus (SLE), coronary artery disease in type
2 diabetes, psoriasis and RA (11,19,35,36). Several
SNPs in the human TNFAIP3 locus are associated with
increased susceptibility to type 1 diabetes, SLE, celiac disease,
Crohn's disease, psoriasis, multiple sclerosis and RA (12), suggesting that defects in
TNFAIP3 expression or activity could be involved in the
development of specific autoimmune diseases (33). TNFAIP3 deficiency in myeloid
cells triggers erosive polyarthritis resembling RA (33). Consequently, the downregulated
expression of TNFAIP3 may give rise to RA disease,
indicating a critical and cell-specific function in the etiology of
RA.
The inverse correlation between TNFAIP3
expression and RA disease severity indicated by RA score, CRP,
anti-CCP antibodies or RF suggest that the insufficient expression
of TNFAIP3 may contribute to diagnosis and severity of the
disease. The RA classification criteria and scoring system used is
a global score, which was developed and validated for diagnostic
purposes in RA. Therefore, the negative correlation between the
TNFAIP3 mRNA expression and score in the RA patients means
that the low expression of TNFAIP3 was associated with the
diagnosis. Additionally, CRP, anti-CCP antibodies or RF are also
indicators of the degree of inflammation and used to help diagnose
disease. The level of TNFAIP3 expression may be an index of
disease diagnosis since the TNFAIP3 mRNA expression was
negatively correlated with the CRP, anti-CCP antibodies or RF in
the RA patients. In addition, there was a significant difference in
TNFAIP3 expression between patients with a positive result
of anti-CCP antibodies and those with a negative result of anti-CCP
antibodies. Antibodies to CCPs have been described in patients with
RA and these appear to be the most specific markers of the disease.
The association of TNFAIP3 expression with anti-CCP
antibodies in RA patients also indicates reduction of
TNFAIP3 expression, and was involved in diagnosis of RA
disease. Taken together, the analysis of the association between
TNFAIP3 expression and score, anti-CCP antibodies, RF and
CRP further suggest a potential role of decreased TNFAIP3
expression in diagnosis of RA.
As for the causes of the low expression of
TNFAIP3 in RA, there are several possible explanations. One
reason may be polymorphisms of the TNFAIP3 gene (18,19). In
peripheral blood cells, no significant difference was detected in
TNFAIP3 expression according to genotype at the intergenic
markers rs6920220 and rs13207033 in RA patients or controls
(37). For rs5029937 and rs7749323,
TNFAIP3 expression was reduced in RA patients carrying the
risk allele compared with G/G in controls (37). For rs5029937 and rs7749423, the trend
demonstrated that TNFAIP3 expression was reduced in healthy
controls, suggesting a possible small allele-specific effect on
TNFAIP3 expression (37).
Therefore, the difference of TNFAIP3 expression may be
partially attributed to polymorphism of the TNFAIP3 gene. By
contrast, to identify susceptibility alleles associated with RA, a
genome-wide association study was carried out to identify genetic
regions with potential DNA variants determining susceptibility to
RA (18,38). Two SNPs were unequivocally identified
at 6q23. Although these variants are not located in a gene, they
are thought to influence TNFAIP3 as its nearest gene (~150
kb downstream of TNFAIP3), possibly by the presence of
potential regulatory DNA elements in this region (12).
The other possible reason is promoter methylation of
the TNFAIP3 gene. TNFAIP3 is targeted by promoter
methylation in certain hematological malignancies (39). Thus, the low expression of the
TNFAIP3 gene in RA may be due in part to its aberrant
methylation. This requires further investigation. The present study
is limited by the sole RT-qPCR method in the measurement of
TNFAIP3 expression and lacks protein and mechanistic data.
Quantification of the TNFAIP3 protein by flow cytometry or
western blot analysis, as well as demonstration of the mechanism,
will be an emphasis in future studies.
In conclusion, TNFAIP3 mRNA expression was
downregulated and inversely correlated with certain disease
parameters in RA patients. The results suggest that reduction of
TNFAIP3 expression may correlate with the diagnosis of RA.
The data provide the support that the TNFAIP3 gene may be a
target for gene therapy or pharmacological agents of RA
disease.
References
|
1
|
Firestein GS: Evolving concepts of
rheumatoid arthritis. Nature. 423:356–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Iwamoto T, Okamoto H, Toyama Y and
Momohara S: Molecular aspects of rheumatoid arthritis: Chemokines
in the joints of patients. FEBS J. 275:4448–4455. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li D, Wang L, Fan Y, Song L, Guo C, Zhu F,
Zhang L and Shi Y: Down-regulation of A20 mRNA expression in
peripheral blood mononuclear cells from patients with systemic
lupus erythematosus. J Clin Immunol. 32:1287–1291. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Apostolidis SA, Lieberman LA, Kis-Toth K,
Crispín JC and Tsokos GC: The dysregulation of cytokine networks in
systemic lupus erythematosus. J Interferon Cytokine Res.
31:769–779. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Geiler J, Buch M and McDermott MF:
Anti-TNF treatment in rheumatoid arthritis. Curr Pharm Des.
17:3141–3154. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Silverman N and Maniatis T: NF-kappaB
signaling pathways in mammalian and insect innate immunity. Genes
Dev. 15:2321–2342. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Verma IM, Stevenson JK, Schwarz EM, Van
Antwerp D and Miyamoto S: Rel/NF-kappa B/I kappa B family: Intimate
tales of association and dissociation. Genes Dev. 9:2723–2735.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cavalcante LO, Melo MR, Dinis VG, Castro
RB, Souza BD and Longui CA: Quantitation of glucocorticoid receptor
alpha and NF-κB pathway mRNA and its correlation with disease
activity in rheumatoid arthritis patients. Genet Mol Res.
9:2300–2310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Criswell LA: Gene discovery in rheumatoid
arthritis highlights the CD40/NF-kappaB signaling pathway in
disease pathogenesis. Immunol Rev. 233:55–61. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Simmonds RE and Foxwell BM: Signalling,
inflammation and arthritis: NF-kappaB and its relevance to
arthritis and inflammation. Rheumatology (Oxford). 47:584–590.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Elsby LM, Orozco G, Denton J, Worthington
J, Ray DW and Donn RP: Functional evaluation of TNFAIP3 (A20) in
rheumatoid arthritis. Clin Exp Rheumatol. 28:708–714.
2010.PubMed/NCBI
|
|
12
|
Vereecke L, Beyaert R and van Loo G: The
ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of
immunopathology. Trends Immunol. 30:383–391. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Coornaert B, Carpentier I and Beyaert R:
A20: Central gatekeeper in inflammation and immunity. J Biol Chem.
284:8217–8221. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kingeter LM, Paul S, Maynard SK,
Cartwright NG and Schaefer BC: Cutting edge: TCR ligation triggers
digital activation of NF-kappaB. J Immunol. 185:4520–4524. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Düwel M, Welteke V, Oeckinghaus A, Baens
M, Kloo B, Ferch U, Darnay BG, Ruland J, Marynen P and Krappmann D:
A20 negatively regulates T cell receptor signaling to NF-kappaB by
cleaving Malt1 ubiquitin chains. J Immunol. 182:7718–7728. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee EG, Boone DL, Chai S, Libby SL, Chien
M, Lodolce JP and Ma A: Failure to regulate TNF-induced NF-kappaB
and cell death responses in A20-deficient mice. Science.
289:2350–2354. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Orozco G, Hinks A, Eyre S, et al Wellcome
Trust Case Control Consortium; YEAR consortium: Combined effects of
three independent SNPs greatly increase the risk estimate for RA at
6q23. Hum Mol Genet. 18:2693–2699. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Musone SL, Taylor KE, Lu TT, Nititham J,
Ferreira RC, Ortmann W, Shifrin N, Petri MA, Kamboh MI, Manzi S, et
al: Multiple polymorphisms in the TNFAIP3 region are independently
associated with systemic lupus erythematosus. Nat Genet.
40:1062–1064. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Thomson W, Barton A, Ke X, Eyre S, Hinks
A, Bowes J, Donn R, Symmons D, Hider S, Bruce IN, et al Wellcome
Trust Case Control Consortium; YEAR Consortium: Rheumatoid
arthritis association at 6q23. Nat Genet. 39:1431–1433. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Aletaha D, Neogi T, Silman AJ, Funovits J,
Felson DT, Bingham CO III, Birnbaum NS, Burmester GR, Bykerk VP,
Cohen MD, et al: 2010 Rheumatoid arthritis classification criteria:
An American College of Rheumatology/European League Against
Rheumatism collaborative initiative. Arthritis Rheum. 62:2569–2581.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hah YS, Lee YR, Jun JS, Lim HS, Kim HO,
Jeong YG, Hur GM, Lee SY, Chung MJ, Park JW, et al: A20 suppresses
inflammatory responses and bone destruction in human
fibroblast-like synoviocytes and in mice with collagen-induced
arthritis. Arthritis Rheum. 62:2313–2321. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Boone DL, Turer EE, Lee EG, Ahmad RC,
Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, et
al: The ubiquitin-modifying enzyme A20 is required for termination
of Toll-like receptor responses. Nat Immunol. 5:1052–1060. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Renner F and Schmitz ML: Autoregulatory
feedback loops terminating the NF-kappaB response. Trends Biochem
Sci. 34:128–135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kato M, Sanada M, Kato I, et al: Frequent
inactivation of A20 in B-cell lymphomas. Nature. 459:712–716. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schmitz R, Hansmann ML, Bohle V,
Martin-Subero JI, Hartmann S, Mechtersheimer G, Klapper W, Vater I,
Giefing M, Gesk S, et al: TNFAIP3 (A20) is a tumor suppressor gene
in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp
Med. 206:981–989. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Heyninck K and Beyaert R: A20 inhibits
NF-kappaB activation by dual ubiquitin-editing functions. Trends
Biochem Sci. 30:1–4. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tavares RM, Turer EE, Liu CL, Advincula R,
Scapini P, Rhee L, Barrera J, Lowell CA, Utz PJ, Malynn BA, et al:
The ubiquitin modifying enzyme A20 restricts B cell survival and
prevents autoimmunity. Immunity. 33:181–191. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kool M, van Loo G, Waelput W, De Prijck S,
Muskens F, Sze M, van Praet J, Branco-Madeira F, Janssens S, Reizis
B, et al: The ubiquitin-editing protein A20 prevents dendritic cell
activation, recognition of apoptotic cells, and systemic
autoimmunity. Immunity. 35:82–96. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hövelmeyer N, Reissig S, Xuan NT,
Adams-Quack P, Lukas D, Nikolaev A, Schlüter D and Waisman A: A20
deficiency in B cells enhances B-cell proliferation and results in
the development of autoantibodies. Eur J Immunol. 41:595–601. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Handel ML, McMorrow LB and Gravallese EM:
Nuclear factor-kappa B in rheumatoid synovium. Localization of p50
and p65. Arthritis Rheum. 38:1762–1770. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Marok R, Winyard PG, Coumbe A, Kus ML,
Gaffney K, Blades S, Mapp PI, Morris CJ, Blake DR, Kaltschmidt C,
et al: Activation of the transcription factor nuclear factor-kappaB
in human inflamed synovial tissue. Arthritis Rheum. 39:583–591.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brown KD, Claudio E and Siebenlist U: The
roles of the classical and alternative nuclear factor-kappaB
pathways: Potential implications for autoimmunity and rheumatoid
arthritis. Arthritis Res Ther. 10:2122008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vereecke L, Beyaert R and van Loo G:
Genetic relationships between A20/TNFAIP3, chronic inflammation and
autoimmune disease. Biochem Soc Trans. 39:1086–1091. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Matmati M, Jacques P, Maelfait J,
Verheugen E, Kool M, Sze M, Geboes L, Louagie E, Mc Guire C,
Vereecke L, et al: A20 (TNFAIP3) deficiency in myeloid cells
triggers erosive polyarthritis resembling rheumatoid arthritis. Nat
Genet. 43:908–912. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Boonyasrisawat W, Eberle D, Bacci S, Zhang
YY, Nolan D, Gervino EV, Johnstone MT, Trischitta V, Shoelson SE
and Doria A: Tag polymorphisms at the A20 (TNFAIP3) locus are
associated with lower gene expression and increased risk of
coronary artery disease in type 2 diabetes. Diabetes. 56:499–505.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tejasvi T, Stuart PE, Chandran V, Voorhees
JJ, Gladman DD, Rahman P, Elder JT and Nair RP: TNFAIP3 gene
polymorphisms are associated with response to TNF blockade in
psoriasis. J Invest Dermatol. 132:593–600. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Maxwell JR, Gowers IR, Kuet KP, Barton A,
Worthington J and Wilson AG: Expression of the autoimmunity
associated TNFAIP3 is increased in rheumatoid arthritis but does
not differ according to genotype at 6q23. Rheumatology (Oxford).
51:1514–1515. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Plenge RM, Cotsapas C, Davies L, Price AL,
de Bakker PI, Maller J, Pe'er I, Burtt NP, Blumenstiel B, DeFelice
M, et al: Two independent alleles at 6q23 associated with risk of
rheumatoid arthritis. Nat Genet. 39:1477–1482. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Frenzel LP, Claus R, Plume N, Schwamb J,
Konermann C, Pallasch CP, Claasen J, Brinker R, Wollnik B, Plass C,
et al: Sustained NF-kappaB activity in chronic lymphocytic leukemia
is independent of genetic and epigenetic alterations in the TNFAIP3
(A20) locus. Int J Cancer. 128:2495–2500. 2011. View Article : Google Scholar : PubMed/NCBI
|