Introduction
Clinical cancer lesions are masses of heterologous
cancer cells. The mainstream hypothesis of cancer metastasis is
that ‘a population of highly malignant cells acquires metastatic
ability due to the accumulation of gene mutations forming
metastatic lesions’ (1), i.e., it is
hypothesized that highly malignant cancer cells with metastatic
ability and low-malignant cancer cells with no metastatic ability
are present in the same cancer lesion, and metastatic lesions are
mainly formed by the former. Certain established cancer cell lines
have a high metastatic ability (highly metastatic cell lines) that
is similar to clinical cancers. To establish a highly metastatic
cell line, an animal model of metastasis is prepared and cells
collected from metastatic lesions are transplanted into another
animal, and this procedure is repeated. A highly metastatic cell
line is established by passaging only a cell population that has
metastasized in the metastasis model, however, why such a highly
malignant cell line is established from a genetically cloned cell
line through this passaging method is questionable. It is unlikely
that a new change in a gene sequence, such as gene mutation, occurs
through only several passages even in a metastasis model. Cancer
cell lines are manufactured on the assumption that cells will be
passaged a specific number of times. When a gene mutation occurs
within a small number of passages, this contradicts the stability
of the cancer cell line. Therefore, the difference between a highly
metastatic cell line and its parent cell line with no metastatic
ability is of interest and we hypothesize that the difference is
not a genetic mutation.
Recent studies identified the presence of the
alternation of cell phenotypes, termed epigenetic abnormality, in
cancers, in addition to genetic mutation. The representative
epigenetic change is DNA methylation, particularly the methylation
of gene promoter regions. The state of methylation is inherited
even after cell division, and its abnormality (hypermethylation or
genome-wide hypomethylation) in various types of cancer has been
reported (2). Highly malignant human
cancer cell lines established by the passage method include a
highly metastatic human colon cancer cell line, KM12SM, and its
parent cell line, KM12C, with no metastatic ability (3). We hypothesized that abnormal methylation
is not present in the parent cell line KM12C, and it has occurred
in highly metastatic KM12SM and altered certain gene and protein
expression levels (increase or decrease), resulting in the
difference in biological behavior between the two cell lines.
Therefore, the differences in the frequency of methylation of the
gene promoter regions were extensively analyzed between the highly
metastatic and parent cell lines.
Materials and methods
Cell lines and assay
Two human colon cancer cell lines, KM12C and KM12SM,
derived from human colon cancer, which were kindly provided from Dr
Nakajima at SBI Pharmaceuticals Co., Ltd. (Tokyo, Japan) were used.
Cells were cultured at 37°C in RPMI-1640 medium (Life Technologies,
Carlsbad, CA, USA) supplemented with 10% fetal calf serum (Sigma,
St. Louis, MO, USA) and 1% penicillin and streptomycin (Life
Technologies) under a humidified atmosphere containing 5%
CO2. DNA sample preparation was as follows: Genomic DNA
was isolated from the cell lines using the DNeasy® Blood and Tissue
kit (Qiagen, Hilden, Germany). To confirm the quality of the
samples, the concentration of DNA (µg/µl) and absorbance ratio
(260/280 nm) were measured by a NanoDrop (Thermo Fisher Scientific,
Waltham, MA, USA). DNA of 81.8 µg/µl with a ratio of 1.98 for
KM12C, and 81.1 µg/µl with a ratio of 1.93 for KM12SM was prepared.
These samples were confirmed under electrophoresis on a 1% agarose
gel to check the size of the fragments at 200 V of constant power
for 25 min. The extent of abnormal methylation of the promoter
region was assessed by bead array analysis. Bead array analysis was
as follows: analysis was performed following the protocol of the
Illumina Infinium HD Methylation assay. As the software,
GenomeStudio version V2011.1 and Methylation Module version 1.9.0
were used. Regarding KM12SM and KM12C as comparative and control
groups, respectively, the level of methylation was determined at
~480,000 sites throughout the genome, grading no methylation and
methylation of only one and both alleles as 0, 0.5 and 1,
respectively, and the average (AVG) was calculated for each gene.
In addition, the difference between KM12SM-AVG and KM12C-AVG was
calculated using the following formula: Log-ratio = (log2
KM12SM-AVG) - (log2 KM12C-AVG). When the difference was <0.6,
the gene was regarded as equally methylated (EM), and when it was
≥0.6, the gene was regarded as differently methylated (DM).
Western blot analysis of
activated-RhoA
For analysis, the RhoA Activation Assay Biochem kit™
(Cytoskeleton, Inc., Denver, CO, USA) was used following the
manufacturer's protocol. The cell lysates for KM12SM and KM12C were
seeded in culture flasks at 5×104 cells/ml and cultured
in 5% CO2 at 37°C. After 3 days, cells were lysed with
500 µl of cell lysis buffer [50 mM Tris (pH 7.5), 10 mM
MgCl2, 0.5 M NaCl and 2% IPEGAL] and centrifuged at
10,000 × g, and the supernatant was collected as a lysate. The
protein level was measured using the BCA™ Protein Assay
kit-Reducing Agent Compatible (Thermo Fisher Scientific).
For the sample preparation, the KM12C and KM12SM
lysates were aliquoted into 3 portions, adjusting the protein
content to 700 µg. To 2 of the 3 aliquots, 1/100 volumes of GTPγS
(200 mM solution) and GDP (100 mM solution) were added as positive-
and negative-control samples, respectively. The remaining sample
was used as an untreated sample. For the pulldown assay, to each
sample 50 µg (60 µl) of Rhotekin-Rho-binding domain beads was
added, the sample was incubated at 4°C for 1 h, and the supernatant
was removed. Following washing, the precipitate was resuspended
with 20 µl of 2X Laemmli sample buffer [125 mM Tris (pH 6.8), 20%
glycerol, 4% SDS, 0.005% bromophenol blue and 5% β-mercaptoethanol]
and boiled for 2 min. For the western blot assay, following the
pulldown assay, 15 µl of each sample was applied on 4–20%
Mini-PROTEAN® TGX™ gel (Bio-Rad Laboratories, Hercules, CA, USA)
and electrophoresed at 200 V for 32 min, followed by transfer to a
PVDF membrane at 75 V for 45 min. Following blocking, the membrane
was reacted with an anti-RhoA monoclonal antibody as a primary
antibody (ARH03; Cytoskeleton, Inc.), and chemiluminescence emitted
by the target band using electrochemiluminescence was exposed to an
X-ray film. The area of bands detected on the X-ray film was
calculated using Manual ROI Selection (Draw Polygons) of Tissue
Studio 4.0 (Definiens, München, Germany).
Analysis of activated small
G-protein
Activated-RhoA was quantified using the G-LISA® RhoA
Activation Assay Biochem kit™ (Cytoskeleton, Inc.). Preparation of
the cell lysates and protein assay was performed as described
previously. For the G-LISA assay, the assay was performed following
the manufacturer's protocol. Briefly, 120 µl of cell lysis buffer
combined with 1.2 µl of the protease inhibitor cocktail and 120 µl
of the binding buffer were mixed and used as a buffer blank, and a
mixture of 24 µl of the RhoA control protein, 96 µl of the cell
lysis buffer, and 120 µl of the binding buffer was used as a
RhoA-positive control. KM12SM and KM12C lysates were adjusted to
0.5, 1.0 and 1.5 mg/ml, and 90 µl of each lysate sample was mixed
with 90 µl of the binding buffer. These samples, the buffer blank
and the RhoA-positive control (50 µl) were distributed to 3 wells
each. The anti-RhoA monoclonal antibody was added as a primary
antibody and detected using horseradish peroxidase, and the
absorbance was measured at 490 nm.
Results
Methylation in KM12SM and KM12C
cells
In the overall investigated regions, EM and DM in
KM12SM compared to methylation in KM12C were 78.44 and 21.56%,
respectively. Limiting to promoter regions, EM and DM were 84.15
and 15.85%, respectively. DM exhibited hypermethylation and
hypomethylation in KM12SM compared with those in KM12C in 37.70 and
62.30% of the overall regions, respectively, and 25.98 and 74.00%
of the promoter regions, respectively. The promoter regions with
the top 15 methylation levels, including the two overlap in KM12SM
and in its parent cell line KM12C, are shown in Fig. 1. Of these, the highest methylation
level in KM12SM was detected in the Rho GTPase-activating protein
28 (ARHGAP28) gene.
 | Figure 1.Methylation of the promoter region.
The y-axis shows the average (AVG) methylation in each gene. The
x-axis shows the top 15 methylation levels of the promoter regions,
including the two overlap. AMELY, amelogenin, Y-linked;
UDP2A2, glucuronosyl transferase 2 family, polypeptide A2;
ARHGAP28, Rho GTPase-activating protein 28; KLHL13,
kelch-like 13 (Drosophila); IDO1, indoleamine
2,3-dioxygenase 1; OR5T3, olfactory receptor, family 5,
subfamily T, member 3; TP53I13, tumor protein p53-inducible
protein 13; RARRES1, retinoic acid receptor responder
(tazarotene induced) 1; LEF1-AS1, LEF1 antisense RNA 1;
CSRNP3, cysteine-serine-rich nuclear protein 3;
IFNGR2, interferon-γ receptor 2 (interferon-γ transducer 1);
GLYATL1, glycine-N-acyltransferase-like 1. |
ARHGAP28 was identified as a GTPase-activating
protein (GAP) of a low molecular weight G-protein, RhoA protein
(4). Activated RhoA that has bound to
guanosine triphosphate (GTP) is converted to inactivated RhoA bound
to guanosine diphosphate (GDP) through the action of a GAP,
ARHGAP28. The hypermethylated state of the ARHGAP28 gene
indicates that activated-RhoA cannot return to inactivated-RhoA, as
the GAP of RhoA, ARHGAP28, is not functioning, which results in the
persistence of activated-RhoA (hyperactivated-RhoA).
The cDNA sequence of RhoA was analyzed to
investigate whether or not mutation of the RhoA gene occurred prior
to activated-RhoA expression in the two cell lines. Briefly, the
method and findings were as follows: RNA was extracted from KM12SM
and KM12C, converted to cDNA, and exons 1–5 were exhaustively
sequenced. The base sequence of RhoA cDNA was identical between
KM12SM and KM12C, but the sequences with G and T at position 464
were mixed in the two cell lines, and due to this mutation, the
amino acid sequences with Gly (GGG) and Val (GTG) at position 155
were simultaneously present.
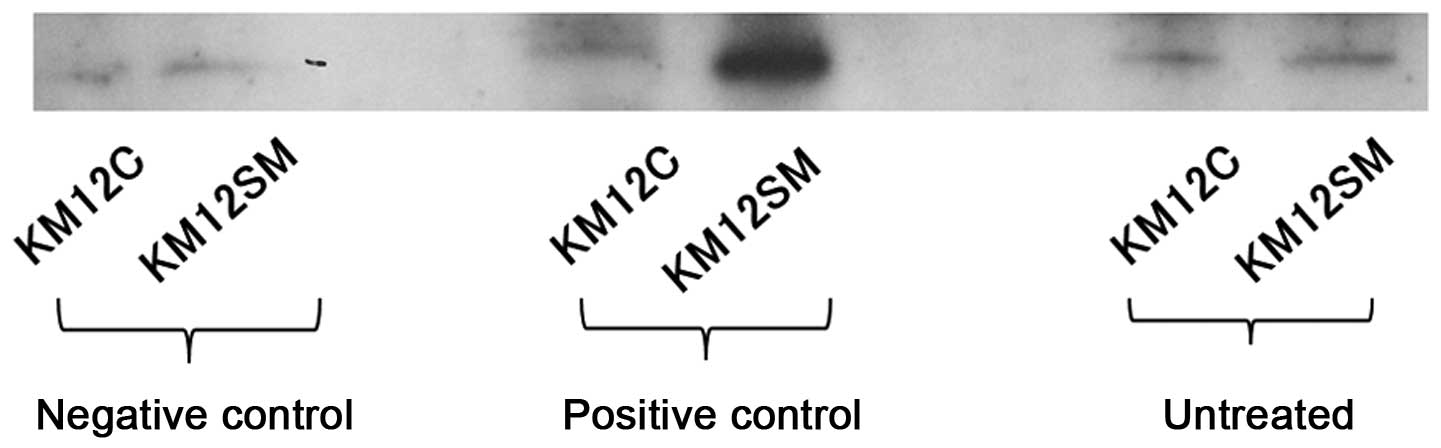
Western blot analysis
The western blot analysis of RhoA-GTP in the parent
cell line, KM12C, and KM12SM is shown in Fig. 2. Bands were detected at the target
molecular weight, 21 kDa. The band area measured using the Tissue
Studio was 13,713.47 µm2 in KM12SM and 11,781.86
µm2 in KM12C, showing that it was ~1.2-fold wider in
KM12SM compared to KM12C.
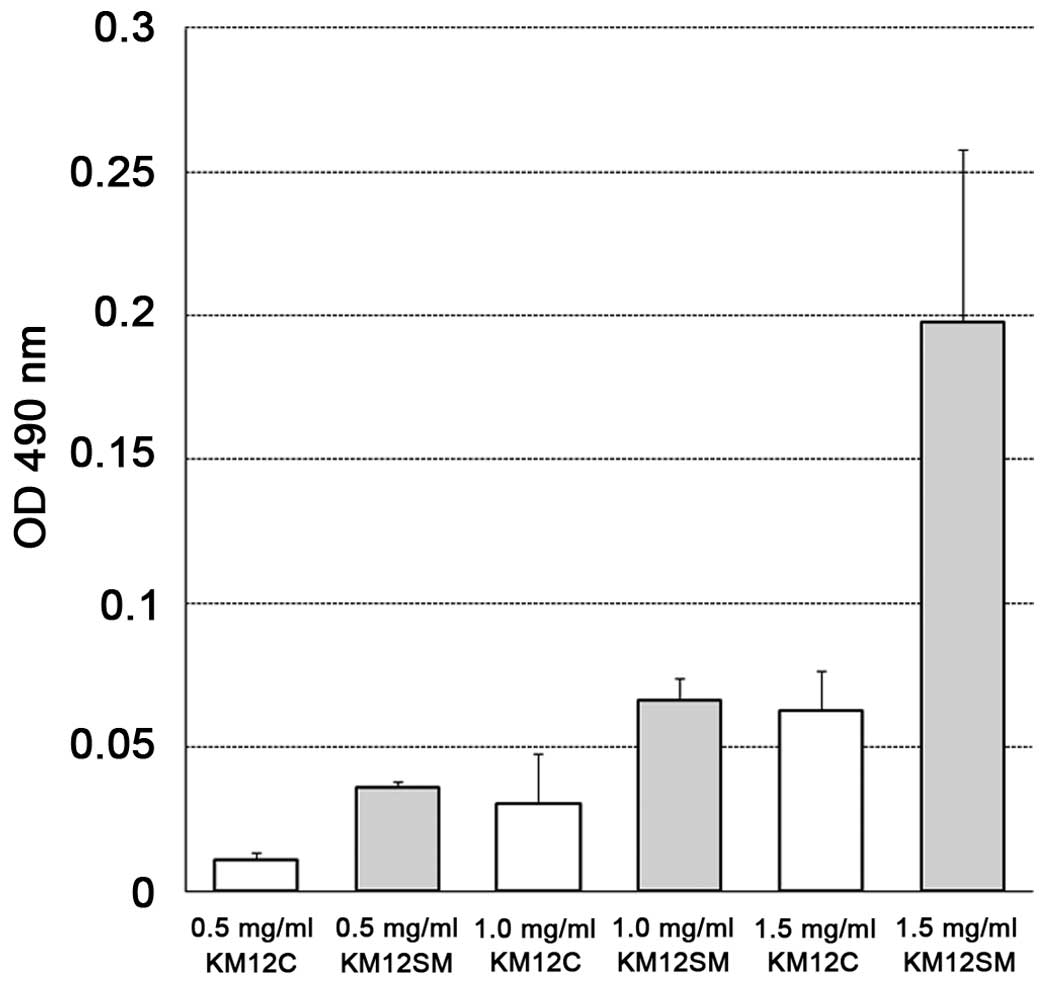
G-LISA analysis
The optical density at 490 nm on the G-LISA assay
was compared between KM12C and KM12SM by the concentration of the
cell lysate. The absorbances were 0.011±0.005 vs. 0.036±0.004 at
0.5 mg/ml, 0.030±0.035 vs. 0.066±0.016 at 1.0 mg/ml, and
0.063±0.028 vs. 0.198±0.120 at 1.5 mg/ml, respectively, showing
that RhoA-GTP, i.e., the activated-RhoA level, was higher in KM12SM
compared to KM12C at all 3 concentrations (Fig. 3).
Discussion
Epigenetic changes are originally mechanisms that
efficiently use genes, and DNA methylation and histone
modifications are representative mechanisms. In DNA methylation, a
methyl group (−CH3) is added to a cytosine of a CpG sequence, which
strongly inhibits the transcription of downstream genes. In histone
modification, a specific site of the DNA scaffold protein, termed
histone, is methylated, which inhibits DNA transcription. It has
long been known that exposure to various environmental factors with
aging is a cause of abnormality of these mechanisms; however,
bacterial infection, cigarette smoking and hormones are also
environmental factors inducing epigenetic abnormalities. Abnormal
DNA methylation is roughly determined by what tissue is exposed to
what environmental factor. For example, DNA methylation is
different between esophageal cancer exposed to cigarette smoking
and gastric cancer induced by H. pylori infection. In
colorectal cancers, the cancer phenotype with augmentation of
enhanced DNA methylation in CpG islands in numerous genes is termed
the CpG island methylator phenotype (CIMP), and gene instability
due to inactivated BRAF and MLH1 genes in CIMP-high
colon cancers, KRAS gene abnormality in CIMP-low colon
cancers, and the TP53 gene abnormality in CIMP-negative
colon cancers have been reported (5).
A merit of investigating DNA methylation is the
superior stability of methylated states. mRNA and protein
expression levels markedly vary depending on the environment of
cancers and cell cycle. For example, slight changes in temperature
and the composition and condition of culture medium in ex
vivo alter mRNA and protein expression levels. By contrast, DNA
methylation is mostly stable anytime it is measured, and the cell
condition can be determined based on it. In addition, the number of
methylations is 0 (no methylation), 0.5 (only one allele is
methylated) or 1 (both alleles are methylated). Thus, the value is
unlikely to be biased in cancer cell populations, which are
heterologous. This is useful for the pathological diagnosis of
cancer. Extensive analysis of DNA methylation has recently been
performed for early discovery and identifying a risk group of
cancer. Regarding the benefit of extensive analysis, previous
studies clarified that DNA was non-methylated in normal tissue and
hypermethylated in cancers (silencing by methylation), i.e.,
non-expressed genes were identified. The genes silenced by
methylation are considered as tumor suppressor genes. However, the
identification of genes expressed at a low level in normal cells
even though they are not methylated has become possible with
technological advancements. Analysis of various types of cancer
using Infinium HumanMethylation is the most rapidly progressing.
The present study used Infinium HumanMethylation450 of Illumina for
extensive analysis, which simultaneously measured the methylation
level at ~480,000 sites throughout the genome. The methylation
state was most markedly different in the ARHGAP28 gene in
the highly metastatic cell line, KM12SM, compared with the parent
cell line, KM12C. The methylation frequency of the ARHGAP28
gene was nearly 1.0 in KM12SM, showing that both alleles were
methylated in the majority of KM12SM cells. By contrast, the
methylation level was mostly 0 in this region in KM12C, clarifying
that the promoter region of the ARHGAP28 gene is
hypermethylated in the highly metastatic human colon cancer cell
line KM12SM compared with that in the parent cell line KM12C.
ARHGAP28 is not a direct cause of carcinogenesis or
malignancy, i.e., the abnormality is not a driver gene mutation.
Thus, it is likely that methylation of the ARHGAP28 gene is
a passenger methylation due to the phenomenon of metastasis.
Low molecular weight G-proteins are classified into
5 groups: the Ras, Rho, Rab, Ran and Sar/Arf families. The Rho
family includes 3 typical molecules: RhoA, Rac1 and Cdc42, and
several molecules exit downstream of each. ARHGAP28 is involved in
a Rho family, RhoA, as discussed in the following. RhoA is a 21-kDa
protein consisting of 193 amino acids, and it is involved in
cytoskeleton formation and cell migration (6). The core effector domains of RhoA, Y42 and
G17, are highly conserved among the Rho family proteins. G17 is a
pocket that GNP enters. Y42 and R5 are present on the external
surface of RhoA, and Y42 is important as the binding site of
RhoA-activity regulatory protein. RhoA is normally bound to GDP:
RhoA-GDP = inactivated-RhoA; and GDP is replaced with GTP by the
GDP/GTP exchange factor in response to certain stimulation,
converting the molecule to RhoA-GTP = activated-RhoA. When the
active form becomes unnecessary, GTPase activity of RhoA itself is
enhanced by ARHGAP28 and the bound GTP is hydrolyzed to GDP,
converting the molecule to inactivated-RhoA. A highly
activated-RhoA level in KM12SM with a highly metastatic ability
observed in the western blot analysis and G-LISA analysis, and a
hypermethylated state of the gene of ARHGAP28, which is GAP
of RhoA, in the methylation analysis do not contradict each other.
Thus, we considered that the RhoA activity level increased in a
cell population with a hypermethylated ARHGAP28 gene
(possibly a small number) in KM12C, and the population acquired
metastatic ability. Therefore, the RhoA gene may be involved in
metastasis, and there are studies supporting this. It has been
reported that a constantly active mutant of the RhoA gene may be a
driver gene, and cases with RhoA gene mutation showed typical
features of diffuse gastric cancer with an alveolar structure of
lesions comprised of one to several cells in the infiltrated region
(7). Cells were scattered in diffuse
gastric cancer, and a budding cell group was present in the deep
infiltrated region of colon cancer, being involved in metastasis
(8). If these findings were involved
in the cell scattering process, these do not contradict our
consideration. RhoA kinase is present downstream of RhoA, and the
inhibition of colon cancer cell migration by RhoA kinase inhibitor
has been reported (9), but the absence
of an association between RhoA activity and infiltration of colon
cancer has also been reported (10).
In either case, a question arises as to whether there is merit in
analyzing only one driver gene. In 2004, Müller et al
(11) reported a study using
methylation abnormality observed in CpG islands of the promoter
region of the secreted frizzled-related protein 2 (SFRP2)
gene as a biomarker, in which the sensitivity was high (77%,
10/13), although the number of cases was only 13. Takeda et
al (12) performed an additional
study on these findings, in which when a ≥1% methylation rate was
judged as methylated, it was observed in 30 and 85% of normal
mucosa and colon cancer, respectively. ARHGAP28 gene
methylation observed in the highly metastatic cell line KM12SM may
lead to findings similar to those observed in SFRP2. To investigate
this, future studies should screen clinical samples for the
activity level of RhoA, on which ARHGAP28 acts as a GAP, as a liver
metastasis-predicting factor.
In conclusion, a high level of RhoA, which is
involved in cell migration, is due to hypermethylation of the gene
of ARHGAP28, which is a GAP of RhoA, and it may be
associated with a highly metastatic ability.
Acknowledgements
The authors would like to thank Mrs. Yukiko Iwahara
and Mrs. Aimi Uchida, and the University students belonging to the
Department of Clinical Pharmacy of Tokyo University of Pharmacy and
Life Sciences for their valuable technical assistance.
References
|
1
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yamashita S, Hosoya K, Gyobu K, Takeshima
H and Ushijima T: Development of a novel output value for
quantitative assessment in methylated DNA immunoprecipitation-CpG
island microarray analysis. DNA Res. 16:275–286. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morikawa K, Walker SM, Nakajima M, Pathak
S, Jessup JM and Fidler IJ: Influence of organ environment on the
growth, selection, and metastasis of human colon carcinoma cells in
nude mice. Cancer Res. 48:6863–6871. 1988.PubMed/NCBI
|
|
4
|
Yeung CY, Taylor SH, Garva R, Holmes DF,
Zeef LA, Soininen R, Boot-Handford RP and Kadler KE: Arhgap28 is a
RhoGAP that inactivates RhoA and downregulates stress fibers. PLoS
One. 9:e1070362014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yamamoto E, Suzuki H, Yamano HO, Maruyama
R, Nojima M, Kamimae S, Sawada T, Ashida M, Yoshikawa K, Kimura T,
et al: Molecular dissection of premalignant colorectal lesions
reveals early onset of the CpG island methylator phenotype. Am J
Pathol. 181:1847–1861. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ishizaki T, Morishima Y, Okamoto M,
Furuyashiki T, Kato T and Narumiya S: Coordination of microtubules
and the actin cytoskeleton by the Rho effector mDia1. Nat Cell
Biol. 3:8–14. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kakiuchi M, Nishizawa T, Ueda H, Gotoh K,
Tanaka A, Hayashi A, Yamamoto S, Tatsuno K, Katoh H, Watanabe Y, et
al: Recurrent gain-of-function mutations of RHOA in diffuse-type
gastric carcinoma. Nat Genet. 46:583–587. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ueno H, Murphy J, Jass JR, Mochizuki H and
Talbot IC: Tumour ‘budding’ as an index to estimate the potential
of aggressiveness in rectal cancer. Gastroenterology. 127:385–394.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Al-haidari AA, Syk I, Jirström K and
Thorlacius H: CCR4 mediates CCL17 (TARC)-induced migration of human
colon cancer cells via RhoA/Rho-kinase signaling. Int J Colorectal
Dis. 28:1479–1487. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
de Toledo M, Anguille C, Roger L, Roux P
and Gadea G: Cooperative anti-invasive effect of Cdc42/Rac1
activation and ROCK inhibition in SW620 colorectal cancer cells
with elevated blebbing activity. PLoS One. 7:e483442012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Müller HM, Oberwalder M, Fiegl H,
Morandell M, Goebel G, Zitt M, Mühlthaler M, Ofner D, Margreiter R
and Widschwendter M: Methylation changes in faecal DNA: A marker
for colorectal cancer screening? Lancet. 363:1283–1285. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Takeda M, Nagasaka T, Dong-Sheng S, Nishie
H, Oka T, Yamada E, Mori Y, Shigeyasu K, Morikawa T, Mizobuchi S,
et al: Expansion of CpG methylation in the SFRP2 promoter region
during colorectal tumorigenesis. Acta Med Okayama. 65:169–177.
2011.PubMed/NCBI
|