Introduction
Although splenectomy is always followed by a
significant elevation in hemoglobin (Hb) levels in hemoglobin H
(HbH) disease, also known as α-thalassemia intermedia, this
procedure is not generally recommended as the majority of patients
do not experience adverse effects with Hb levels in a steady state.
However, in certain circumstances, such as a baseline Hb <7
g/dl, significant hepatosplenomegaly with or without hypersplenism,
abdominal discomfort or growth retardation requiring frequent
transfusions (10–12 transfusions per year), splenectomy may be
considered (1). Thromboembolic events
(TEE) are frequently reported in patients with thalassemia
following splenectomy. In a survey involving 56 tertiary referral
centers in eight countries, venous thromboembolism (VTE) was
observed in 146 of 8,860 patients with thalassemia, of which 134
underwent splenectomy (2). These
pathogenic mechanisms could explain the chronic hypercoagulable
state observed in thalassemia patients without a spleen, including
thrombocytosis and chronic platelet activation, perturbation of the
red blood cell membrane, and protein C (PC) and protein S (PS)
deficiencies that increase following splenectomy (3,4). Several
studies have demonstrated that genetic mutations do not appear to
have an important role in the pathogenesis of thrombosis observed
in thalassemia (5–8). However, PC and PS mutations are frequent
genetic risk factors involved in VTE in the Chinese population.
Therefore, the presence of these mutations may aggravate the risk
of thrombosis in thalassemia. In support of this hypothesis, the
present study reports a case of a patient with HbH disease bearing
mutations associated with thrombosis who experienced recurrent
VTE.
Case report
The patient was a 22-year-old Chinese female of Han
nationality with no siblings, and was diagnosed with HbH disease at
0.5-years old. In recent years, the Hb level of the patient
fluctuated between 40 and 60 g/l, and thus the red blood cell
transfusions were frequent, approximately once every 1–2 weeks. The
ferritin level ranged from 3,500–6,800 ng/ml (female normal range:
13–150 ng/ml), and therefore, iron chelation therapy was also
applied periodically. Since 2012, the patient experienced inflation
pain on the epigastrium and an abdominal ultrasonogram repeatedly
showed apparent splenomegaly. Subsequently, the patient underwent a
splenectomy in 2013. Following this, the Hb level ranged from 90 to
110 g/l postoperation. However, recurrent infection, particularly
cellulitis of the lower limbs, appeared following the surgery. The
first thrombotic event, portal vein thrombus, occurred in 2013 at
the age of 20 years, only 1 month after the splenectomy. Despite
receiving anticoagulant treatment, the patient suffered from
another four thrombotic events at set intervals. The details of
each TEE are presented in Table I.
 | Table I.Summary of the clinical features of
patients with postsplenectomy TE. |
Table I.
Summary of the clinical features of
patients with postsplenectomy TE.
| Interval between
splenectomy and each TE | Type of TE | Postsplenectomy
PLT | Postsplenectomy
RBC |
|---|
| 1 month | Portal vein
thrombus |
710×109/l |
5.28×1012/l |
| 1 year and 3
months | PE |
619×109/l |
5.04×1012/l |
| 1 year and 7
months | Superficial
thrombosis of the left long saphenous vein |
5.02×1012/l
420×109/l |
|
| 1 year and 9
months | Superficial
thrombosis of both long saphenous vein; recurrent PE |
357.3×109/l |
5.05×1012/l |
| 1 year and 10
months | Thrombosis of the
right cephalic vein and thrombophlebitis |
330×109/l |
4.96×1012/l |
Computed tomography of the different sections of the
body showed thrombosis of the portal vein, bilateral pulmonary
artery, both long saphenous veins and the right cephalic vein
(Fig. 1; certain sections are not
shown). Recent laboratory studies demonstrated that the PC antigen
and activity were 68.2% (normal range, 70–150%) and 51.0% (normal
range, 60–140%), and the corresponding values for PS were 61.4%
(normal range, 65–135%) and 50.2% (normal range, 76–135%),
respectively. Activated partial thromboplastin time, prothrombin
time, antithrombin activity, fibrinogen and anticardiolipin were
normal, and only D-dimer was elevated (Table II). PC and PS antigens were measured
by an enzyme-linked immunosorbent assay using the Human Protein C
ELISA kit (AssayPro, St. Charles, MO, USA) and Zymutest Protein S
ELISA kit (Hyphen Biomed, sur-Oise, France). PC and antithrombin
activities were measured by a substrate method using STA-Stachrom
Protein C and STA-Stachrom AT III (Beckman Coulter, Fullerton, CA,
USA). PS activity was tested by coagulation method using
STA-Staclot Protein S (Beckman Coulter). Other parameters were all
detected by automatic coagulation Analyzer in Grade A Tertiary
Hopital.
| Table II.Laboratory data of the patient. |
Table II.
Laboratory data of the patient.
| Variables | Patient | Normal range |
|---|
| Protein C activity,
% | 51 |
60–140 |
| Protein C antigen,
% | 68.2 |
70–150 |
| Protein S activity,
% | 50.2 |
76–135 |
| Protein S antigen,
% | 61.4 |
65–135 |
| Antithrombin
(AT-III), % | 125 |
83–128 |
| Fibrinogen, g/l | 4.84 | 2.00–5.00 |
| D-dimer, ng/ml | 856 |
0–450 |
| APTT, sec | 40 | 25.0–45.0 |
| PT, sec | 12.6 | 10–15 |
| Anticardiolipin
antibody | (−) | (−) |
Initial intravenous administration of low molecular
weight heparin calcium was followed by warfarin sodium.
Subsequently, an oral anticoagulant was continued to maintain the
International normalized ratio within a range of 2–3.
Genetic analysis was performed. Isolation of genomic
DNA from whole blood was conducted using the QIAamp DNA mini kit
(Qiagen, Hilden, Germany) according to the manufacturer's protocol
and was followed by polymerase chain reaction (PCR) sequencing
analysis of the PC gene (PROC), PS gene (PROS1), antithrombin gene
(SERPINC1), and several candidate single-nucleotide polymorphisms
(SNPs) associated with deep-vein thrombosis (DVT) identified by
genome-wide association studies. The detected mutations were
confirmed by reverse sequencing and were verified using a second
amplicon. The amplified fragments were sequenced on an ABI 3730XL
DNA automated sequencer (Applied Biosystems, Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The variants were designated
according to current nomenclature and the recommendations of the
Human Genome Variation Society (HGVS, http://www.hgvs.org/mutnomen/). These SNPs were rs6025
(FV c.1691G>A), rs1799963 (FII g.20210G>A), rs5361 (SELE),
rs1613662 (GP6) and rs13146272 [cytochrome P450, family 4,
subfamily V, polypeptide 2 (CYP4V2)]. Subsequent to sequencing all
nine exons of PROC, the following three SNPs were identified in
exons 2, 6 and 7: rs14430087 (66T>C), rs5936 (423G>T), and
rs146922325 (565C>T, Arg189Trp, R147W; Fig. 2). The former two SNPs, rarely reported,
are synonymous mutations, while the missense mutation, Arg189Trp,
has increasingly been observed in previous studies (9–11). In
addition, PROS1 contained an SNP in exon 9, rs747259055 (947G>A,
Arg316His). Finally, SERPINC1 had a synonymous SNP in exon 5,
rs5877 (981A>G; Fig. 2). Among the
candidate SNPs, rs13146272 was identified in CYP4V2 (Table III).
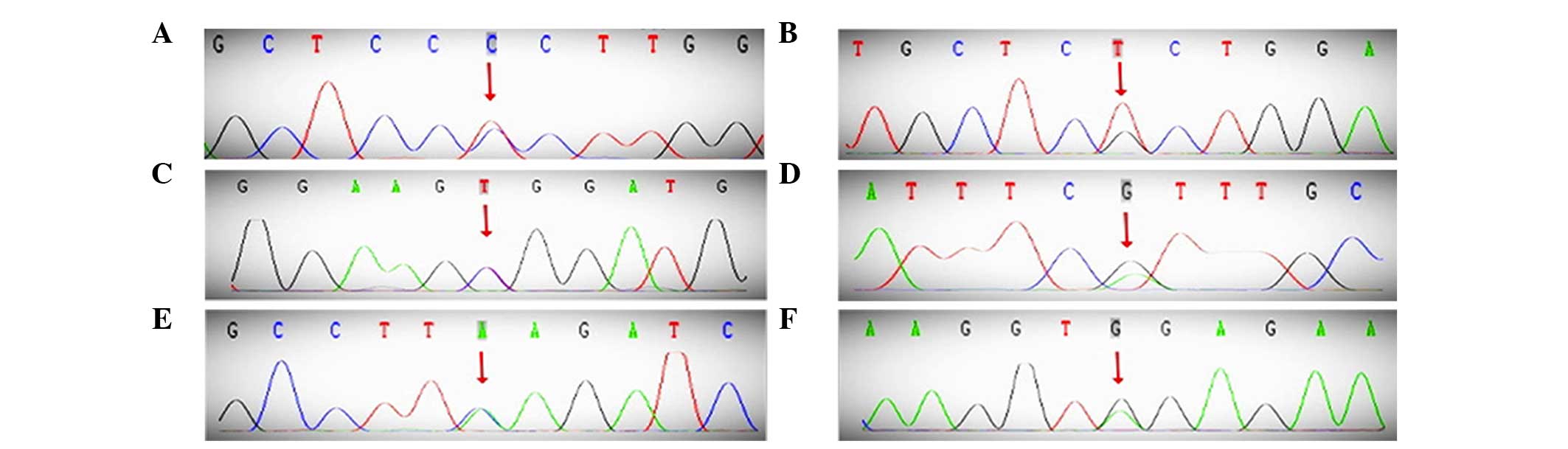 | Figure 2.DNA sequence of the PROC, PROS1,
SERPINC1 and CYP4V2 genes. (A) 66T>C, (B) 423G>T, (C)
565C>T. (D) PROS1, 947G>A. (E) SERPINC1, 981A>G. (F)
CYP4V2, 775C>A. PROC, protein C; PROS1, protein S; SERPINC1,
antithrombin; CYP4V2, cytochrome P450, family 4, subfamily V,
polypeptide 2. |
| Table III.Sequence analysis of the various
SNPs. |
Table III.
Sequence analysis of the various
SNPs.
| Gene | Chromosome | Region | SNP | Nucleotide position
and mutation | Amino acid
change | Functional
consequence |
|---|
| PROC | 2 | Exon 2 | rs14430087 | 66
T>C | Pro22= | Synonymous |
|
| 2 | Exon 6 | rs5936 | 423 G>T | Ser141= | Synonymous |
|
| 2 | Exon 7 | rs146922325 | 565 C>T | Arg189Trp | Missense |
| PROS1 | 3 | Exon 9 | rs747259055 | 947 G>A | Arg316His | Missense |
| SERPINC1 | 1 | Exon 5 | rs5877 | 981 A>G | Val327= | Synonymous |
| CYP4V2 | 4 | – | rs13146272 | 775 C>A | GIn259Lys | Missense |
Discussion
It is well known that several risk factors have a
synergistic effect on the pathogenesis of TEE in thalassemia,
however, studies of recurrent TEE in thalassemia, particularly in
HbH related to congenital thrombophilic mutations, are limited. In
2002, Kahn et al (12) first
described a β-thalassemia major patient with recurrent VTE in
association with factor V R506Q and prothrombin G20210A mutations.
However, these mutations, which are reported frequently in
Caucasians, were not detected in the Chinese Han patient of the
present study. PROC, PROS1 and SERPINC1, as well as candidate SNPs,
were sequenced in DNA samples from the patient and three missense
mutations and three synonymous mutations were identified.
There have been several studies on the inherited
anomalies of anticoagulation factors, such as antithrombin, PS and
PC, in patients with venous thrombosis in the Chinese population.
PC deficiency is associated with an increased risk of VTE, and its
genetic background has been analyzed in several populations
(13,14). In the present study, rs14430087
(66T>C), rs5936 (423G>T) and rs146922325 (c.565C>T,
p.Arg189Trp, R147W) in PROC were identified in the patient. The
p.Arg189Trp variant has been described in the Chinese population in
patients with PC deficiency (Table
IV) (9–11). Tang et al (9) described a recurrent mutation
(c.565C>T) that appear in 17 of the 34 probands (50%) and was
not only the most frequent variant for PC deficiency, but also a
significant risk factor for venous thrombosis in Chinese
individuals. The study revealed that first-degree relatives bearing
this variant had an 8.8-fold increased risk of venous thrombosis
(9). In addition, Tsay et al
(10) tested PROC in the families of
21 unrelated probands with symptomatic PC deficiency, and reported
that the heterozygous R147W mutation by itself remained a
significant thrombotic risk factor. Furthermore, they discovered
that among the 21 families, nine symptomatic propositi shared the
R147W missense mutation in exon 7 plus a silent T66C mutation in
exon 2, which was not identified in other family members or in the
healthy control group that did not carry the R147W mutation
(10). The present study concurred
that this was the case in the patient. A study on the clinical and
genetic features of PC deficiency in 23 unrelated subjects in the
Chinese population demonstrated that the Arg189Trp mutation
occurred in 43.5% of all subjects. This study also illustrated that
complex genotypes in PROC deficiency are mainly responsible for the
increased risk for VTE, particularly for recurrent VTE (15).
| Table IV.Associated citations with the gene
variant (rs146922325, c.565C>T, p.Arg189Trp or p.Arg147Trp,
R147W) in the protein C gene. |
Table IV.
Associated citations with the gene
variant (rs146922325, c.565C>T, p.Arg189Trp or p.Arg147Trp,
R147W) in the protein C gene.
|
| No. of
deficiencies/total (%) |
|
|
|
|
|---|
|
|
|
|
|
|
|
|---|
| Study, year | Cases | Controls | Odds ratio (95%
CI) | P-value | Population | Refs. |
|---|
| Tsay et al,
2004 | 5/116
(4.31) | 11/1,292 (0.85) | 5.10 (1.7–14.8) | – | Chinese | (10) |
| Tang et al,
2012 | 59/1,003 (5.88) | 9/1,031
(0.87) | 7.10
(3.50–14.39); adjusted: 7.34 (3.61–14.94) or 7.13 (3.49–14.56) |
3.31×10−10; adjusted:
3.88×10−8 or 6.88×10−8 | Chinese | (9) |
| Tang et al,
2013 | 68/1,304 (5.21) | 12/1,334 (0.90) | 6.06
(3.26–11.25) |
1.03×10−10 | Chinese | (11) |
Additionally, the present patient also suffered from
PS deficiency. However, only one novel mutation, Arg316His
(947G>A), was identified in exon 9 of PROS1. Furthermore, the
antithrombin level was normal, however, g.981A>G (rs5877) was
detected in exon 5 of SERPINC1. Although silent mutations (such as
g.981A>G) do not influence protein structure expression, they
may still affect protein function and, therefore, the sensitivity
of a patient to heparin (16). In
addition, rs13146272 in the CYP4V2 gene, which has been reported to
be associated with DVT (17,18), was found in the present patient;
however, whether this variant is associated with TEE in the Chinese
population remains to be elucidated.
In the present study, the patient experienced the
first thrombosis at the age of 20 years, and four recurrent
thrombotic events occurred in an extremely short time interval (~2
years). Several risk factors with respect to the hypercoagulable
state in thalassemia, as well as acquired and congenital
thrombophilia, could result in an increased risk of VTE or
recurrent VTE. Therefore, this case illustrates a patient with HbH
who underwent a splenectomy with congenital thrombophilic mutation
and a requirement for long-term anticoagulant treatment.
In conclusion, when thrombotic events repeatedly
occur in a patient with thalassemia, risk factors associated with
the hypercoagulable state, as well as acquired and congenital
thrombophilia, should be screened for. In addition, prophylactic
anticoagulation therapy is also recommended in thalassemia
intermedia patients who are undergoing certain types of surgery, as
well as those who have a previous history of deep vein thrombosis
or pulmonary embolism. In patients with TEE life-long
anticoagulation appears to be rational and effective in prevention
of recurrent TEE.
Glossary
Abbreviations
Abbreviations:
|
Hb
|
hemoglobin
|
|
HbH
|
hemoglobin H
|
|
TEE
|
thromboembolic events
|
|
VTE
|
venous thromboembolism
|
|
PC
|
protein C
|
|
PS
|
protein S
|
|
APTT
|
activated partial thromboplastin
time
|
|
PT
|
prothrombin time
|
|
PROC
|
protein C gene
|
|
PROS1
|
protein S gene
|
|
SERPINC1
|
antithrombin gene
|
|
SNPs
|
single-nucleotide polymorphisms
|
|
DVT
|
deep-vein thrombosis
|
References
|
1
|
Fucharoen S and Viprakasit V: Hb H
disease: Clinical course and disease modifiers. Hematology Am Soc
Hematol Educ Program. 2009:26–34. 2009.
|
|
2
|
Taher A, Isma'eel H, Mehio G, Bignamini D,
Kattamis A, Rachmilewitz EA and Cappellini MD: Prevalence of
thromboembolic events among 8,860 patients with thalassaemia major
and intermedia in the Mediterranean area and Iran. Thromb Haemost.
96:488–491. 2006.PubMed/NCBI
|
|
3
|
Sirachainan N: Thalassemia and the
hypercoagulable state. Thromb Res. 132:637–641. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cappellini MD, Poggiali E, Taher AT and
Musallam KM: Hypercoagulability in β-thalassemia: A status quo.
Expert Rev Hematol. 5:505–511; quiz 512. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Taher AT, Otrock ZK, Uthman I and
Cappellini MD: Thalassemia and hypercoagulability. Blood Rev.
22:283–292. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cappellini MD, Musallam KM, Marcon A and
Taher AT: Coagulopathy in Beta-thalassemia: Current understanding
and future perspectives. Mediterr J Hematol Infect Dis.
1:e20090292009.PubMed/NCBI
|
|
7
|
Cappellini MD, Motta I, Musallam KM and
Taher AT: Redefining thalassemia as a hypercoagulable state. Ann N
Y Acad Sci. 1202:231–236. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Succar J, Musallam KM and Taher AT:
Thalassemia and venous thromboembolism. Mediterr J Hematol Infect
Dis. 3:e20110252011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tang L, Guo T, Yang R, Mei H, Wang H, Lu
X, Yu J, Wang Q and Hu Y: Genetic background analysis of protein C
deficiency demonstrates a recurrent mutation associated with venous
thrombosis in Chinese population. PLoS One. 7:e357732012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsay W and Shen MC: R147W mutation of PROC
gene is common in venous thrombotic patients in Taiwanese Chinese.
Am J Hematol. 76:8–13. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tang L, Wang HF, Lu X, Jian XR, Jin B,
Zheng H, Li YQ, Wang QY, Wu TC, Guo H, et al: Common genetic risk
factors for venous thrombosis in the Chinese population. Am J Hum
Genet. 92:177–187. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kahn JE, Veyssier-Belot C, Renier JL, de
Mazancourt P, Peltier JY and de Raucourt E: Recurrent
thromboembolism in a patient with beta-thalassemia major associated
with double heterozygosity for factor V R506Q and prothrombin
G20210A mutations. Blood Coagul Fibrinolysis. 13:461–463. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reitsma PH, Poort SR, Allaart CF, Briët E
and Bertina RM: The spectrum of genetic defects in a panel of 40
Dutch families with symptomatic protein C deficiency type I:
Heterogeneity and founder effects. Blood. 78:890–894.
1991.PubMed/NCBI
|
|
14
|
Gandrille S and Aiach M: Identification of
mutations in 90 of 121 consecutive symptomatic French patients with
a type I protein C deficiency. The French INSERM Network on
Molecular Abnormalities Responsible for Protein C and Protein S
deficiencies. Blood. 86:2598–2605. 1995.PubMed/NCBI
|
|
15
|
Ding Q, Shen W, Ye X, Wu Y, Wang X and
Wang H: Clinical and genetic features of protein C deficiency in 23
unrelated Chinese patients. Blood Cells Mol Dis. 50:53–58. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang J, Ma HP, Ti AL, Zhang YQ and Zheng
H: Prothrombotic SERPINC1 gene polymorphism may affect heparin
sensitivity among different ethnicities of Chinese patients
receiving heart surgery. Clin Appl Thromb Hemost. 21:760–767. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bezemer ID, Bare LA, Doggen CJ, et al:
Gene variants associated with deep vein thrombosis. JAMA.
299:1306–1314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Austin H, De Staercke C, Lally C, Bezemer
ID, Rosendaal FR and Hooper WC: New gene variants associated with
venous thrombosis: A replication study in White and Black
Americans. J Thromb Haemost. 9:489–495. 2011. View Article : Google Scholar : PubMed/NCBI
|















