Introduction
Genetic damage is largely random and therefore tends
to affect the larger, functional regions of the human genome more
frequently than the smaller regions (1). For example, a systematic study has
revealed that cancer fusion genes, on average, are statistically,
significantly larger than other human genes (2,3). The large
introns of potential cancer fusion genes presumably allow for many
different productive recombination opportunities, i.e., many
recombinations that would allow for exon juxtaposition and the
generation of hybrid proteins. Smaller cancer fusion genes tend to
be associated with the rare types of cancer, for example EWS RNA
binding protein 1 in Ewing's sarcoma.
Cytoskeleton-related protein coding regions (CPCRs),
including extracellular matrix (ECM) proteins, are among the
largest coding regions in the human genome and are heavily mutated
in various types of cancer (1,4,5). The
possibility that these coding regions would also be commonly
vulnerable to de novo mutations, or mutations occurring in
relatively recent past-generations, was considered, and therefore
had not been included in the conventional single nucleotide
polymorphism (SNP) databases. In the present study, CPCR single
nucleotide variants (SNVs) that appeared in The Cancer Genome Atlas
(TCGA) normal blood and breast cancer (BRCA) sample datasets were
identified. ‘Non-unique’ SNVs were discovered to be relatively
common among a sample of 31 individuals, and SNVs specifically
present in single individuals (i.e., private variations) were also
common.
Materials and methods
Basic algorithm
The basic approach is indicated in Fig. 1 and the detailed steps are provided in
the supporting online material (SOM) file, ‘Stoll 2016 SOM Figure 1, detailed protocol’; all of the SOM
files are hosted at http://www.universityseminarassociates.com/Supporting_online_material_for_scholarly_pubs.php).
Whole exome sequence (WXS) sample manifest files were downloaded
from the Cancer Genomics Hub Browser (browser.cghub.ucsc.edu; approved NIH dbGaP project
number 6300) using the following filters: ‘Breast invasive
carcinoma’ under ‘By Disease’; ‘Blood Derived Normal’ and ‘Primary
Solid Tumor’ under ‘By Sample Type’; ‘WXS’ under ‘By Library Type’;
and ‘GRCH37/HG19’ under ‘By Assembly’. Search results were sorted
by ‘Barcode’ so that solid tumor samples were immediately followed
by the matched blood sample. The manifest files for matched BRCA
and normal blood samples were downloaded and placed into a file
labeled ‘Blood’ to be consistent with the language of the
BreastBlood.sh coding program (Stoll 2016 SOM Figure 1A-BreastBlood) written specifically
for the current study. The manifest files were subsequently
processed with the BreastBlood.sh to create .csv output files (from
the original .xml manifest files). The original programming code,
BreastBlood.sh, and output files (Stoll 2016 SOM Figure 1E-BreastBlood Variants) are provided
in the SOM. Note that the output files have been further processed
using various macros, also provided in the SOM, to eliminate
previously identified variants that will not be included in
analysis. BreastBlood.sh was designed to identify variants in
cytoskeletal protein-related coding regions (CPCRs). The CPCR list
was generated from the study by Parry and Blanck (5), representing the most commonly mutated
CPCRs in five cancer datasets that were investigated plus two
additional CPCRs that are commonly mutated in the melanoma (skin
cutaneous melanoma) dataset, which were not evaluated in the
previous study (5). The CPCRs are
presented in Table I.
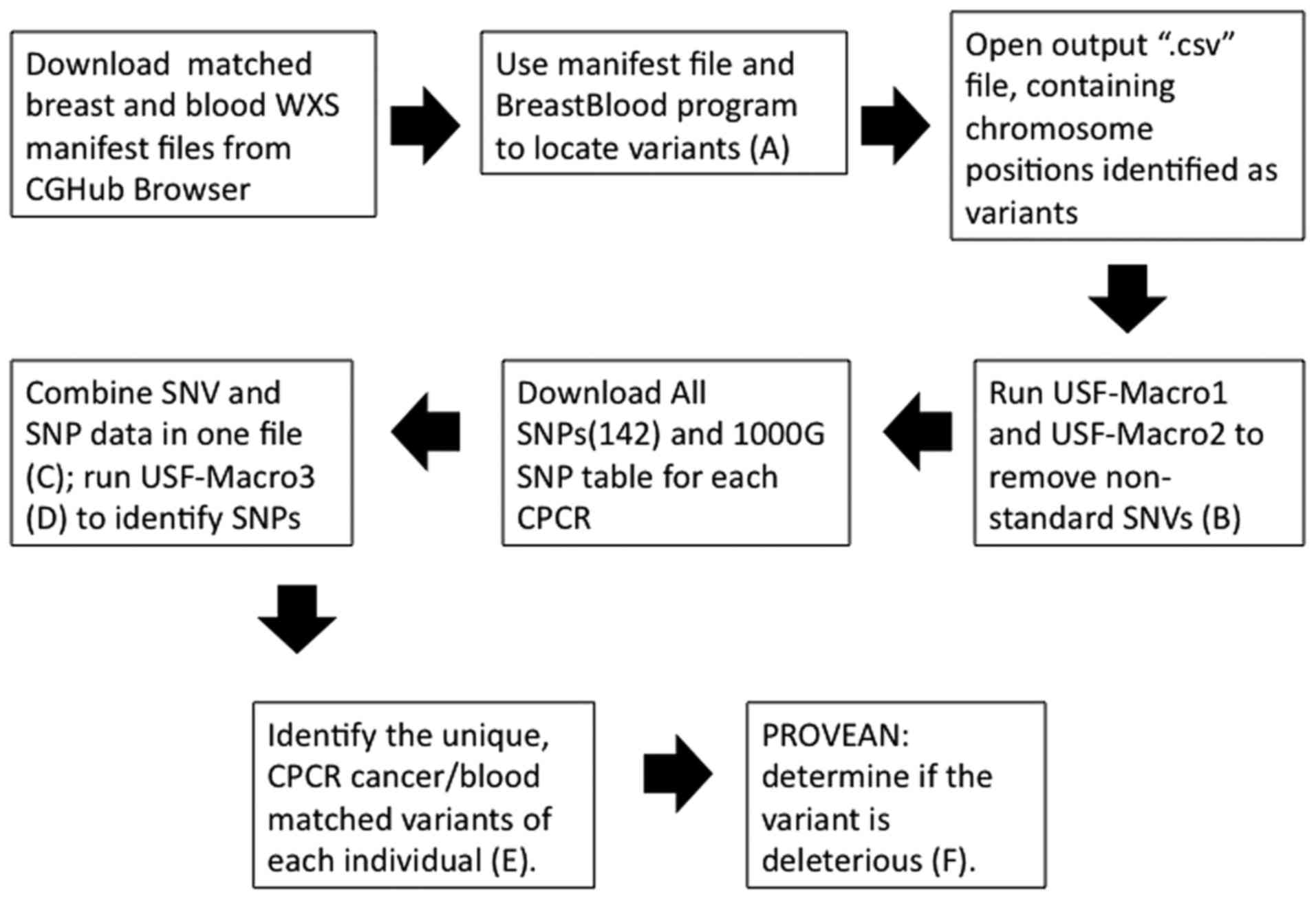 | Figure 1.Flow chart representing the processing
steps for generating Tables II and
III, and Figs. 2 and 3.
The upper case letters in parentheses refer to Excel files in the
SOM produced by the indicated step in the flow chart. However, (C)
in the SOM represents an example file, i.e., SNP removal using the
All SNPs(142) database. SOM, supporting online material; WXS, whole
exome sequence; SNV, single nucleotide variant; SNP, single
nucleotide polymorphism; USF, University of South Florida; CPCR,
cytoskeleton-related protein coding regions; PROVEAN, Protein
Variation Effect Analyzer. |
 | Table I.HUGO symbols for the cytoskeletal
protein-related coding regions set. |
Table I.
HUGO symbols for the cytoskeletal
protein-related coding regions set.
| HUGO symbol | Gene name |
|---|
| ANK2 | Ankyrin 2,
neuronal |
| APC | Adenomatous polyposis
coli |
| COL11A1 | Collagen, type XI,
α1 |
| DNAH10 | Dynein, axonemal,
heavy chain 10 |
| DNAH11 | Dynein, axonemal,
heavy chain 11 |
| DNAH3 | Dynein, axonemal,
heavy chain 3 |
| DNAH5 | Dynein, axonemal,
heavy chain 5 |
| DNAH7 | Dynein, axonemal,
heavy chain 7 |
| DNAH8 | Dynein, axonemal,
heavy chain 8 |
| DSCAM | Down syndrome cell
adhesion molecule |
| DST | Dystonin |
| FAT3 | FAT atypical cadherin
3 |
| FAT4 | FAT atypical cadherin
4 |
| FBN2 | Fibrillin 2 |
| FGFR1 | Fibroblast growth
factor receptor 1 |
| FLG | Filaggrin |
| MUC16 | Mucin 16, cell
surface associated |
| MUC17 | Mucin 17, cell
surface associated |
| MUC4 | Mucin 4, cell surface
associated |
| NEB | Nebulin |
| NEFH | Neurofilament, heavy
polypeptide |
| NF1 | Neurofibromin 1 |
| PCDH15 | Protocadherin-related
15 |
| PCDHAC2 | Protocadherin alpha
subfamily C, 2 |
| PCDHGC5 | Protocadherin gamma
subfamily C, 5 |
| PCLO | Piccolo presynaptic
cytomatrix protein |
| PKHD1 | Polycystic kidney and
hepatic disease 1 (autosomal recessive) |
| PLEC | Plectin |
| RELN | Reelin |
| SPTA1 | Spectrin, alpha,
erythrocytic 1 |
| SPTAN1 | Spectrin, alpha,
non-erythrocytic 1 |
| SSPO | SCO-spondin |
| SYNE1 | Spectrin repeat
containing, nuclear envelope 1 |
| SYNE2 | Spectrin repeat
containing, nuclear envelope 2 |
| TTN | Titin |
| XIRP2 | Xin actin binding
repeat containing 2 |
Use of SNP databases and the PROVEAN
web tool
Two SNP databases [All SNPs(142) and 1000 Genomes;
genome.ucsc.edu] were used to filter the
previously identified variants in the raw sequence files
representing the matched BRCA and normal blood barcodes, as
indicated in Fig. 1. An example of
this process is provided in two SOM files, ‘Stoll 2016 SOM Figure 1C (All SNPs(142) Example)’ and ‘Stoll
2016 SOM Figure 1C (All SNPs(142)
Example, Excel)’. Variants not identified by either SNP database
were classified by frequency. The classification of variants can be
found in SOM Excel file, ‘Stoll 2016 SOM Figure 1E-BreastBlood Variants’ in sheet
‘Variant Color-Coding.’ Unique matched variants were defined as
SNVs that were present in the BRCA and normal blood sample matched
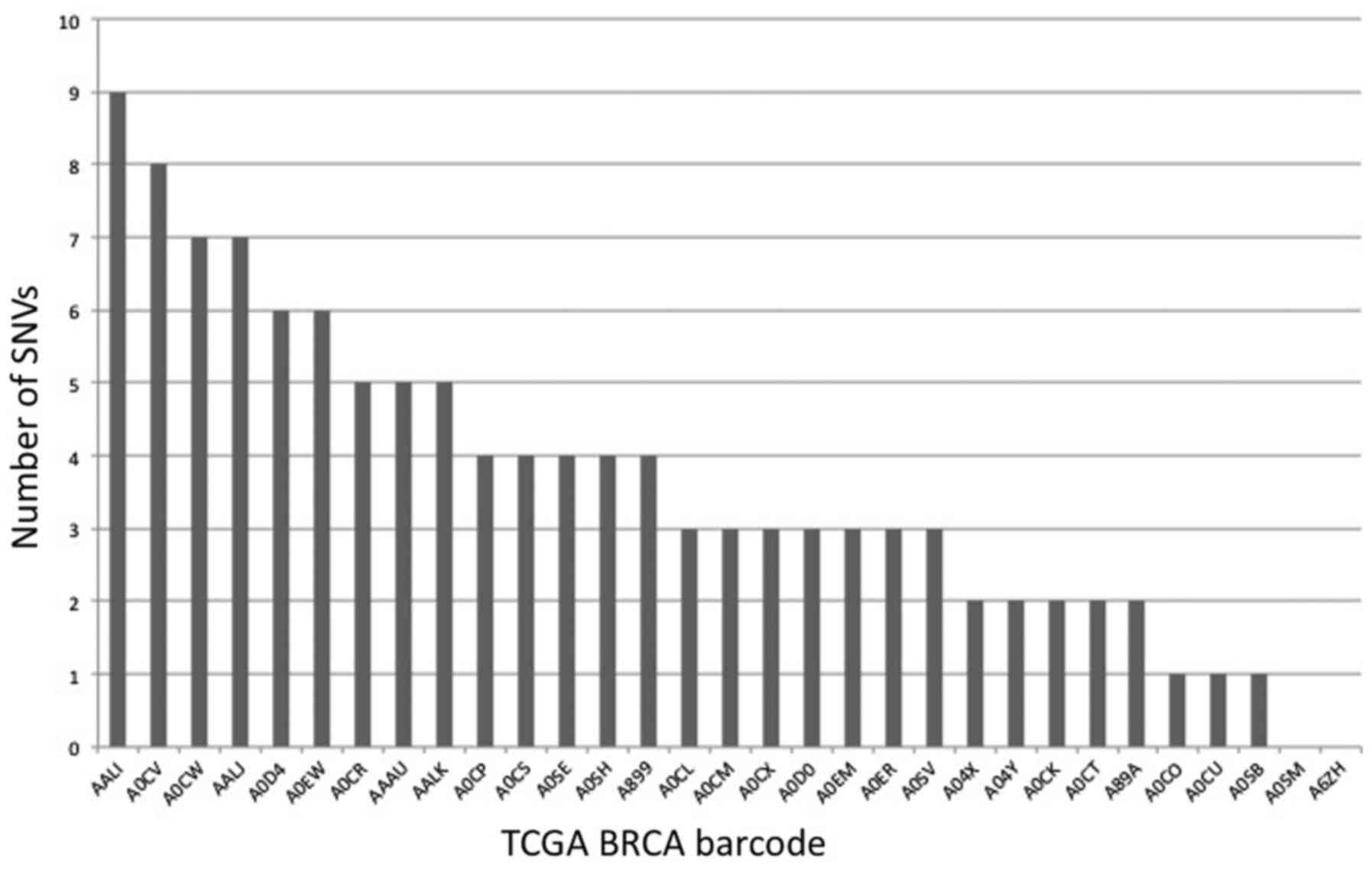
barcodes. A histogram of the number of cancer-/blood-matched CPCR
SNVs limited to a single individual is presented in Fig. 2. These SNVs, as well as the non-unique
SNVs identified among the investigated set of 31 individuals in
this report (but not present in the SNP databases) served as input
for the Protein Variation Effect Analyzer (PROVEAN) human genome
variants program (http://provean.jcvi.org/genome_submit_2.php). This
tool was used to determine if the matched mutations caused damaging
amino acid changes. A summary of the PROVEAN results is presented
in Table II. In addition, the SIFT
analysis (http://provean.jcvi.org/genome_submit_2.php) of amino
acid substitutions is available at the indicated web site and the
results for the current project for the SIFT and PROVEAN analyses
are present in the SOM files labeled ‘Stoll 2016 SOM Figure 1E-Provean Results, aa variants unique
to a barcode’ and ‘Stoll 2016 SOM Figure
1F-Provean Results, aa variants unique to a barcode’.
| Table II.Deleterious amino acids identified in
the cancer-/blood-matched cytoskeletal protein-related coding
region single nucleotide variants and limited to one barcode
(individual). |
Table II.
Deleterious amino acids identified in
the cancer-/blood-matched cytoskeletal protein-related coding
region single nucleotide variants and limited to one barcode
(individual).
| Residue
(wild-type) | Residue (mutant) | PROVEAN
prediction | Gene name | Amino acid
length |
|---|
| Barcode: AAAU |
|
|
|
|
| A | V | Deleterious | TTN | 27118a |
| Barcode: AALI |
|
|
|
|
| G | C | Deleterious | FLG | 4061 |
| R | C | Deleterious | DST | 5171 |
| Barcode: A899 |
|
|
|
|
| P | L | Deleterious | DNAH7 | 4024 |
| Barcode: AALJ |
|
|
|
|
| H | Y | Deleterious | TTN | 27118a |
| Barcode: A0SE |
|
|
|
|
| G | S | Deleterious | MUC4 | 1176 |
| Barcode: A0CL |
|
|
|
|
| L | S | Deleterious | FAT3 | 892 |
| Barcode: A0CP |
|
|
|
|
| P | T | Deleterious | TTN | 33423a |
| Barcode: A0CS |
|
|
|
|
| E | K | Deleterious | TTN | 27118a |
| Barcode: A0CT |
|
|
|
|
| K | E | Deleterious | DNAH10 | 4471 |
| Barcode: A0CV |
|
|
|
|
| C | Y | Deleterious | RELN | 3458 |
| Barcode: A0CW |
|
|
|
|
| L | R | Deleterious | MUC16 | 14507 |
| D | Y | Deleterious | ANK2 | 3924 |
| Barcode: A0ER |
|
|
|
|
| R | C | Deleterious | SYNE2 | 6818 |
| Barcode: A0EM |
|
|
|
|
| V | A | Deleterious | TTN | 27118 |
| Barcode: AALK |
|
|
|
|
| S | L | Deleterious | NEB | 6669 |
Statistical analysis
Statistical significance was determined using
Student's t-test with Microsoft Excel (version 2010). P<0.05 was
considered to indicate a statistically significant difference.
Results
SNVs unique to individuals of the BRCA
study set and representing deleterious amino acids
To assess SNVs potentially present in CPCRs
(Table I) of BRCA and normal blood
samples of a single individual, a series of processing steps,
indicated in Fig. 1 were performed.
Any SNVs that were present in the All SNPs(142) and 1000 Genome SNP
databases (Fig. 1) were eliminated
from further consideration. In addition, any SNVs that were not
verified by 20 reads or more were eliminated from consideration.
Results indicated that 29 barcodes had CPCR SNVs, i.e., nucleotides
not matching the reference human genome (Fig. 2). Of these, 14 barcodes represented
deleterious amino acid replacements, according to PROVEAN analysis,
in the following coding regions: TTN, MUC4, FLG, DST, DNAH7, FAT3,
DNAH10, RELN, MUC16, ANK2, SYNE2 and NEB (Table II). The SIFT analysis indicated 21
damaging mutations (SOM Excel file labeled, ‘Stoll 2016 SOM
Figure 1F-Provean Results, aa variants
unique to a barcode’).
SNVs shared among members of the BRCA
study set, but not present in common SNP databases
The results of the current SNV analyses indicated
that a number cancer-/blood-matched CPCR SNVs were shared between
barcodes in this analysis group of 31 barcodes, despite using the
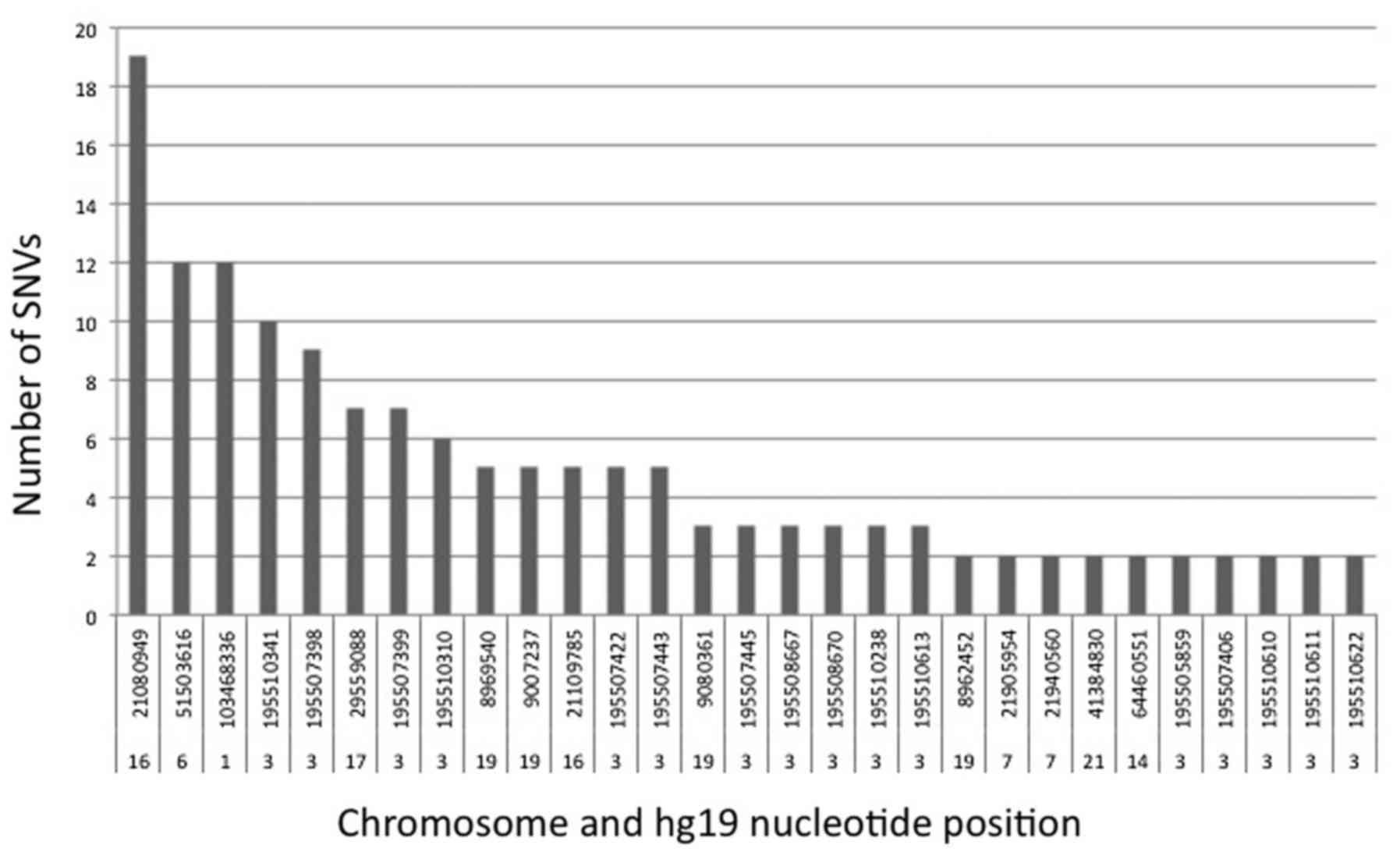
above two SNP databases to clear the known SNPs. Fig. 3 demonstrates the chromosome and
nucleotide positions of the cancer-/blood-matched SNVs identified
in more than one barcode, as well as the number of barcodes with a
given variant. These SNVs were also analyzed using the PROVEAN tool
(Table III; Stoll 2016 SOM Figure 1G-Provean Results, aa variants present
in more than one barcode). A summary of the specific, non-unique
CPCR SNVs is provided in Table III.
Thus, 112 SNVs unique to one individual (Fig. 2) indicated 16 deleterious AA
substitutions (Table II), whereas 29
SNVs shared among the barcodes represented zero deleterious AA
substitutions, using the PROVEAN analysis (P<0.00002; Student's
t-test). The SIFT analysis detected 21 deleterious AA among the
unique, matched SNVs, and there were three deleterious AA
substitutions among the shared, matched SNVs. The SIFT analysis
maintains the trend indicated by the PROVEAN analysis, but was not
statistically significant.
| Table III.Amino acid changes identified in the
cancer-/blood-matched cytoskeletal protein-related coding region
single nucleotide variants of multiple individuals. |
Table III.
Amino acid changes identified in the
cancer-/blood-matched cytoskeletal protein-related coding region
single nucleotide variants of multiple individuals.
| Residue (wild
type) | Residue
(mutant) | PROVEAN
prediction | Gene name | Amino acid
length |
|---|
| 3,195505859;
Barcodes: A0SE, A0CR |
|
|
|
|
| T | A | Neutral | MUC4 | 4442 |
| 3, 195507398;
Barcodes: A89A, AALJ, A0SB, A0SE, A0CL, A0CR, A0CO, A0EW, A0CS |
|
|
|
|
| D | N | Neutral | MUC4 | 4442 |
| 3, 195507399;
Barcodes: A0SB, A0SM, AALK, A0CV, A0CR, A0CO, A0EW |
|
|
|
|
| T | T | Neutral | MUC4 | 4442 |
| 3,195507406;
Barcodes: A0CL, AALJ |
|
|
|
|
| P | L | Neutral | MUC4 | 4442 |
| 3, 195507422;
Barcodes: A89A, A0SE, A0CL, A0CR, A0CO |
|
|
|
|
| D | H | Neutral | MUC4 | 4442 |
| 3, 195507443;
Barcodes: AAAU, A89A, AALJ, A0SB, A0CL |
|
|
|
|
| T | P | Neutral | MUC4 | 4442 |
| 3,195507445;
Barcodes: A0SB, A0CL, A0CS |
|
|
|
|
| D | V | Neutral | MUC4 | 4442 |
| 3,195508667;
Barcodes: A89A, AALK, A0CR |
|
|
|
|
| T | A | Neutral | MUC4 | 4442 |
| 3,195508670;
Barcodes: A89A, AALK, A0CR |
|
|
|
|
| D | H | Neutral | MUC4 | 4442 |
| 3,195510238;
Barcodes: A0SB, AALK, A0CL |
|
|
|
|
| L | P | Neutral | MUC4 | 4442 |
| 3, 195510310;
Barcodes: A89A, A0SB, A0CV, A0CO, A0CR, A0EW |
|
|
|
|
| Y | S | Neutral | MUC4 | 4442 |
| 3, 195510341;
Barcodes: A89A, A0SB, A0SE, A0CW, A0CV, AALK, A0CO, A0CR, A0EW,
A0CS |
|
|
|
|
| S | P | Neutral | MUC4 | 4442 |
| 3,195510610;
Barcodes: A0SE, A0CR |
|
|
|
|
| L | P | Neutral | MUC4 | 4442 |
| 3, 195510611;
Barcodes: A0SB, A0SE |
|
|
|
|
| L | I | Neutral | MUC4 | 4442 |
| 3, 195510613;
Barcodes: A0SB, A0SE, A0CO |
|
|
|
|
| S | N | Neutral | MUC4 | 4442 |
| 3,195510622;
Barcodes: A0CW, A0EW |
|
|
|
|
| P | H | Neutral | MUC4 | 4442 |
| 1, 103468336;
Barcodes: A899, A89A, A6ZH, AALJ, A0CU, A0CT, A0EM, A0CM, A04Y,
A04X, A0D0, A0EW |
|
|
|
|
| G | G | Neutral | COL11A1 | 1767 |
Discussion
The above data and analyses indicate that systemic
mutations in CPCRs are relatively common, consistent with the large
size of these coding regions. In general, systemic, de novo
mutations are considered sporadic and unpredictable, as in the case
of Rett's syndrome, which can be caused by a point mutation in
methyl CpG binding protein-2 (6).
However, the large size of the CPCRs renders it more likely that
de novo, or relatively recently generated SNVs, will be
present in the CPCRs. Furthermore, the above analyses indicate that
SNVs that lead to deleterious amino acid substitutions are more
common when not shared.
The relatively common occurrences of these de
novo, systemic deleterious amino acid substitutions in CPCRs
raises questions regarding the value of evaluating these coding
regions. For example, small coding regions are unlikely to have
de novo mutations and, thus, there may not be a practical
justification for routine analysis. However, if de novo CPCR
SNVs are identified to be essentially inevitable, investigating
them would be valuable, particularly with regard to potentially
identifying deleterious amino acid alternations. Such routine
analysis may have value in better understanding a range of medical
conditions, for example heart disease, due to the major role of
various CPCRs in the formation of the sarcomere cytoskeleton
(7).
In addition, the results cause us to query other
coding region groups, where individual group members may be too
small to regularly reveal systemic mutations. The entire group
collection, however, may be large enough such that de novo
systemic mutations would alter one member of the group often enough
to justify screening. For example, a comprehensive collection of
tumor suppressor genes would likely have a member that represents a
systemic mutation, due to random chance, fairly often. While such a
collection is not likely to be as large as a CPCR collection, it is
likely a large enough portion of genome, with mutations affecting a
large enough number of individuals, to justify screening.
Furthermore, due to the degeneracy of numerous tumor suppressor
signaling pathways (8), a mutation in
any one tumor suppressor, among a set, may represent
pre-disposition to cancer.
In conclusion, SNPs not present in the SNP databases
used here, but that were present in more than one of the
individuals investigated above, and potentially present in other
SNP databases, were detected. However, as all of the individuals
analyzed in the current study were BRCA patients, the outcome of
multiple shared SNVs results in the hypothesis that certain de
novo, systemic CPCR mutations may facilitate BRCA development.
Future studies are required to address the occurrence of potential,
cancer specific SNVs in the CPCR set by repeating the above
analyses for multiple types of cancer.
Acknowledgements
Authors would like to acknowledge support from the
Anna Valentine program and the University of South Florida research
computing facility, in particular, Dr Tony Green.
Glossary
Abbreviations
Abbreviations:
|
CPCR
|
cytoskeleton-related protein coding
regions
|
|
ECM
|
extracellular matrix
|
|
BRCA
|
breast cancer
|
|
TCGA
|
The Cancer Genome Atlas
|
|
SNV
|
single nucleotide variant
|
|
PROVEAN
|
Protein Variation Effect Analyzer
|
|
WXS
|
whole exome sequence
|
|
SNP
|
single nucleotide polymorphism
|
|
SOM
|
supporting online material
|
References
|
1
|
Parry ML, Ramsamooj M and Blanck G: Big
genes are big mutagen targets: A connection to cancerous, spherical
cells? Cancer Lett. 356(2)B479–482. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Narsing S, Jelsovsky Z, Mbah A and Blanck
G: Genes that contribute to cancer fusion genes are large and
evolutionarily conserved. Cancer Genet Cytogenet. 191:78–84. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pava LM, Morton DT, Chen R and Blanck G:
Unifying the genomics-based classes of cancer fusion gene partners:
Large cancer fusion genes are evolutionarily conserved. Cancer
Genomics Proteomics. 9:389–395. 2012.PubMed/NCBI
|
|
4
|
Fawcett TJ, Parry ML and Blanck G: A Novel
Approach to Evaluating Cancer Driver Gene Mutation Densities:
Cytoskeleton-related Gene Candidates. Cancer Genomics Proteomics.
12:283–290. 2015.PubMed/NCBI
|
|
5
|
Parry ML and Blanck G: Flat cells come
full sphere: Are mutant cytoskeletal-related proteins
oncoprotein-monsters or useful immunogens? Hum Vaccin Immunother.
12:120–123. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ballestar E, Yusufzai TM and Wolffe AP:
Effects of Rett syndrome mutations of the methyl-CpG binding domain
of the transcriptional repressor MeCP2 on selectivity for
association with methylated DNA. Biochemistry. 39:7100–7106. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kontrogianni-Konstantopoulos A, Ackermann
MA, Bowman AL, Yap SV and Bloch RJ: Muscle giants: Molecular
scaffolds in sarcomerogenesis. Physiol Rev. 89:1217–1267. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ford SA and Blanck G: Signal persistence
and amplification in cancer development and possible, related
opportunities for novel therapies. Biochim Biophys Acta.
1855:18–23. 2014.PubMed/NCBI
|