Introduction
The incidence of intracranial aneurysm (IA) is high
at approximately 2–3% in the general population worldwide (1). IA rupture is the most common reason for
life-threatening subarachnoid hemorrhages (SAH), which accounts for
~85% of overall SAH (2). Despite
advances in diagnosis and treatment, the rates of mortality and
morbidity of SAH associated with a ruptured IA remain high
(3). Therefore, it is important to
understand the molecular pathogenesis of IA to reduce the
occurrence of SAH.
There is evidence to indicate that environmental and
genetic factors are associated with the pathogenesis of IAs
(4,5).
The modifiable factors, such as smoking, hypertension, and
excessive alcohol consumption have been reported to lead to IA
formation (6). In addition,
accumulating evidence indicates that genetics are significant in
the pathogenesis of IA. Familial IA is not rare, accounting for
7–20% of patients with aneurysmal SAH (7). The risk of experiencing unruptured IAs is
significantly higher (~4 times) in familial IA families than in the
general population (8). Furthermore,
certain genetic factors have been investigated and identified to be
associated with IA (9–13), including angiotensin-converting enzyme
(ACE).
ACE is the key enzyme in the
renin-angiotensin-aldosterone system (RAAS), as an important
modulator of cerebrovascular disease (CVD) (14). The ACE gene, which consists of
26 exons and 25 introns is located on chromosome 17q23 (14). The ACE gene contains functional
insertion/deletion (I/D) polymorphism of a 287-bp Alu sequence
within intron 16 (15). Individuals
with the DD genotype exhibit increased serum ACE levels and
activity when compared with individual with the ID and II genotypes
(15,16). In addition, increased serum ACE levels
may contribute to vascular injury, which is hypothesized to confer
increased risk of IA.
Certain previous studies investigated the influence
of ACE I/D gene polymorphisms on IA susceptibility (17–23);
however, the results of these studies remain inconsistent. As a
single study might not be powered to demonstrate the overall
effects, a meta-analysis using currently available data was
performed in the present study to detect the potential association
of ACE I/D gene polymorphisms on IA risk.
Materials and methods
Publication search
The search was performed using PubMed (https://www.ncbi.nlm.nih.gov/pubmed), Embase
(www.embase.com), and Wanfang databases (updated
to January 6th, 2016; http://www.wanfangdata.com.cn) with the following
terms: ‘Polymorphism’, ‘genotype’, ‘allele’, ‘mutation’, ‘variant’,
in combination with ‘cerebral aneurysm’, ‘brain aneurysm’,
‘intracranial aneurysm’, ‘SAH‘, ‘subarachnoid hemorrhage’, in
combination with ‘ACE’, ‘angiotensin converting enzyme’, and
‘insertion/deletion’. The search was conducted without a limitation
on language. Reference lists of eligible studies were also
retrieved to find additional articles.
Inclusion criteria
Two reviewers independently screened the literature
for relevance and disagreements were resolved by consensus. Studies
included in the current meta-analysis met the following criteria:
The studies i) evaluated the ACE gene polymorphisms and IA
risk; ii) were case-control studies; iii) contained available
genotype frequencies for calculating odds ratios (ORs) with their
95% confidence interval (CI). The exclusion criteria were as
follows: i) Duplicated studies; ii) limited sample size; iii)
reviews, editorials or comments.
Data extraction
Information was carefully extracted from all
eligible publications independently by two authors of the present
paper. For conflicting evaluations, agreement was reached by
discussion. For each study, the following characteristics were
considered: First author surname, year of publication, country of
origin, ethnicity of study subjects, source of control groups
(population- or hospital-based controls), total number of cases and
controls, genotyping method, evidence of Hardy-Weinberg equilibrium
(HWE) and numbers of cases and controls with the II, ID, and DD
genotypes for ACE. Subjects were categorized as East Asian and
Caucasian.
Statistical analysis
Deviation from HWE was examined by χ2
test and P<0.05 was considered to indicate a statistically
significant difference. The strength of associations between
ACE gene polymorphisms and IA risk were measured by OR with
95%CI. The pooled ORs were performed for I allele contrast (I vs.
D), homozygote comparison of codominant, homozygote comparison of
codominant (II vs. DD) and heterozygote comparison of codominant
(ID vs. DD), dominant model (II+ID vs. DD), and recessive model (II
vs. ID+DD), respectively. Heterogeneity assumption was verified by
χ2-based Q-test and quantified using the I2
value. If the studies lacked heterogeneity
(Ph>0.1 and I2<50%), the
fixed-effects model (the Mantel-Haenszel method) was adopted to
calculate the overall ORs value (24).
Otherwise, the random-effects model (the DerSimonian and Laird
method) were used (25). To assess the
stability of the results, sensitivity analyses were performed by
removing each study individually, and recalculating the OR and the
95% CI. Publication bias was assessed using Begg's funnel plots and
Egger's linear regression test, and P<0.05 was considered to
indicate a statistically significant difference (26). All statistical analyses were performed
with Stata software (version 12.0; StataCorp LP, College Station,
TX, USA), using two-sided P-values.
Results
Study characteristics
Our initial search identified 43 studies according
to the search terms. After removing replicates and screening of
titles and abstracts, 13 articles remained for further detailed
evaluation (Fig. 1). Finally, a total
of seven case-control studies were included in our meta-analysis,
involving 1,074 IA cases and 1,500 control subjects. The main study
characteristics are presented in Table
I (19–23). Three studies involving 390 cases and
432 control subjects were from East Asian populations and four
involved 684 cases and 1,068 control participants from Caucasian
populations. In terms of the source of controls, threes studies
included population-based studies, and four studies included
hospital-based studies. Four studies were conducted in large
samples and three in small samples. The genotype distributions
among the controls of all studies were consistent with HWE except
one study by Yu et al (23).
 | Table I.Characteristics of studies included
in the current metaanalysis. |
Table I.
Characteristics of studies included
in the current metaanalysis.
|
|
|
|
|
|
|
| Case | Control |
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|---|
| Author year | Ethnicity | Country | Sample size
(case/control) | SC | Genotyping
methods | HWE | II | ID | DD | II | ID | DD | (Refs.) |
|---|
| Liu, 2013 | East Asian | China | 220/220 | HB | PCR-RFLP | Yes | 64 | 106 | 50 | 44 | 99 | 77 | (17) |
| Staalsø, 2011 | Caucasian | Denmark | 174/498 | HB | TaqMan | Yes | 39 | 96 | 39 | 104 | 270 | 124 | (18) |
| Pannu, 2005 | Caucasian | USA | 162/143 | PB | ASO-PCR | Yes | 33 | 85 | 44 | 27 | 73 | 43 | (19) |
| Slowik, 2004 | Caucasian | Poland | 90/128 | PB | ASO-PCR | Yes | 47 | 14 | 29 | 30 | 65 | 33 | (20) |
| Keramatipour,
2000 | Caucasian | UK | 258/299 | PB | Triple-primer
PCR | Yes | 78 | 126 | 54 | 71 | 146 | 82 | (21) |
| Takenaka, 1998 | East Asian | Japan | 83/104 | HB | ASO-PCR | Yes | 38 | 40 | 5 | 43 | 45 | 16 | (22) |
| Yu, 2005 | East Asian | China | 87/108 | HB | Quantitative PCR,
Sequencing | No | 31 | 47 | 9 | 47 | 38 | 23 | (23) |
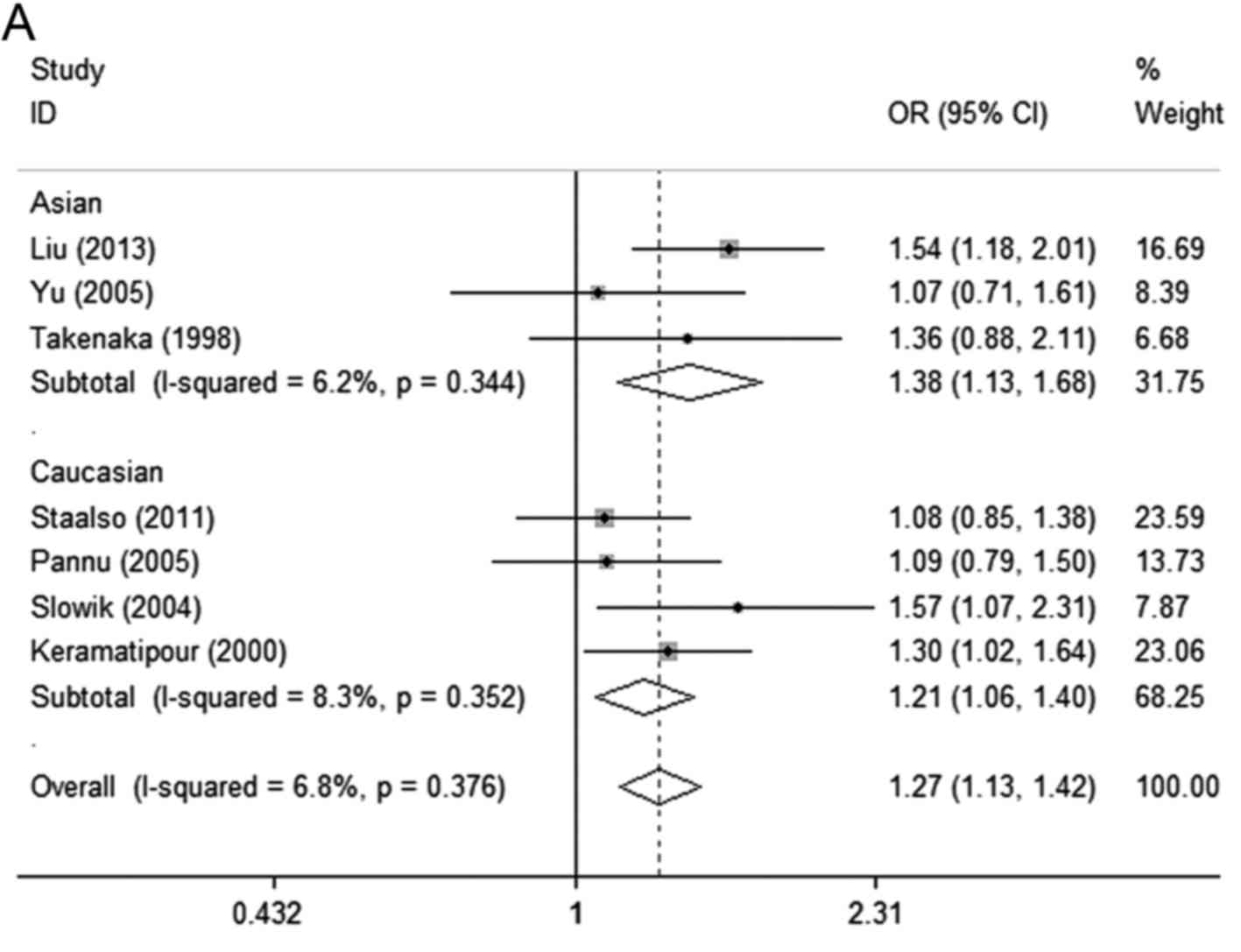
Meta-analysis
Table II summarizes
the key results of the meta-analysis. In the overall analysis, a
significant strong association between the ACE polymorphism and an
increased risk of IA was identified in the allele (I vs. D;
OR=1.27, 95% CI=1.13–1.42; POR=0.000), homozygote comparison
of codominant (II vs. DD; OR=1.64, 95% CI=1.30–2.07;
POR=0.000) and dominant (II+ID vs. DD; OR=1.38, 95%
CI=1.05–1.80; POR=0.021) models. By contrast, no
statistically significant association was detected under the
heterozygote comparison of the codominant model (ID vs. DD;
OR=1.25, 95% CI=0.79–1.96; POR=0.343) and the recessive
model (II vs. ID+DD; OR=1.35, 95% CI=0.98–1.87;
POR=0.064).
| Table II.Meta-analysis results for the
angiotensin-converting enzyme polymorphisms and IA risk. |
Table II.
Meta-analysis results for the
angiotensin-converting enzyme polymorphisms and IA risk.
|
|
|
|
|
|
|
| Codominant
model |
|
|
|
|
|
|
|
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|---|
|
|
|
| Allele I vs. D | Homozygote II vs.
DD | Heterozygote ID vs.
DD | Dominant model
(II+ID) vs. DD | Recessive model II
vs. (ID+DD) |
|---|
|
|
|
|
|
|
|
|
|
|---|
| Variables | Cases n | Cases/control | I2
(%) |
Ph | OR (95%CI) |
POR | I2
(%) |
Ph | OR (95%CI) |
POR | I2
(%) |
Ph | OR (95%CI) |
POR | I2
(%) |
Ph | OR (95%CI) |
POR | I2
(%) |
Ph | OR (95%CI) |
POR |
|---|
| Total | 7 | 1074/1500 | 6.8 | 0.376 | 1.27 (1.13,
1.42) | 0 | 0 | 0.577 | 1.64 (1.30,
2.07) | 0 | 76.7 | 0 | 1.25 (0.79,
1.96) | 0.343 | 44.5 | 0.094 | 1.38 (1.05,
1.80) | 0.021 | 66.1 | 0.007 | 1.35 (0.98,
1.87) | 0.064 |
| Ethnicity |
| East
Asian | 3 | 390/432 | 6.2 | 0.344 | 1.38
(1.13,1.68) | 0.002 | 0 | 0.761 | 2.18
(1.44,3.31) | 0 | 6.8 | 0.342 | 2.02
(1.35,3.03) | 0.001 | 0 | 0.691 | 2.01
(1.41,2.86) | 0 | 59.5 | 0.085 | 1.15
(0.71,1.87) | 0.561 |
|
Caucasian | 4 | 684/1068 | 8.3 | 0.352 | 1.21
(1.06,1.40) | 0.006 | 0 | 0.663 | 1.44
(1.09,1.90) | 0.011 | 80.2 | 0.002 | 0.86
(0.49,1.53) | 0.615 | 11.8 | 0.334 | 1.15
(0.90,1.47) | 0.264 | 74.9 | 0.008 | 1.53
(0.95,2.45) | 0.08 |
| Source of
control |
| HB | 4 | 564/930 | 31.1 | 0.226 | 1.25
(1.07,1.46) | 0.004 | 19.3 | 0.293 | 1.72
(1.25,2.38) | 0.001 | 48.8 | 0.119 | 1.72
(1.11,2.68) | 0.015 | 38.4 | 0.181 | 1.68
(1.15,2.44) | 0.007 | 41 | 0.166 | 1.15
(0.83,1.59) | 0.401 |
| PB | 3 | 510/570 | 2.1 | 0.36 | 1.28
(1.08,1.52) | 0.004 | 0 | 0.652 | 1.56
(1.12,2.17) | 0.009 | 86.5 | 0.001 | 0.76
(0.32,1.80) | 0.526 | 41.2 | 0.183 | 1.12
(0.78,1.62) | 0.543 | 79 | 0.009 | 1.73
(0.92,3.27) | 0.089 |
| Sample size |
|
>300 | 4 | 814/1160 | 31.9 | 0.221 | 1.25
(1.09,1.42) | 0.001 | 16.4 | 0.309 | 1.56
(1.20,2.03) | 0.001 | 0 | 0.629 | 1.30
(1.04,1.63) | 0.023 | 1.5 | 0.384 | 1.38
(1.11,1.71) | 0.003 | 0 | 0.534 | 1.31
(1.05,1.63) | 0.015 |
|
<300 | 3 | 260/340 | 0 | 0.4 | 1.33
(1.05,1.68) | 0.019 | 0 | 0.738 | 1.93
(1.19,3.13) | 0.007 | 91.4 | 0 | 1.27
(0.22,7.48) | 0.79 | 74 | 0.021 | 1.58
(0.63,3.97) | 0.325 | 86.9 | 0 | 1.45
(0.58,3.67) | 0.429 |
| HWE |
|
Yes | 6 | 987/1392 | 12.6 | 0.334 | 1.28
(1.14,1.45) | 0 | 0 | 0.449 | 1.64
(1.29,2.08) | 0 | 76.4 | 0 | 1.11
(0.70,1.75) | 0.669 | 44.9 | 0.106 | 1.31
(1.00,1.73) | 0.054 | 60.5 | 0.027 | 1.48
(1.08,2.03) | 0.014 |
| No | 1 | 87/108 |
|
| 1.07
(0.71,1.61) | 0.757 |
|
| 1.69
(0.69,4.12) | 0.252 |
|
| 3.16
(1.31,7.63) | 0.01 |
|
| 2.35
(1.02,5.38) | 0.044 |
|
| 0.72
(0.40,1.28) | 0.264 |
When stratified by ethnicity, the ACE polymorphism
showed a significant contribution to IA risk in the Asian
population in all genetic models except in the recessive model (II
vs. ID+DD; OR=1.15, 95% CI=0.71–1.87; POR=0.561).
In addition, a significant strong association was detected under
the allele model (I vs. D; OR=1.21, 95% CI=1.06–1.40;
POR=0.006) and homozygote comparison of
codominant model (II vs. DD; OR=1.44, 95% CI=1.09–1.90;
POR=0.011) in the Caucasian population (Table II; Fig.
2A-D).
In the subgroup analysis for the HB group, a
significantly increased risk in all genetic models was observed
except for in the recessive model (II vs. ID+DD; OR=1.15, 95%
CI=0.83–1.59; POR=0.401). Similar findings were
revealed in the allele model (I vs. D; OR=1.28, 95% CI=1.08–1.52;
POR=0.004) and homozygote comparison of
codominant model (II vs. DD; OR=1.56, 95% CI=1.12–2.17;
POR=0.009) in the PB subgroup (Table II).
When stratified by sample size, a statistically
significant association was found in the large group in all of the
genetic models. However, only the allele and homozygote comparison
of codominant models showed significant associations in the small
subgroup (OR=1.33, 95% CI=1.05–1.68; POR=0.019
and OR=1.93, 95% CI=1.19–3.13; POR=0.007,
respectively) (Table II).
Heterogeneity analysis
Significant heterogeneity in the ACE polymorphism
was observed in the heterozygote comparison of codominant model
(I2=76.7%; Ph=0.000), dominant model
(I2=44.5%; Ph=0.094) and recessive model
(I2=66.1%; Ph=0.007). To investigate the
potential sources of heterogeneity across studies, the pooled ORs
under all comparisons were assessed via subgroup and sensitivity
analyses. In the subgroup analysis by ethnicity, the heterogeneity
of ACE was significant in the Caucasian studies in the heterozygote
comparison of the codominant and recessive models
(I2=80.2%; Ph=0.002 and I2=74.9%;
Ph=0.008, respectively). When stratified by source of
control, the heterogeneity of ACE in the population-based studies
was significant in the heterozygote comparison of codominant and
recessive models (I2=86.5% and Ph=0.001;
I2=79% and Ph=0.009, respectively). When
stratified by sample size, the heterogeneity of ACE in the small
subgroup studies was significant in the heterozygote comparison of
the codominant, dominant and recessive models (I2=91.4%
and Ph=0.000; I2=74% and Ph=0.021; and
I2=86.9% and Ph=0.000, respectively) (Table II).
Notably, when the Caucasian population-based control
study with <300 cases by Slowik et al (20) was excluded, the heterogeneity
significantly decreased in the heterozygote comparison of
codominant and recessive models (Table
III). Therefore, the study by Slowik et al (20) may have contributed to the substantial
heterogeneity of the ACE polymorphism.
| Table III.Sensitivity analysis of the
angiotensin-converting enzyme polymorphism on IA risk. |
Table III.
Sensitivity analysis of the
angiotensin-converting enzyme polymorphism on IA risk.
|
|
|
|
|
| Codominant
model |
|
|
|
|
|
|
|
|
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|---|
|
| Allele I vs. D | Homozygote II vs.
DD | Heterozygote ID vs.
DD | Dominant model
(II+ID) vs. DD | Recessive model II
vs. (ID+DD) |
|
|---|
|
|
|
|
|
|
|
|
|---|
| Author, year | I2
(%) |
Ph | ORs (95%CIs) |
POR | I2
(%) |
Ph | ORs (95%CIs) |
POR | I2
(%) |
Ph | ORs (95%CIs) |
POR | I2
(%) |
Ph | ORs (95%CIs) |
POR | I2
(%) |
Ph | ORs (95%CIs) |
POR | (Refs.) |
|---|
| Liu, 2013 |
|
| 0 | 0.561 | 1.21
(1.07,1.37) | 0.003 | 0 | 0.697 | 1.52
(1.18,1.97) | 0.001 | 79 | 0.000 | 1.18
(0.69,2.04) | 0.542 | 40.5 | 0.135 | 1.27
(1.03,1.57) | 0.028 | 70.2 | 0.005 | 1.31
(0.90,1.91) | 0.163 | (17) |
| Staalsø, 2011 |
|
| 0 | 0.486 | 1.32
(1.16,1.50) | 0.000 | 0 | 0.713 | 1.78
(1.37,2.31) | 0.000 | 80.4 | 0.000 | 1.28
(0.72,2.26) | 0.398 | 49.9 | 0.076 | 1.44
(1.04,2.00) | 0.028 | 69.7 | 0.006 | 1.41
(0.97,2.06) | 0.075 | (18) |
| Pannu, 2005 |
|
| 9.2 | 0.357 | 1.29
(1.14,1.46) | 0.000 | 0 | 0.587 | 1.72
(1.34,2.20) | 0.000 | 80.5 | 0.000 | 1.27
(0.74,2.20) | 0.387 | 51.5 | 0.067 | 1.43
(1.04,1.97) | 0.028 | 70.8 | 0.004 | 1.40
(0.97,2.02) | 0.072 | (19) |
| Slowik, 2004 |
|
| 2.1 | 0.403 | 1.24
(1.10,1.40) | 0.000 | 0 | 0.457 | 1.62
(1.27,2.08) | 0.000 | 29.2 | 0.216 | 1.42
(1.15,1.76) | 0.001 | 18.2 | 0.296 | 1.47
(1.20,1.80) | 0.000 | 13.8 | 0.326 | 1.21
(1.00,1.47) | 0.046 | (20) |
| Keramatipour,
2000 |
|
| 21.7 | 0.271 | 1.26
(1.10,1.43) | 0.001 | 0 | 0.449 | 1.63
(1.25,2.13) | 0.000 | 80.5 | 0.000 | 1.24
(0.70,2.20) | 0.459 | 53.5 | 0.057 | 1.38
(0.98,1.94) | 0.067 | 71.7 | 0.003 | 1.35
(0.90,2.01) | 0.142 | (21) |
| Takenaka, 1998 |
|
| 20.8 | 0.277 | 1.26
(1.12,1.42) | 0.000 | 0 | 0.588 | 1.59
(1.26,2.02) | 0.000 | 78.7 | 0.000 | 1.14
(0.71,1.84) | 0.576 | 43.6 | 0.114 | 1.33
(1.10,1.62) | 0.004 | 71.4 | 0.004 | 1.38
(0.95,1.99) | 0.086 | (22) |
| Yu, 2005 |
|
| 12.6 | 0.334 | 1.28
(1.14,1.45) | 0.000 | 0 | 0.449 | 1.64
(1.29,2.08) | 0.000 | 76.4 | 0.001 | 1.11
(0.70,1.75) | 0.669 | 44.9 | 0.106 | 1.33
(1.09,1.61) | 0.005 | 60.5 | 0.027 | 1.48
(1.08,2.03) | 0.014 | (23) |
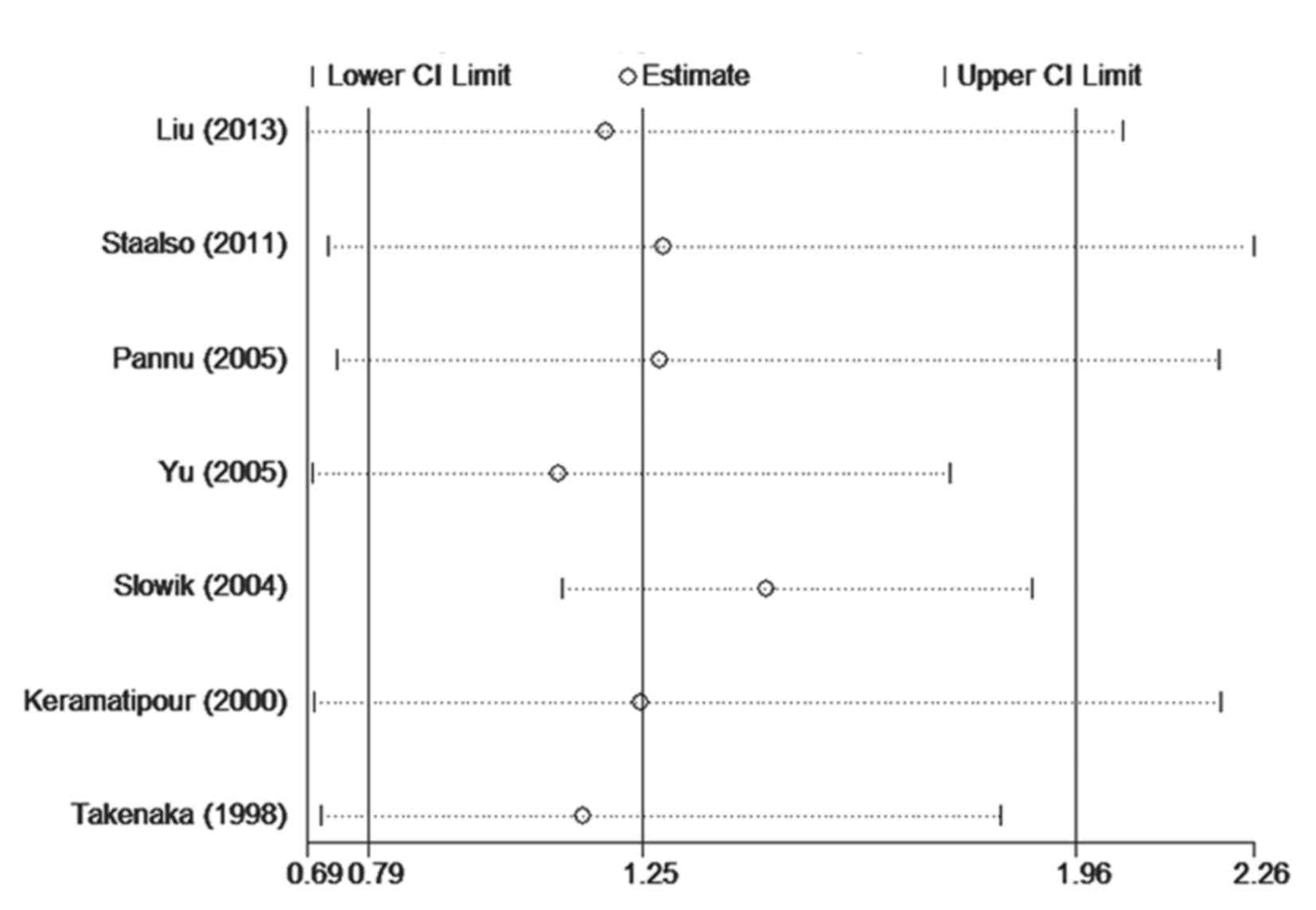
Sensitivity analyses
Sensitivity analyses were performed by sequential
deletion of single studies for all subjects to determine the
influence of its individual data sets to the pooled OR, and the
corresponding pooled ORs were not materially altered in the allele
and homozygote comparison of the codominant models. The pooled ORs
ranged from 1.11 to 1.42 in the heterozygote comparison of
codominant model, 1.27 to 1.47 in the dominant model, and 1.21 to
1.48 in the recessive model (Table
III). When excluding the study by Slowik et al (20), a significantly increased risk was
identified with the ACE polymorphism in the heterozygous comparison
of the codominant (OR=1.42, 95% CI=1.15–1.76; POR=0.001) and
recessive (OR=1.21, 95% CI=1.00–1.47; POR=0.046) models
(Table III; Fig. 3A and B). In addition, a significantly
increased risk was detected after excluding the study by Yu et
al (23) in the recessive model
(OR=1.48, 95% CI=1.08–2.03; POR=0.01) (Table III and Fig.
3B). Furthermore, a significantly decreased risk was observed
after excluding the study by Keramatipour et al (21) in the dominant model (OR=1.38, 95%
CI=0.98–1.94; POR=0.067) (Table
III and Fig. 3C). Thus, the
overall association between the ACE polymorphism and IA risk was
significantly influenced by these three studies.
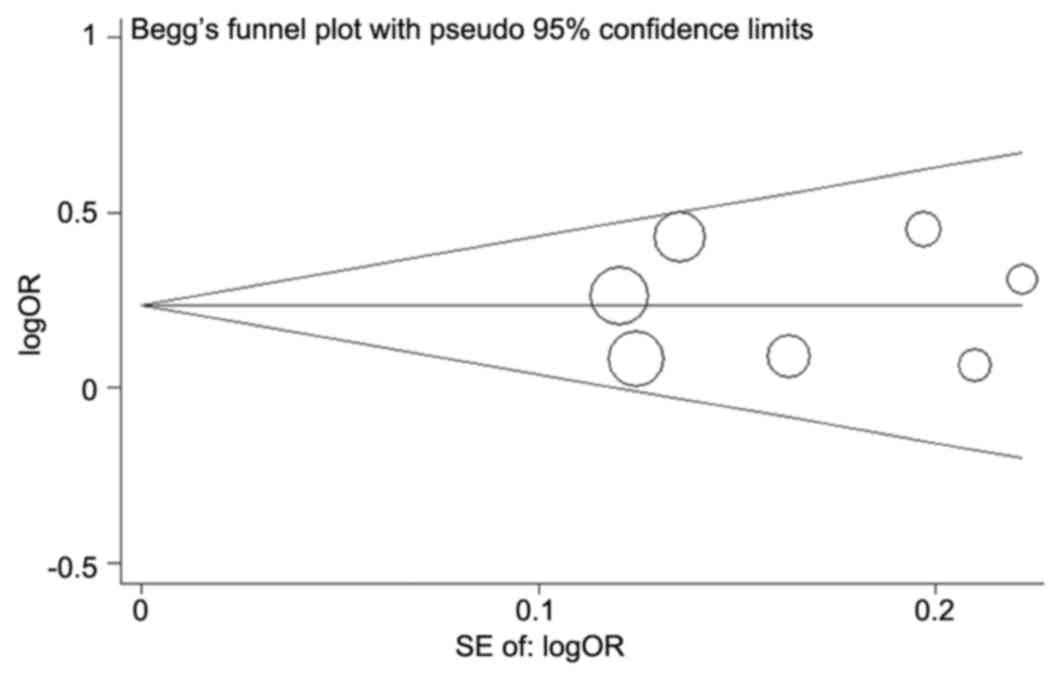
Publication bias
Begg's funnel plot and Egger's test were performed
to access the potential publication bias of the literature. The
funnel plot shapes of these polymorphisms were symmetrical in each
genetic model (Fig. 4 for the allele
model). Subsequently, the Egger's test was used to provide
statistical evidence of funnel plot symmetry. The results indicated
that the current meta-analysis demonstrated a lack of publication
bias in all of the genetic models (I vs. D: P=0.865; II vs. DD:
P=0.542; ID vs. DD: P=0.945; II+ID vs. DD: P=0.520; II vs. ID+DD:
P=0.956. data not shown). The calculation results are consistent
with the shape of the Begg's funnel plot.
Discussion
IA is a growing problem and leads to devastating
consequences, such as SAH, hemiplegia and epilepsy. Rupture of IA
is the most common cause of SAH, which is associated with
particularly high mortality and morbidity rates (3). Thus, it is important to identify the
characteristics of individuals who are at high risk of IA and focus
on regularly screening for IA. Previous studies indicate that
genetics contribute to the development of IA (27–29).
Furthermore, it is of great value to investigate the genetic
architecture and establish the underlying molecular mechanism of
IA. Until now, various polymorphisms in candidate genes have been
considered as an important risk contributor to the development of
IA.
Hypertension, which is a polygenic and
multifactorial disorder, has been demonstrated to be a risk factor
for IA (30). The RAAS plays a
critical role in the development of hypertension and CVD (31). ACE is a key circulating enzyme in the
RAAS, which catalyzes the conversion of angiotensin I to
angiotensin II, and further degrades bradykinin (14). The D allele of the I/D polymorphism of
a 287-bp Alu sequence within intron 16 of the ACE gene has
been reported to be correlated with increased circulating ACE
levels in humans (15,16). Furthermore, the ACE I/D polymorphisms
have been extensively evaluated in numerous types of vascular
disease (32). Therefore, ACE may
present as a candidate gene for IA development.
A meta-analysis of two case-control studies by
Keramatipour et al (21) showed
that the I allele of the ACE gene maybe a risk factor for
IA. Subsequently, various studies with conflicting opinions have
been reported (18,19,23). A
previous meta-analysis failed to detect an association between the
ACE I/D polymorphism and IA susceptibility in the dominant
(OR=1.23, 95% CI=0.82–1.85; POR=0.31) or the
recessive (OR=1.58, 95% CI=0.98–2.57; POR=0.06)
models (11). By contrast, another
meta-analysis demonstrated a close association between the ACE I/D
polymorphism and IA risk. In the current study, a comprehensive
meta-analysis was conducted with a markedly larger sample size to
derive a more precise estimation of the association.
By combining data from seven case-control studies,
including 1,074 IA patients and 1,500 control subjects, the present
meta-analysis evaluated the association between the I/D
polymorphisms of the ACE gene and IA susceptibility. The
pooled result showed a significant association between the I allele
of the ACE gene and IA risk (OR=1.27, 95% CI=1.13–1.42;
POR=0.000). The II genotype was associated with
an increased risk for IA when compared with the DD genotype
(OR=1.64, 95% CI=1.30–2.07; POR=0.000). The ACEII
+ ID genotype was correlated with a significantly increased risk
for IA (OR=1.38, 95% CI=1.05–1.80;
POR=0.021).
In the subgroup analysis by ethnicity, an increased
risk between ACE and IA was identified among Asian individuals (ID
vs. DD: OR=2.02, 95%CI=1.35–3.03, POR=0.001;
II+ID vs. DD: OR=2.01, 95%CI=1.41–2.86,
POR=0.000), although not in Caucasian individuals
(ID vs. DD: OR=0.86, 95%CI=0.49–1.53, POR=0.615;
II+ID vs. DD: OR=1.15, 95%CI=0.90–1.47,
POR=0.264). A number of factors may also be
involved in the underlying mechanism for this difference between
ethnic groups. In addition to the genetic backgrounds and the
living environments, other factors, such as selection bias and
different matching criteria may account for the different genetic
effects. Thus, additional studies using different populations are
warranted to further validate ethnic differences on the impact of
the ACE polymorphism on IA risk.
Subgroup analysis by source of controls revealed a
significantly increased risk among studies using hospital-based
controls, but not population-based controls in the heterozygote
comparison of codominant model and the dominant model, suggesting
that the controls in the hospital-based studies may be sufficient
to represent the general population. Thus, the use of proper and
representative cancer-free control subjects is important in
reducing bias in such genotype association studies.
Subgroup analysis by sample size revealed a
significantly increased risk among studies using large samples in
each genetic model, although no significance was found in the small
sample subgroup in the heterozygote comparison of codominant,
dominant and recessive models, suggesting that studies with large
samples are required.
The statistical significance of genotype
distributions was also detected in male and female groups (males:
OR=3.56, 95% CI=1.43–8.86; POR=0.0006; females:
OR=3.86, 95% CI=1.75–8.51; POR=0.0005) in the
study by Slowik et al (20). In
addition, Liu et al (17)
reported that no statistically significant differences were
identified between genotypes in patients with IA, when stratified
by the site, shape, size and Fisher Grade of aneurysms (17). In future studies, greater focus on
clinical characteristics, such as gender, alcohol consumption,
smoking, family history, and site, shape, size and Fisher Grade of
IA should be taken into consideration to provide a more powerful
analytical framework.
There were certain limitations of the current
meta-analysis. First, the number of eligible studies and subjects
of studies were relatively small, particularly for the subgroup
analyses, which may result in insufficient power to detect a
slight, although real effect of the ACE polymorphisms on IA risk.
In addition, the study by Yu et al (23) whose genotype distribution in the
control group was not consistent with HWE may contribute to the
bias of the meta-analysis, as the results were affected after
excluding this study in the sensitivity analysis. Finally, the
results were based on single-factor estimates without adjustments
for other risk factors.
In conclusion, this meta-analysis identified that
the ACE polymorphism is associated with an increased risk of IA.
However, large, well-designed multicenter studies are required to
verify the present findings. In addition, further evaluation of the
ACE polymorphism on IA risk should focus on the effect of
gene-to-gene and gene-to-environment interactions.
Acknowledgements
The present study was supported by the National
Science Foundation of China (grant nos. 81571141 and 81401802).
References
|
1
|
Rinkel GJ, Djibuti M, Algra A and van Gijn
J: Prevalence and risk of rupture of intracranial aneurysms: A
systematic review. Stroke. 29:251–256. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Korja M and Kaprio J: Controversies in
epidemiology of intracranial aneurysms and SAH. Nat Rev Neurol.
12:50–55. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hop JW, Rinkel GJ, Algra A and van Gijn J:
Case-fatality rates and functional outcome after subarachnoid
hemorrhage: A systematic review. Stroke. 28:660–664. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yan J, Hitomi T, Takenaka K, Kato M,
Kobayashi H, Okuda H, Harada KH and Koizumi A: Genetic study of
intracranial aneurysms. Stroke. 46:620–626. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tromp G, Weinsheimer S, Ronkainen A and
Kuivaniemi H: Molecular basis and genetic predisposition to
intracranial aneurysm. Ann Med. 46:597–606. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rinkel GJ: Intracranial aneurysm
screening: Indications and advice for practice. Lancet Neurol.
4:122–128. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schievink WI: Genetics of intracranial
aneurysms. Neurosurgery. 40:651–663. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ronkainen A, Hernesniemi J, Puranen M,
Niemitukia L, Vanninen R, Ryynänen M, Kuivaniemi H and Tromp G:
Familial intracranial aneurysms. Lancet. 349:380–384. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Akiyama K, Narita A, Nakaoka H, Cui T,
Takahashi T, Yasuno K, Tajima A, Krischek B, Yamamoto K, Kasuya H,
et al: Genome-wide association study to identify genetic variants
present in Japanese patients harboring intracranial aneurysms. J
Hum Genet. 55:656–661. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Marchese E, Vignati A, Albanese A, Nucci
CG, Sabatino G, Tirpakova B, Lofrese G, Zelano G and Maira G:
Comparative evaluation of genome-wide gene expression profiles in
ruptured and unruptured human intracranial aneurysms. J Biol Regul
Homeost Agents. 24:185–195. 2010.PubMed/NCBI
|
|
11
|
McColgan P, Thant KZ and Sharma P: The
genetics of sporadic ruptured and unruptured intracranial
aneurysms: A genetic meta-analysis of 8 genes and 13 polymorphisms
in approximately 20,000 individuals. J Neurosurg. 112:714–721.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ruigrok YM and Rinkel GJ: From GWAS to the
clinic: Risk factors for intracranial aneurysms. Genome Med.
2:612010. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mohan D, Munteanu V, Coman T and Ciurea
AV: Genetic factors involves in intracranial aneurysms-actualities.
J Med Life. 8:336–341. 2015.PubMed/NCBI
|
|
14
|
Skeggs LT, Dorer FE, Kahn JR, Lentz KE and
Levine M: The biochemistry of the renin-angiotensin system and its
role in hypertension. Am J Med. 60:737–748. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rigat B, Hubert C, Alhenc-Gelas F, Cambien
F, Corvol P and Soubrier F: An insertion/deletion polymorphism in
the angiotensin I-converting enzyme gene accounting for half the
variance of serum enzyme levels. J Clin Invest. 86:1343–1346. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tiret L, Rigat B, Visvikis S, Breda C,
Corvol P, Cambien F and Soubrier F: Evidence, from combined
segregation and linkage analysis, that a variant of the angiotensin
I-converting enzyme (ACE) gene controls plasma ACE levels. Am J Hum
Genet. 51:197–205. 1992.PubMed/NCBI
|
|
17
|
Liu Y, Li P, Hu X, Hu Y, Sun HG, Ma WC,
Qiao F, He M and You C: Angiotensin-converting enzyme
insertion/deletion gene polymorphism and risk of intracranial
aneurysm in a Chinese population. J Int Med Res. 41:1079–1087.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Staalsø JM, Nielsen M, Edsen T, Koefoed P,
Springborg JB, Moltke FB, Laursen H, Nielsen HB and Olsen NV:
Common variants of the ACE gene and aneurysmal subarachnoid
hemorrhage in a Danish population: A case-control study. J
Neurosurg Anesthesiol. 23:304–309. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pannu H, Kim DH, Seaman CR, Van Ginhoven
G, Shete S and Milewicz DM: Lack of an association between the
angiotensin-converting enzyme insertion/deletion polymorphism and
intracranial aneurysms in a Caucasian population in the United
States. J Neurosurg. 103:92–96. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Slowik A, Borratynska A, Pera J, Betlej M,
Dziedzic T, Krzyszkowski T, Czepko R, Figlewicz DA and Szczudlik A:
II genotype of the angiotensin-converting enzyme gene increases the
risk for subarachnoid hemorrhage from ruptured aneurysm. Stroke.
35:1594–1597. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Keramatipour M, McConnell RS, Kirkpatrick
P, Tebbs S, Furlong RA and Rubinsztein DC: The ACE I allele is
associated with increased risk for ruptured intracranial aneurysms.
J Med Genet. 37:498–500. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Takenaka K, Yamakawa H, Sakai H, Yoshimura
S, Murase S, Okumura A, Nakatani K, Kimura T, Nishimura Y, Yoshimi
N and Sakai N: Angiotensin I-converting enzyme gene polymorphism in
intracranial saccular aneurysm individuals. Neurol Res. 20:607–611.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu S, Zhao J, Zhang D, Bi Z and Kang S: A
study of the relationship between polymorphism of the angiotensin
converting enzyme gene insertion/delection and intracranial
aneurysm. J Cap Univ Med Sci. 4:380–382. 2005.
|
|
24
|
Mantel N and Haenszel W: Statistical
aspects of the analysis of data from retrospective studies of
disease. J Natl Cancer Inst. 22:719–748. 1959.PubMed/NCBI
|
|
25
|
DerSimonian R and Kacker R: Random-effects
model for meta-analysis of clinical trials: An update. Contemp Clin
Trials. 28:105–114. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Egger M, Smith G Davey, Schneider M and
Minder C: Bias in meta-analysis detected by a simple, graphical
test. BMJ. 315:629–634. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kojima M, Nagasawa S, Lee YE, Takeichi Y,
Tsuda E and Mabuchi N: Asymptomatic familial cerebral aneurysms.
Neurosurgery. 43:776–781. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nakagawa T and Hashi K: The incidence and
treatment of asymptomatic, unruptured cerebral aneurysms. J
Neurosurg. 80:217–223. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Caranci F, Briganti F, Cirillo L, Leonardi
M and Muto M: Epidemiology and genetics of intracranial aneurysms.
Eur J Radiol. 82:1598–1605. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Inci S and Spetzler RF: Intracranial
aneurysms and arterial hypertension: A review and hypothesis. Surg
Neurol. 53:530–542. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lavoie JL and Sigmund CD: Minireview:
Overview of the renin-angiotensin system-an endocrine and paracrine
system. Endocrinology. 144:2179–2183. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sayed-Tabatabaei FA, Oostra BA, Isaacs A,
van Duijn CM and Witteman JC: ACE polymorphisms. Circ Res.
98:1123–1133. 2006. View Article : Google Scholar : PubMed/NCBI
|