Introduction
New medical treatments in oncology depend upon
identifying novel prognostic markers and therapeutic targets.
Well-studied malignancies, such as breast cancer, have defined
mechanisms based on presence or absence of estrogen or progesterone
receptors, which can be used as genetic prognostic markers to
determine treatment methods (1,2). Non-small
cell lung cancer treatments have developed therapies catering
towards patients' unique genetic mutations in proto-oncogenes, such
as anaplastic lymphoma kinase or in epidermal growth factor
receptor genes (3–5). However, molecular pathways and genetic
alterations in bladder cancer need to be studied further to have
effective treatment options as well. Urinary system tumors are the
5th most common tumor type in the United States, with bladder
cancer comprising 54% of new urinary system-based tumor cases
(6). Bladder cancer mortality rates
have remained relatively unchanged over the past 25 years, while
treatment options with low efficacy are in continued use (7,8). More
biological data must be collected to develop proper therapeutic
targets and prognostic markers for bladder cancer to improve
treatment methods and patient mortality.
Identification of glycogen debranching
enzyme (AGL) as a new player in bladder cancer
To identify new genes responsible for aggressive
bladder cancer growth, an in vivo RNA interference library
screening was conducted using a whole genome shRNA library
(9). This screen identified
amylo-α-1,6-glucosidase,4-α-glucanotransferase (AGL) as a potential
new regulator of bladder cancer growth (9).
AGL, or glycogen debranching enzyme, is involved in
glycogen breakdown (glycogenolysis) (10). Germline mutations identified in AGL
have been associated with Cori disease, otherwise known as glycogen
storage disease III (GSD III), characterized by irregular
glycogenolysis and accumulation of abnormally branched glycogen
(limit dextrin) in the liver (10,11). The
symptomatic diagnosis of GSD III primarily includes poor liver
function and, in some cases, poor skeletal muscle function
(11). Despite the understanding of
AGL in both glycogenolysis and in GSD III, the present authors were
the first to report a connection between AGL and bladder cancer
(9,12).
Analysis of bladder tumors from patients demonstrated that loss of
AGL expression (gene and protein) in cancer has been associated
with clinically aggressive tumor prognosis and poor patient
mortality (9). These studies have
developed investigative interest in characterizing AGL loss in
bladder cancer.
AGL, glycogen metabolism and regulation of
glycine synthesis
Following identifying AGL as a potential regulator
of bladder cancer growth from the functional genomic screen, and
demonstrating that loss of AGL expression in bladder tumors leads
to poor patient outcome, the authors carried out various cell based
functional assays to validate whether AGL serves any functional
role in bladder cancer (9). Tumor
cells expressing low levels of AGL displayed enhanced anchorage
independent and dependent growth, while AGL overexpression
decreased anchorage independent and dependent growth (9). Further analysis of AGL low tumor cells
presented increased production of limit dextrin, or abnormally
branched glycogen, similar to patients with GSD III. To understand
in detail whether a connection between inhibition of glycogen
breakdown and aggressive bladder cancer growth existed, the authors
investigated the role of glycogen phosphorylase (PYG), the other
enzyme known to interplay with AGL in glycogen breakdown (9,10,12). In addition, experiments using enzymatic
null mutants of AGL were conducted to understand AGL's enzymatic
activity in bladder tumor growth (9).
Results from these experiments indicated that depletion of PYG did
not affect anchorage independent growth of bladder cancer, nor did
alteration of the AGL's enzymatic region involved in glycogenolysis
have any impact on bladder tumor growth (9). Thus, it was confirmed that neither AGL's
known enzymatic function, nor inhibition of glycogen breakdown,
serves a role in bladder cancer progression. This confirmed that
AGL plays a role in bladder cancer via an unknown mechanism.
Furthermore, low AGL expression was observed to make
bladder cancer cells dependent upon higher levels of glucose for
growth. To obtain an insight as to how glucose may be involved in
aggressive bladder cancer growth with AGL loss, a metabolomics
screen was carried out using 13C-labeled glucose and observed for
intake and distribution of glucose (9). This study indicated that low AGL
expressing cells rely upon glucose to help drive increased glycine
synthesis (9). This evidence was
supported by the amplified serine hydroxymethyltransferase 2
(SHMT2) gene, responsible for glycine synthesis (13,14). In
addition, elevated expression of SHMT2 correlates with poor patient
prognosis in bladder cancer patients with low AGL expression,
verifying the importance of glycine synthesis in AGL low bladder
cancer (9). These findings concluded
AGL's biologically relevant position as a tumor suppressor,
independent of its enzymatic function, and the potential it has for
clinical studies in bladder cancer.
AGL and hyaluronic acid synthesis
The significance of AGL in bladder cancer is clear,
but the mechanisms behind tumor growth in AGL low malignancies
require further investigation. To observe genetic differences in
AGL low bladder cancer, a transcriptional profiling of human
bladder cancer cells with and without AGL expression was performed,
searching for genes differentially regulated with loss of AGL
(15). Hyaluronic acid synthase 2
(HAS2), a gene associated with HA synthesis (16,17), was one
of the overexpressed genes with AGL loss (15). Elevated HAS2 expression is correlated
with high-grade bladder cancer and poor patient mortality, making
HAS2 a gene of interest for further study in the AGL model of
bladder cancer (15).
Hylauronic acid synthases (HAS) are responsible for
making hyaluronan, or HA (17). Known
as non-branching glycosaminoglycans, HA vary in size based on the
number of disaccharide repeats, with glucose serving as the
precursor molecule for HA synthesis (16,17). Located
in the extracellular matrix, HA can bind to cell surface receptors
designed for different sized HA (18).
HA is biologically relevant in angiogenesis, anchorage independent
growth and for allowing cell-to-cell interactions with cancer
tissue and surrounding stromal tissue (16,19). The
significance of HA in cancer biology provided a reason to further
investigate HAS2-mediated HA synthesis in AGL low bladder
cancer.
HAS2 was identified as the HAS isoform of interest,
as the expression of other isoforms, HAS1 or HAS3, were found to be
irrelevant in AGL depleted cells (15). In concordance with increased expression
of HAS2, bladder cancer cells with AGL loss presented increased
levels of HA synthesis (15). To
further validate the significance of HAS2-driven HA synthesis in
AGL low bladder cancer, genetic knockdown of the HAS2 gene and
treatment with HA synthesis inhibitor, 4-methylumbelliferone (4MU)
(20), was carried out in bladder
cancer cells with and without AGL followed by in vitro and
in vivo growth assays. Knockdown of HAS2 resulted in loss of
anchorage-independent growth, reduced proliferation and xenograft
tumor growth of bladder cancer cells without AGL expression
(15). Treatment with 4MU specifically
inhibited growth of AGL low bladder cancer cells, similar to
genetic inhibition of HAS2, in in vitro and xenograft growth
assays (15). Further analysis of
bladder cancer patient datasets demonstrated that patients with
high HAS2 and low AGL mRNA expression exhibit poor outcome, giving
credence that bladder tumors with low AGL expression are dependent
of HAS2 for aggressive growth (15).
These experiments illustrate the importance of HAS2-mediated HA
synthesis in bladder carcinoma cells with AGL loss, and provide a
potential mechanism of action to investigate for prognostic markers
or therapeutic targets.
AGL and hyaluronic acid receptors
The authors have established that aggressive growth
of low AGL expressing bladder cancer relies upon HAS2-driven HA
synthesis. However, the downstream signaling triggered by HA in AGL
low bladder tumors remains unclear. Hyaladherins, or proteins that
bind to extracellular HA, can be either cell surface receptors or
extracellular matrix proteins (21).
Two of these cell surface hyaladherins, RHAMM and CD44, are
clinically significant in various cancer models (16,19,22). These hyaladherins have been
demonstrated to provide multi-drug resistance, assist with
anchorage independent growth, and cell motility of cancer cells
(23–25). Since both RHAMM and CD44 serve a major
role in tumor progression, the authors investigated whether these
proteins were involved in HA driven rapid growth of bladder cancer
cells with AGL loss.
RHAMM and CD44 expressed by cancer cells can be
changed by manipulating the amount of HA the cancer cells are
exposed to (26,27). This has been used as a measure of HA
signaling by these receptors. Analysis of CD44 and RHAMM expression
in AGL low bladder cancer cells indicated that neither knockdown of
HAS2, nor treatment with HA synthesis inhibitor 4MU, was able to
reduce expression of CD44 or RHAMM (28). The addition of superfluous HA also made
little change to RHAMM and CD44 expression (28). This suggested that changes in HA levels
do not affect RHAMM or CD44 expression in AGL low bladder tumors.
However, it was shown that genetic inhibition of HAS2-, CD44- or
RHAMM-induced apoptosis in AGL low bladder cancer cells, where as
they had minimal effect on apoptosis of bladder cancer cells
expressing AGL (28). Apoptosis was
measured by analyzing caspase signaling and by TUNEL assays
analyzing the number of cells undergoing apoptosis (28). The percentage of AGL low bladder cancer
cells undergoing apoptosis with knockdown of HAS2, CD44 or RHAMM
was similar, suggesting they are part of the same pathway.
Moreover, loss of CD44 or RHAMM reduced expression of HAS2 and HA
synthesis in AGL low bladder cancer cells, validating that CD44 and
RHAMM interaction with HA and signaling is relevant in AGL low
bladder tumors (28). Interestingly, a
bladder cancer cell line with AGL loss is either dependent on loss
of CD44 (UMUC3) or RHAMM (T24T, MGHU4) for induction of apoptosis
(28). The reason for cell line based
preference for one receptor over the other is unknown. Loss of CD44
and RHAMM predominantly inhibited anchorage dependent and
independent growth of AGL low bladder cancer cells as also reported
with loss of HAS2 (28). The current
research clearly demonstrates that AGL loss promotes aggressive
growth of bladder cancer through downstream signaling of
hyaladherins CD44 and RHAMM driven by HAS2-mediated HA
synthesis.
Analysis of CD44 and RHAMM mRNA expression as a
prognostic marker for bladder cancer patients demonstrated that
RHAMM alone and in combination with AGL is prognostic of poor
bladder cancer patient outcome, while CD44 mRNA was not prognostic
in the bladder cancer patient datasets analyzed by us (28). Bladder cancer patients with high RHAMM
and low AGL mRNA expression were demonstrated to present poor
outcome compared to other groups (28). Thus, bladder cancer patients with low
tumor AGL expression and high RHAMM expression can be subjected to
personalized treatment with inhibition of HA synthesis or HA
signaling driven by RHAMM.
Conclusion
Through extensive experimentation, the authors have
reported that the loss of AGL results in aggressive growth of
bladder cancer cells and AGL mRNA and protein expression serves as
a prognostic marker for bladder cancer patients (9). In addition, the loss of AGL, SHMT2-driven
glycine synthesis and HAS2-driven HA synthesis serves a major role
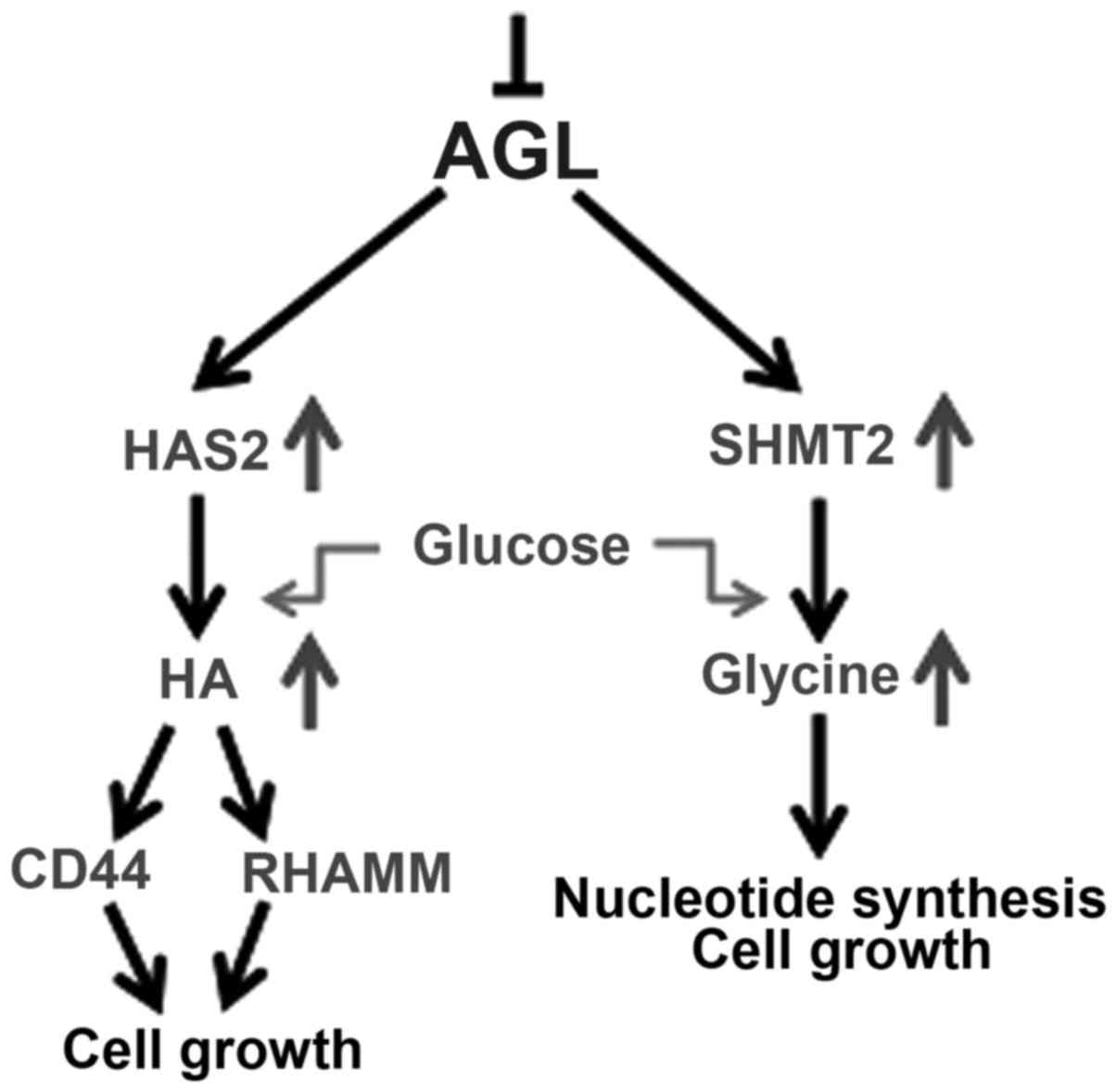
in driving bladder tumor growth (9,15,28). It has been concluded that glucose is a
major driver of glycine synthesis with AGL loss (9), and the authors have unpublished data
indicating that glucose is also the major driver of HA synthesis in
AGL low bladder cancer cells. Thus, it is believed that the
increase in SHMT2 and HAS2 expression with AGL loss drives
increased glucose uptake by cells to make more glycine and HA which
promotes rapid growth of these cells (Fig.
1).
Very little is known about AGL biology, other than
its involvement in glycogen metabolism. It is unknown how HAS2 and
SHMT2 levels increase as a response to low AGL expression. AGL
could interact with these genes directly, or interact with
intermediary biomolecules, which results in overexpression of SHMT2
and HAS2 with loss of AGL in bladder cancer cells. Experiments need
to be carried out to understand how AGL regulates the expression of
SHMT2, HAS2 and other genes or pathways that influence tumor
growth. It is important to investigate and understand which
biomolecules interact directly with AGL and are responsible for
regulating tumor growth. Understanding how AGL functions as a tumor
suppressor, and the different mechanisms of action by which it
regulates aggressive bladder cancer growth, would be beneficial in
the future for identification of better therapeutic options based
of AGL expression status.
Acknowledgements
The present study was supported by the Bladder
Cancer Advocacy Network (BCAN) Young Investigator Award to S.G.,
and by the Gundersen Medical Foundation.
References
|
1
|
Bauer KR, Brown M, Cress RD, Parise CA and
Caggiano V: Descriptive analysis of estrogen receptor
(ER)-negative, progesterone receptor (PR)-negative, and
HER2-negative invasive breast cancer, the so-called triple-negative
phenotype: A population-based study from the California cancer
Registry. Cancer. 109:1721–1728. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ariazi EA, Ariazi JL, Cordera F and Jordan
VC: Estrogen receptors as therapeutic targets in breast cancer.
Curr Top Med Chem. 6:181–202. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Waqar SN and Morgensztern D: Precision
medicine in lung cancer: The battle continues. J Thorac Dis.
8:2991–2993. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa
K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, et al:
PROFILE 1014 Investigators: First-line crizotinib versus
chemotherapy in ALK-positive lung cancer. N Engl J Med.
371:2167–2177. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhuo C, Li X, Zhuang H, Tian S, Cui H,
Jiang R, Liu C, Tao R and Lin X: Evaluating the efficacy and safety
of intravesical chemotherapies for non-muscle invasive bladder
cancer: A network meta-analysis. Oncotarget. 7:82567–82579.
2016.PubMed/NCBI
|
|
8
|
National Cancer Institute, . A Snapshot of
Bladder Cancer. https://www.cancer.gov/research/progress/snapshots/bladderNovember
5–2014
|
|
9
|
Guin S, Pollard C, Ru Y, Lew C Ritterson,
Duex JE, Dancik G, Owens C, Spencer A, Knight S and Holemon H: Role
in tumor growth of a glycogen debranching enzyme lost in glycogen
storage disease. J Natl Cancer Inst. 106:pii: dju0622014.
View Article : Google Scholar
|
|
10
|
Adeva-Andany MM, González-Lucán M,
Donapetry-García C, Fernández-Fernández C and Ameneiros-Rodríguez
E: Glycogen metabolism in humans. BBA Clin. 5:85–100. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dagli A, Sentner CP and Weinstein DA:
Glycogen Storage Disease Type IIIGeneReviews®
[Internet]. Pagon RA, Adam MP, Ardinger HH, et al: University of
Washington; Seattle, WA: pp. 1993–2017
|
|
12
|
Lew C Ritterson, Guin S and Theodorescu D:
Targeting glycogen metabolism in bladder cancer. Nat Rev Urol.
12:383–391. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hebbring SJ, Chai Y, Ji Y, Abo RP, Jenkins
GD, Fridley B, Zhang J, Eckloff BW, Wieben ED and Weinshilboum RM:
Serine hydroxymethyltransferase 1 and 2: Gene sequence variation
and functional genomic characterization. J Neurochem. 120:881–890.
2012.PubMed/NCBI
|
|
14
|
Jain M, Nilsson R, Sharma S, Madhusudhan
N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB and Mootha
VK: Metabolite profiling identifies a key role for glycine in rapid
cancer cell proliferation. Science. 336:1040–1044. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guin S, Ru Y, Agarwal N, Lew CR, Owens C,
Comi GP and Theodorescu D: Loss of Glycogen Debranching Enzyme AGL
Drives Bladder Tumor Growth via Induction of Hyaluronic Acid
Synthesis. Clin Cancer Res. 22:1274–1283. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Karbownik MS and Nowak JZ: Hyaluronan:
Towards novel anti-cancer therapeutics. Pharmacol Rep.
65:1056–1074. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tammi RH, Passi AG, Rilla K, Karousou E,
Vigetti D, Makkonen K and Tammi MI: Transcriptional and
post-translational regulation of hyaluronan synthesis. FEBS J.
278:1419–1428. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang A, de la Motte C, Lauer M and Hascall
V: Hyaluronan matrices in pathobiological processes. FEBS J.
278:1412–1418. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lokeshwar VB, Mirza S and Jordan A:
Targeting hyaluronic acid family for cancer chemoprevention and
therapy. Adv Cancer Res. 123:35–65. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nagy N, Kuipers HF, Frymoyer AR, Ishak HD,
Bollyky JB, Wight TN and Bollyky PL: 4-methylumbelliferone
treatment and hyaluronan inhibition as a therapeutic strategy in
inflammation, autoimmunity, and cancer. Front Immunol. 6:1232015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nikitovic D, Kouvidi K, Kavasi RM,
Berdiaki A and Tzanakakis GN: Hyaluronan/Hyaladherins - a Promising
Axis for Targeted Drug Delivery in Cancer. Curr Drug Deliv.
13:500–511. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dicker KT, Gurski LA, Pradhan-Bhatt S,
Witt RL, Farach-Carson MC and Jia X: Hyaluronan: A simple
polysaccharide with diverse biological functions. Acta Biomater.
10:1558–1570. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Orian-Rousseau V: CD44 Acts as a Signaling
Platform Controlling Tumor Progression and Metastasis. Front
Immunol. 6:1542015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Misra S, Hascall VC, Markwald RR and
Ghatak S: Interactions between Hyaluronan and Its Receptors (CD44,
RHAMM) Regulate the Activities of Inflammation and Cancer. Front
Immunol. 6:2012015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maxwell CA, McCarthy J and Turley E:
Cell-surface and mitotic-spindle RHAMM: Moonlighting or dual
oncogenic functions? J Cell Sci. 121:925–932. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lokeshwar VB, Lopez LE, Munoz D, Chi A,
Shirodkar SP, Lokeshwar SD, Escudero DO, Dhir N and Altman N:
Antitumor activity of hyaluronic acid synthesis inhibitor
4-methylumbelliferone in prostate cancer cells. Cancer Res.
70:2613–2623. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yates TJ, Lopez LE, Lokeshwar SD, Ortiz N,
Kallifatidis G, Jordan A, Hoye K, Altman N and Lokeshwar VB:
Dietary supplement 4-methylumbelliferone: an effective
chemopreventive and therapeutic agent for prostate cancer. J Natl
Cancer Inst. 107:djv0852015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Oldenburg D, Ru Y, Weinhaus B, Cash S,
Theodorescu D and Guin S: CD44 and RHAMM are essential for rapid
growth of bladder cancer driven by loss of Glycogen Debranching
Enzyme (AGL). BMC Cancer. 16:7132016. View Article : Google Scholar : PubMed/NCBI
|