Introduction
Morphine has been a widely used medicine in clinics
for a long time as an effective treatment for diarrhea and pain.
Several lines of evidence demonstrated that morphine is
neuroprotective. Involvement of opioids, currently used in severe
pain treatments, and in neurodegeneration/neuroprotection
mechanisms, is an important field of study. Previous findings have
shown that opioid receptors are involved in neuroprotection
(1,2).
In rat neonatal hypoxia-ischemia model, morphine
application immediately after hypoxia decreased the infarct volume
in the brain (1). In zebra fish
embryos, morphine at certain concentrations, enhances neuron
proliferation and the number of certain neuronal populations and
also protects against glutamate damage in motor neurons and
Pax-6-positive neurons in vivo (3). In addition, morphine is protective
against microglia-mediated lipopolysaccharide- or
1-methyl-4-phenylpyridinium-induced dopaminergic neurotoxicity in
rat primary mesencephalic neuron/glia cultures (4). In rat neuronal/glial cultures, morphine
was reported to prevent cell death induced by HIV envelope
glycoprotein gp120IIIB or BaL (5).
Exogenous morphine pre-incubation improves the population of spike
amplitudes of evoked field potentials, indicating that
pre-conditioning with morphine may be neuroprotective (6). In addition, morphine pre-conditioning
induces opioid receptor-dependent neuroprotection against ischemia
in rats and reduces ischemia-induced cell death in the CA1 regions
of hippocampal slices (2). In mouse
hippocampus slices with oxygen-glucose deprivation, morphine
pre-conditioning improves the neuronal cell survival rate through
protein kinase C (7). However, many
investigations showed that morphine could protect against neuronal
cell death, there are several lines of evidence suggesting that
morphine induces apoptosis in neurons. The long-term effect of
morphine on cerebral neurons indicates that use of opioids may
induce the structural alteration of neurons. On the other hand,
morphine is associated with increased metabolism and oxidative
damage to cells by reducing the intracellular dopamine level which
leads to neuronal death (8). Previous
findings have also shown that repeated application of morphine is
neurotoxic for the neuronal system and spinal cord and triggers
apoptosis (9). In addition, it was
reported that morphine may cause neuronal apoptosis by altering the
expression of Fas, Bcl-2 and caspase-3 (10). Therefore, because improved
understanding of morphine neuroprotection and neurotoxicity may be
useful to control morphine side-effects in medical applications and
to identify new targets for potential therapies and prevention
strategies to opioid addiction, the present study was conducted to
investigate the pre- and post-conditioning effects of morphine on
hippocampal cell apoptosis in a rat model of homocysteine
(Hcy)-induced oxidative stress.
Materials and methods
Drugs and biochemical reagents
D-L-homocysteine, morphine, butylhydroxytoluene
(BHT), 2-thiobarbituric acid (TBA), 1.1.3.3-tetramethoxypropan
(99%), nitro blue diformazan (NBD), nitro blue tetrazolium (NBT),
trichloroacetic acid (TCA), butanol and ethyloleate, hematoxylin,
eosin, sodium pentobarbital (USP), absolute ethanol, xylene and
formaldehyde were obtained from Sigma-Aldrich, Darmstadt, Germany.
Ketamine and xylazine were also purchased from Alfasan Co.
(Woerden, The Netherlands). Hcy powder was dissolved in
hydrochloric acid (1 M) and dilution was performed using PBS
(Sigma-Aldrich). The regulation of solution pH at 7.4 was carried
out by the addition of 0.1 M NaOH. Hcy solutions were freshly
prepared at a concentration of 0.5 µmol/µl. Morphine was dissolved
in distilled water at a concentration of 10 mg/ml.
Animals
Twenty-one adult male Wistar rats weighing between
220 and 250 g were taken from the animal house. The animals were
kept under controlled temperature, humidity, and lighting (12 h
light/dark cycle) conditions and had free access to food and water.
All experiments were performed according to the National Institutes
of Health Guidelines and were approved by the Ethics Committee of
Babol University of Medical Sciences (Babol, Iran). Seven rats in
the sham group were injected with PBS intrahippocampally. In the
Hcy group, 1 µl Hcy (0.5 µmol/µl) was injected intrahippocampally
and saline was injected (i.p.) 5 days before and after Hcy
injection. In the Hcy-morphine group, morphine (10 mg/kg) was
injected (i.p.) 5 days before and after Hcy injection.
Intrahippocampus injection
(stereotaxic surgery)
After anaesthetization with Ketamine-Xylosin (10
mg/kg i.p.), the rats were placed in a stereotaxic frame and the
skull of the rats was orientated according to Paxinos and Watson
stereotaxic atlas (11). After a
sagittal incision, the bregma suture was located and holes were
drilled with an electrical drill at the following co-ordinates; 3.3
mm posterior to bregma, 2.6 mm lateral to the sagittal suture and
3.6 mm ventral. Care was taken not to damage the meninges. A
Hamilton syringe with a cannula of a diameter of 0.3 mm was used to
inject 1 µl of 0.5 µmol/µl Hcy solution or its vehicle (PBS). The
injection was carried out in the left and right dorsal hippocampus
at a rate of 1 µl per 2 min. The cannula was left in situ
for a further 5 min following Hcy injection to allow passive
diffusion from the cannula tip and to minimize spread into the
injection tract. The cannula was then slowly removed and the scalp
was then closed with sutures. The animals were kept warm until
recovery from the anesthesia.
Hippocampus removal
Five days after Hcy injection, the rats were
sacrificed and the brain was exposed by making an incision through
the bone on either side of the parietal suture, from the foramen
magnum to near the orbit. The calvarium was removed, exposing the
brain; the left and right hippocampus were removed carefully,
immediately inserted in PBS solution (0.1 M) and stored at −70°C
for the use in biochemical or histopathological analysis.
Hippocampus histopathological
analysis
Hippocampal tissues were removed and fixed in 10%
neutral-buffered formaldehyde for 24 h, embedded in paraffin and
cut into 3–4 µm sections by a microtome (Leica SM2000R; Leica
Biosystems, Nussloch, Germany). The tissue sections were
deparaffinised in xylene. The slides were stained with hematoxylin
and eosin (H&E) according to the procedure in Wilson and Gamble
(2002), and viewed under a light microscope (Labomed, Los Angeles,
CA, USA) for the structure and morphology of cells. Microscopic
images were obtained by a CCD camera and Digipro software.
Assay of malondialdehyde (MDA) in
hippocampal homogenate
On the day of experiment, left and right hippocampus
of rat were weighed (estimate 0.7 g), and homogenized (10% w/v) in
0.1 M PBS with Polytron homogenizer at pH 7.4. Homogenates were
used immediately for detection of the biomarkers of lipid
peroxidation. Lipid peroxidation was determined according to the
modified method (11). Hippocampal
homogenates (1 ml) were incubated at 37°C in an oscillating water
bath for 1 h. At the end of the incubation period, 0.5 ml of BHT
(0.5 mg/ml in absolute ethanol) and 1 ml of TCA (25%) were added.
The tubes were sealed and heated for 10 min in a boiling water bath
to release MDA (the end product of lipid peroxidation) from
proteins. To avoid adsorption of MDA to insoluble proteins, the
samples were cooled to 4°C and centrifuged at 2,000 × g for 20 min.
Following centrifugation, 2 ml of the protein-free supernatant was
removed from each tube and 0.5 ml of TBA (0.33%) was added to this
fraction. All the tubes were heated for 1 h at 95°C in a water
bath. After cooling, the TBA-MDA complexes were extracted with 2 ml
butanol. The light absorbance was red at 532 nm on UV/visible
spectrophotometer and MDA levels were determined from standard
curve generated from 1,1,1,3 tetramethoxy propan. The results were
represented as nmol/mg wet tissue.
Assay of superoxide anion (SOA) in
hippocampal homogenate
For detection of SOA the assay procedure was a
modification of the method described by Das et al (12). Hippocampal homogenate (1 ml) was
incubated with 0.4 ml of NBT (0.1%) in an oscillating water bath
for 1 h at 37°C. Termination of the assay and extraction of the
reduced NBT was carried out by centrifuging the samples for 10 min
at 2,000 × g followed by resuspension of the pellets with 2 ml of
glacial acetic acid. The absorbance was measured at 560 nm on a
spectrophotometer and converted to micromoles of Diformazan using a
standard curve generated from NBD. The results were represented as
µmol/mg wet tissue.
Determination of hippocampus
apoptosis
A terminal deoxy nucleotidyl transferase dUTP
nick-end labeling (TUNEL) assay was used to assess hippocampus
apoptosis with an apoptosis detection kit (Boster Biological
Technology, Ltd., Wuhan, China) according to the manufacturer's
instructions. For each slide, 10 fields were randomly chosen, with
TUNEL-positive cells showing brown staining within the nucleus of
apoptotic cells. Measurement of the apoptosis level was carried out
on formalin-fixed, paraffin-embedded sections using an In
situ Direct Fragmentation (TUNEL) Assay kit according to the
manufacturer's instructions. First, the slides were fixed by adding
1% (w/v) paraformaldehyde in PBS and placed on ice for 15 min.
After washing slides with PBS, ice-cold 70% (v/v) ethanol was added
to the slides after which they were allowed to stand for a minimum
of 30 min in the freezer. Following the removal of ethanol, the
slides were washed with washing buffer one more time. The slides
were incubated in the prepared staining solution for 60 min at
37°C. Subsequently, rinse buffer was added to each slide for 5 min.
The rinsing step was repeated one more time. Propidium iodide/RNase
A solution was added to the slides and incubated in the dark for 30
min at room temperature. At the end of the experiments, apoptosis
analysis was carried out using a fluorescence microscopy Ex/Em
wavelength (Ex/Em = 495/519 nm).
Statistical analysis
The apoptosis levels of sham, Hcy, and morphine-Hcy
groups were determined. The percentage of cell death was measured
separately. Data are presented as means ± standard error of the
mean (SEM) and analyzed by one-way analysis of variance (ANOVA).
The data of biochemical studies were expressed as means ± SEM and
analyzed using one-way ANOVA. P<0.05 was considered
statistically significant.
Results
Histopathological analysis and
validation of model
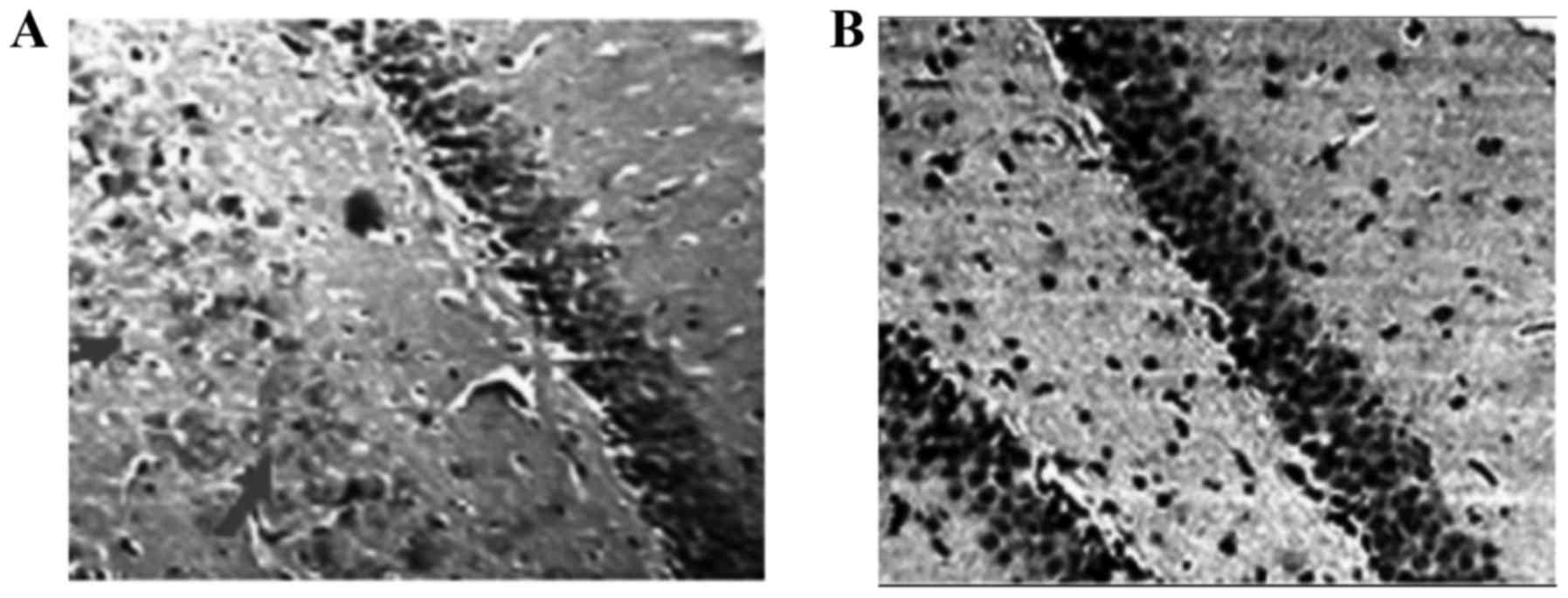
The results showed that there were significant
differences in the morphology and structure of hippocampus cells
between experimental groups. On the other hand, our findings
indicated that the cell density of dentate gyrus (cell count per
mm2) in the Hcy group was significantly (P<0.001)
lower than that of the control group (Fig.
1).
Estimation of oxidative stress
parameters
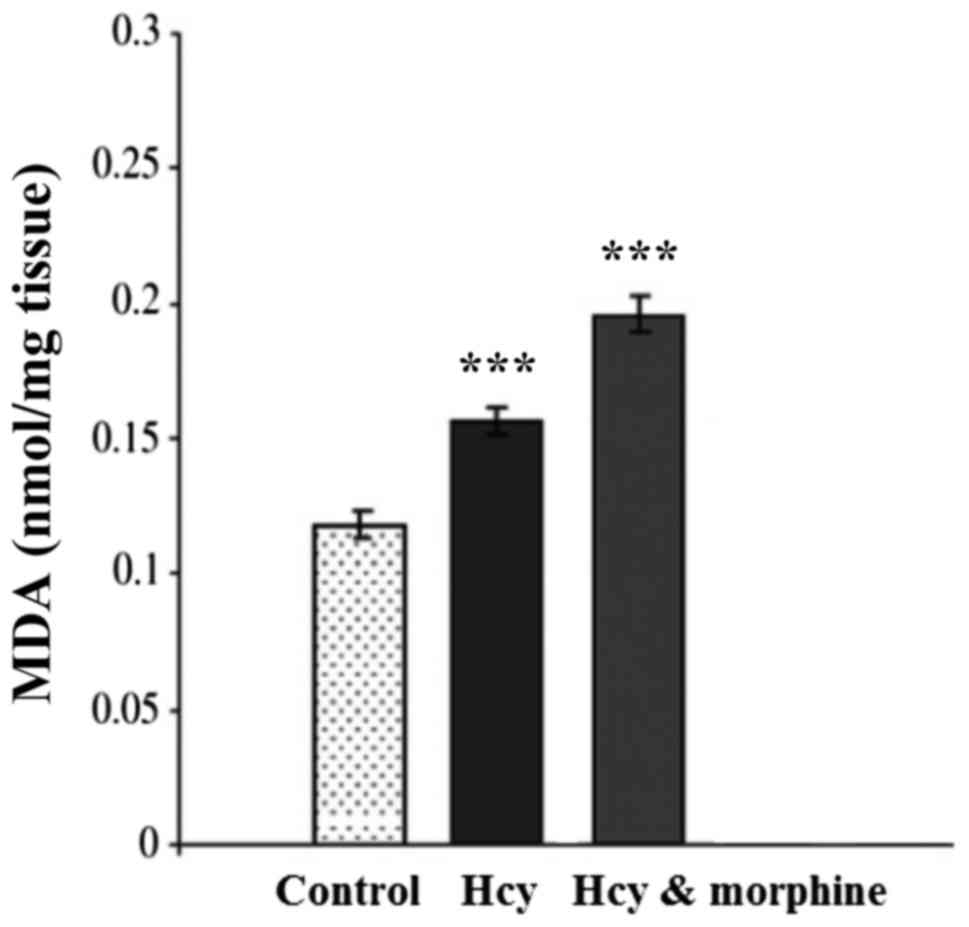
Fig. 2 shows the
effects of drugs on lipid peroxidation. One-way ANOVA indicated
that MDA level in the Hcy group was increased significantly
compared to the sham group (P<0.001). Morphine pre and post
treatment increased the MDA level significantly in rat hippocampus
compared with the other groups (P<0.001).
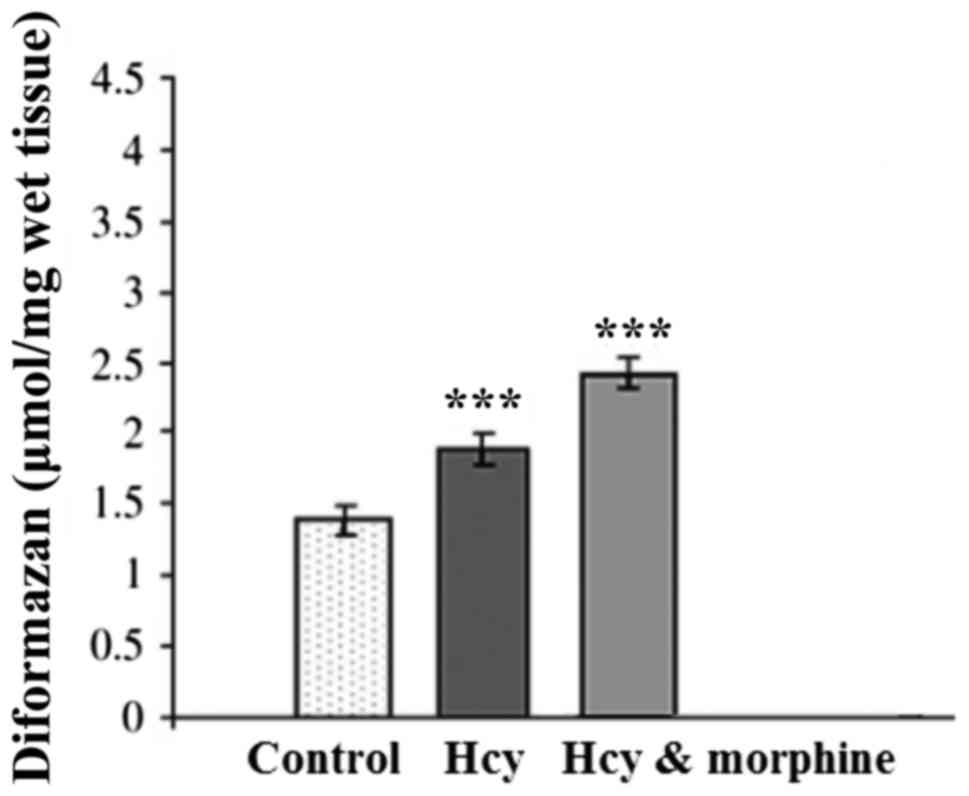
Fig. 3 shows the
effects of drugs on SOA level (µmol/mg wet tissue) in the rat
hippocampal homogenate. One-way ANOVA indicated that SOA level in
the Hcy group was significantly greater than that of the sham
group. In addition, morphine pre- and post-treatment led to a
significant increase in the SOA level (P<0.001).
Evaluation of hippocampus cell
apoptosis
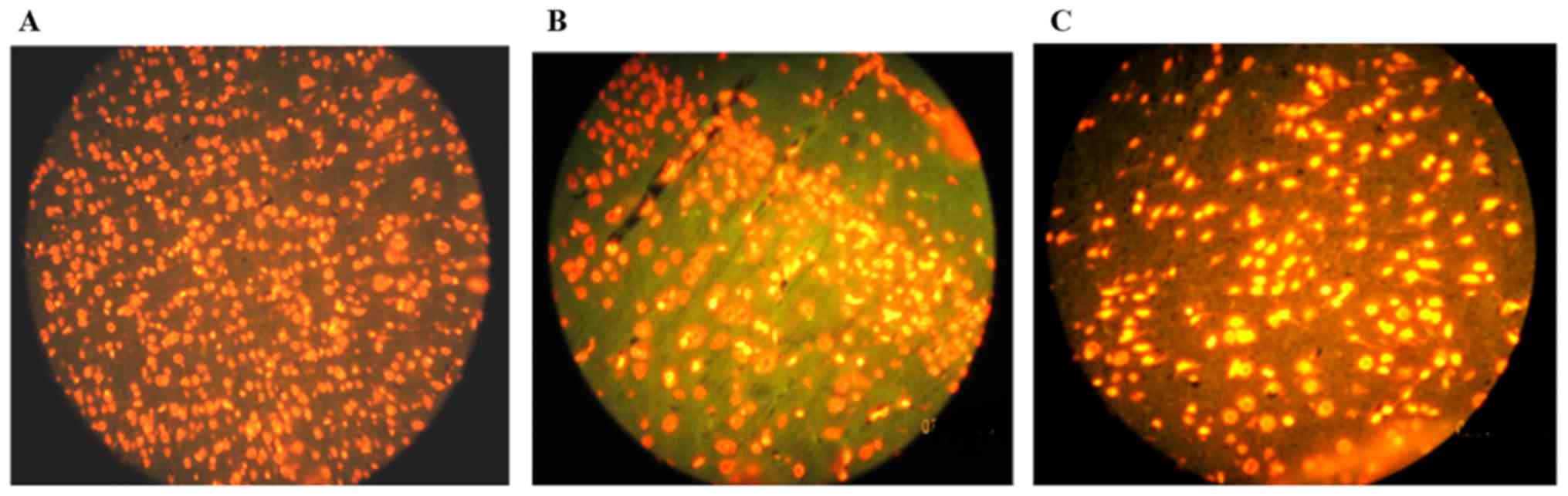
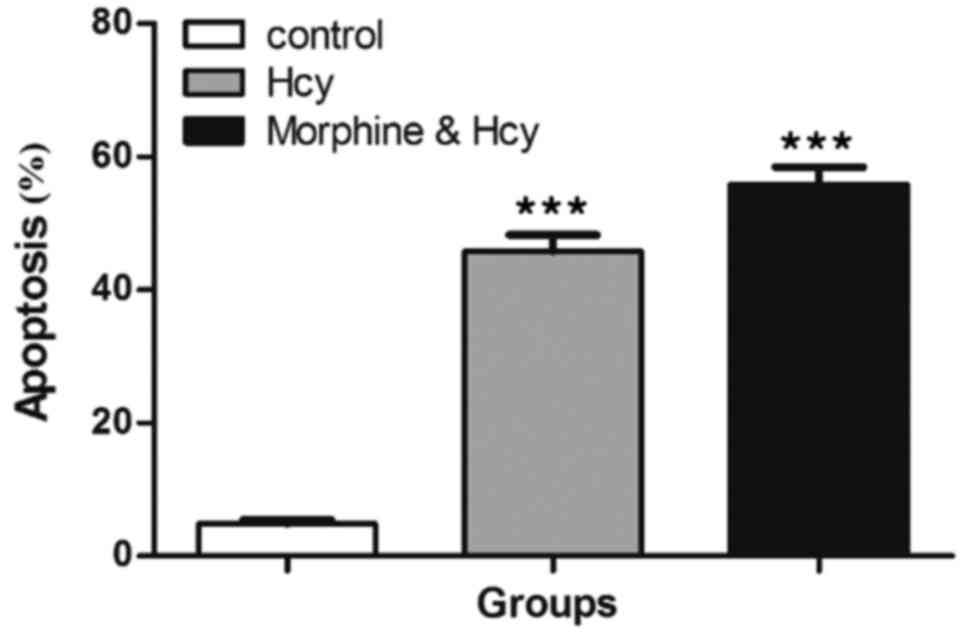
As shown in Fig. 4,
TUNEL staining was used to evaluate hippocampus cell apoptosis. It
was found that Hcy could separately induce apoptosis in hippocampus
cells and significantly increased the number of TUNEL-positive
cells in rat hippocampus compared to the control group
(P<0.001). Of note, our results indicated that morphine pre- and
post-treatment did not decrease TUNEL-positive cells in rat
hippocampus cells compared with the other groups (Fig. 5). Furthermore, pre- and post-treatment
by morphine increased apoptosis in hippocampus cells compared with
the other groups (P<0.001).
Discussion
Opioids as strong analgesics have been used in pain
treatment for more than 100 years (13). Previous results suggested that morphine
possesses some neuroprotective effects in different
ischemia/reperfusion models (14).
However, many investigations showed that morphine could protect
against neuronal cell death, there are several lines of evidence
suggest that morphine could induce apoptosis in neurons. Therefore,
the current study was conducted to investigate the pre- and
post-conditioning effects of morphine on hippocampal cell apoptosis
in a rat model of Hcy-induced oxidative stress. Our results showed
that Hcy separately induced apoptosis in hippocampus cells and
significantly increased the number of TUNEL-positive cells in rat
hippocampus compared to the control group. In addition, MDA and SOA
levels in the Hcy group were increased significantly compared to
the control group. It was in accordance with previous reports that
the expression of apoptosis regulatory proteins, Bax and Bcl-2,
would be altered by Hcy (15). It was
also suggested that Hcy generates reactive oxygen species, which
attacks the polyunsaturated fatty acids of neuronal cell membranes
and induces lipid peroxidation in the hippocampus (16). Hcy is a non-protein amino acid, or
thiol-containing amino, which is derived from methionine (17). Elevated plasma Hcy levels have been
associated with high incidence of atherosclerotic and
neurodegenerative disorders, such as Alzheimer's disease and
dementia (18,19). Evidence shows that Hcy is toxic to
neuronal cells both in vitro and in vivo (20,21) and can
cause calcium influx, oxidative stress, neuroinflammation and
neuronal apoptosis (22). Kruman et
al also demonstrated that Hcy elicits a DNA damage response in
rat hippocampal neurons that promotes apoptosis and
hypersensitivity to excitotoxicity (21). In addition, Maler et al reported
that Hcy induces cell death of rat astrocytes in vitro
(23). Moreover, several animal models
have shown a role of Hcy in cerebrovascular pathology, cognitive
decline and learning disabilities (24). Additionally, in our previous reports,
we demonstrated the neuroprotective effects of the polyphenolic
antioxidant agent, Curcumin, against Hcy-induced cognitive
impairment and oxidative stress in the rat (25). In previous reports by our research
group, Hcy decreased locomotor activities significantly in rats as
well as it could induce apoptosis in substantia nigra cells
(26). Therefore, our previous
experiences and use of Hcy in other published studies as animal
models to induce oxidative stress, neuroinflammation and neuronal
apoptosis can confirm the success of the model. Moreover, the
present study was substantially revised and developed by further
experiments such as analysis of structure and morphology of
hippocampal cells by H&E and assay of MDA and SOA in
hippocampal homogenate which can approve the success of the
model.
Our results have demonstrated that morphine pre- and
post-treatment could not decrease TUNEL-positive cells in rat
hippocampus cells. On the other hand, pre- and post-treatment by
morphine increased apoptosis in hippocampus cells compared with the
other group. Contrast to our results, previous studies suggested
neuroprotective effects of morphine against neuronal cell death. In
rat neuronal/glial cultures, morphine was reported to prevent cell
death induced by HIV envelope glycoprotein gp120IIIB or BaL
(5). In addition, Zhao et al
reported that morphine pre-conditioning induces opioid
receptor-dependent neuroprotection against ischemia in rats and
reduces ischemia-induced cell death in the CA1 regions of
hippocampal slices (2). Several lines
of evidence indicated that the induction of estradiol release in
hippocampal neurons by morphine induces upregulation of heat shock
protein 70 that protects against neuronal damage and cell death
(27). Despite these facts, several
other investigations confirmed our results. Liu et al stated
that long-term use of morphine can induce neuronal apoptosis in the
brain by increasing the expressions of pro-apoptotic Fas and
caspase-3 and decreasing the anti-apoptotic Bcl-2 expression as an
underlying mechanism for the opiate-induced neuronal damage
(8). Moreover, in an investigation
regarding the effect of morphine on apoptosis in the nucleus
accumbens in rat brain, the results showed that apoptotic factors
increased in all the groups treated with morphine (28). It can be hypothesized that these
apoptotic effects may be attributed to effects of opioids on
neuronal structure (cytoskeleton), which can lead to neuronal
damage.
Taken together, our data suggested that morphine
pre- and post-conditioning exacerbates apoptosis and oxidative
stress in hippocampus cells. Although this study achieved its aims,
there were some unavoidable limitations. First, because the
neuroprotective or neurotoxic effect of morphine may be related to
dose, a dose-escalating treatment is suggested to be performed in
future studies. Second, future investigations should consider a
protective or neurotoxic effect of morphine through analysis of the
expression of pro- and anti-apoptotic genes on mRNA and/or protein
level and metabolism genes. Finally, since improved understanding
of morphine neuroprotection and neurotoxicity is useful to control
morphine side-effects in medical applications and to identify new
targets for potential therapies and prevention strategies to opioid
addiction, further studies are needed to reveal the exact mechanism
of morphine in cell death process (apoptosis) or the
neuroprotective properties of morphine should be studied in more
detail.
Acknowledgements
The authors would like to thank Dr Azadmehr, Dr
Zabihi, and Dr Ali Akbar Moghadamnia for their assistance and
effective comments. This study was supported by the Cellular and
Molecular Biology Research Center, Babol University of Medical
Sciences, Babol, Iran.
References
|
1
|
Zhou Q, Krebs JF, Snipas SJ, Price A,
Alnemri ES, Tomaselli KJ and Salvesen GS: Interaction of the
baculovirus anti-apoptotic protein p35 with caspases. Specificity,
kinetics, and characterization of the caspase/p35 complex.
Biochemistry. 37:10757–10765. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhao P, Huang Y and Zuo Z: Opioid
preconditioning induces opioid receptor-dependent delayed
neuroprotection against ischemia in rats. J Neuropathol Exp Neurol.
65:945–952. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sanchez-Simon FM, Arenzana FJ and
Rodriguez RE: In vivo effects of morphine on neuronal fate and
opioid receptor expression in zebrafish embryos. Eur J Neurosci.
32:550–559. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Qian L, Tan KS, Wei SJ, Wu HM, Xu Z,
Wilson B, Lu RB, Hong JS and Flood PM: Microglia-mediated
neurotoxicity is inhibited by morphine through an opioid
receptor-independent reduction of NADPH oxidase activity. J
Immunol. 179:1198–1209. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Avdoshina V, Biggio F, Palchik G, Campbell
LA and Mocchetti I: Morphine induces the release of CCL5 from
astrocytes: Potential neuroprotective mechanism against the HIV
protein gp120. Glia. 58:1630–1639. 2010.PubMed/NCBI
|
|
6
|
Ammon-Treiber S, Stolze D, Schröder H, Loh
H and Höllt V: Effects of opioid antagonists and morphine in a
hippocampal hypoxia/hypoglycemia model. Neuropharmacology.
49:1160–1169. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu Y, Nie YM and Wu WK: The effect of PKC
activation and Smac release on inhibition of myocardial cell
apoptosis by Sini Decoction. Zhong Yao Cai. 31:1675–1678. 2008.(In
Chinese). PubMed/NCBI
|
|
8
|
Liu LW, Lu J, Wang XH, Fu SK, Li Q and Lin
FQ: Neuronal apoptosis in morphine addiction and its molecular
mechanism. Int J Clin Exp Med. 6:540–545. 2013.PubMed/NCBI
|
|
9
|
Mao J, Sung B, Ji RR and Lim G: Neuronal
apoptosis associated with morphine tolerance: Evidence for an
opioid-induced neurotoxic mechanism. J Neurosci. 22:7650–7661.
2002.PubMed/NCBI
|
|
10
|
Yin D, Mufson RA, Wang R and Shi Y:
Fas-mediated cell death promoted by opioids. Nature. 397:2181999.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Placer ZA, Cushman LL and Johnson BC:
Estimation of product of lipid peroxidation (malonyl dialdehyde) in
biochemical systems. Anal Biochem. 16:359–364. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Das UN, Padma M, Sagar PS, Ramesh G and
Koratkar R: Stimulation of free radical generation in human
leukocytes by various agents including tumor necrosis factor is a
calmodulin dependent process. Biochem Biophys Res Commun.
167:1030–1036. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Barry U and Zuo Z: Opioids: Old drugs for
potential new applications. Curr Pharm Des. 11:1343–1350. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lim YJ, Zheng S and Zuo Z: Morphine
preconditions Purkinje cells against cell death under in vitro
simulated ischemia-reperfusion conditions. Anesthesiology.
100:562–568. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ataie A, Ataee R, Shadifar M, Shahabi S,
Aghajanpour SM and Hosseinpour Y: Interaction of Memantine with
homocysteine on the apoptosis in the rat hippocampus cells. Int J
Mol Cell Med. 1:145–152. 2012.PubMed/NCBI
|
|
16
|
Agnati LF, Genedani S, Rasio G, Galantucci
M, Saltini S, Filaferro M, Franco R, Mora F, Ferré S and Fuxe K:
Studies on homocysteine plasma levels in Alzheimer's patients.
Relevance for neurodegeneration. J Neural Transm (Vienna).
112:163–169. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Selhub J: Homocysteine metabolism. Annu
Rev Nutr. 19:217–246. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Biasioli S and Schiavon R: Homocysteine as
a cardiovascular risk factor. Blood Purif. 18:177–182. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sachdev P: Homocysteine, cerebrovascular
disease and brain atrophy. J Neurol Sci. 226:25–29. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lipton SA, Kim WK, Choi YB, Kumar S,
D'Emilia DM, Rayudu PV, Arnelle DR and Stamler JS: Neurotoxicity
associated with dual actions of homocysteine at the
N-methyl-D-aspartate receptor. Proc Natl Acad Sci USA.
94:5923–5928. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kruman II, Culmsee C, Chan SL, Kruman Y,
Guo Z, Penix L and Mattson MP: Homocysteine elicits a DNA damage
response in neurons that promotes apoptosis and hypersensitivity to
excitotoxicity. J Neurosci. 20:6920–6926. 2000.PubMed/NCBI
|
|
22
|
Obeid R and Herrmann W: Mechanisms of
homocysteine neurotoxicity in neurodegenerative diseases with
special reference to dementia. FEBS Lett. 580:2994–3005. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maler JM, Seifert W, Hüther G, Wiltfang J,
Rüther E, Kornhuber J and Bleich S: Homocysteine induces cell death
of rat astrocytes in vitro. Neurosci Lett. 347:85–88. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kamat PK, Vacek JC, Kalani A and Tyagi N:
Homocysteine induced cerebrovascular dysfunction: A link to
Alzheimer's disease etiology. Open Neurol J. 9:9–14. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ataie A, Sabetkasaei M, Haghparast A,
Moghaddam AH and Kazeminejad B: Neuroprotective effects of the
polyphenolic antioxidant agent, Curcumin, against
homocysteine-induced cognitive impairment and oxidative stress in
the rat. Pharmacol Biochem Behav. 96:378–385. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ataie A, Ataee R, Mansoury Z and
Aghajanpour M: Homocysteine intracerebroventricular injection
induces apoptosis in the substantia nigra cells and Parkinson's
disease likebehavior in rats. Int J Mol Cell Med. 2:80–85.
2013.PubMed/NCBI
|
|
27
|
Cui J, Wang Y, Dong Q, Wu S, Xiao X, Hu J,
Chai Z and Zhang Y: Morphine protects against intracellular amyloid
toxicity by inducing estradiol release and upregulation of Hsp70. J
Neurosci. 31:16227–16240. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Katebi N, Razavi Y, Zeighamy Alamdary S,
Irani S, Khodagholi F and Haghparast A: Effect of morphine on
apoptotic factors caspase-3, PARP and Bax/Bcl-2 ratio in nucleus
accumbens in conditioned place preference model in rat. Physiol
Pharmacol. 17:39–50. 2013.
|