Introduction
Corneal dystrophy is a group of hereditary diseases
characterized by corneal opacities resulting from progressive
accumulation of abnormal deposits in corneal stroma, which leads to
visual impairment (1,2). Lattice corneal dystrophy (LCD) is one of
the most common types of corneal disorderand is characterized by
lineal stromal amyloid depositions that usually present bilaterally
and symmetrically. Based on age of onset, severity of the
phenotype, appearance and depth of the lattice lines, LCD was
classified into five subtypes: Type I, IIIA, I/IIIA, IV and IIIB
(3,4).
LCD type I (LCDI), the classic form and most
frequently encountered type of corneal dystrophy, is an early onset
dystrophy exhibiting variable clinical expression (5,6). It is
usually inherited as an autosomal dominant, autosomal recessive or
X-linked recessive Mandelian inheritance trait. Mutations of the
carbohydrate sulfotransferase 6 and transforming growth factor β
induced (TGFBI) gene have been reported to cause macular
corneal dystrophy and corneal disease (4). Currently, LCDI has been associated with a
large number of missense mutations with genotype-phenotype
correlations in the TGFBI gene, and the International
Committee for Classification of Corneal Dystrophies categorized it
as LCD-TGFBI type (3,7).
In 1997, the TGFBI gene was first identified
as a causative gene for corneal dystrophies by Munier et al
(8). This gene, originally termed
β-igh3, comprises 17 exons coding for a unique protein of 683 amino
acids. So far, ~50 different mutations causing LCD have been
identified in the TGFBI gene (www.hgmd.org).
The TGFBI mutation spectrum and their clinical consequences
have been investigated in patients with LCDI in different ethnic
populations (2,7,9–13). For example, the p.Arg124Cys mutation is
reported to be associated with the LCDI phenotype in Greek,
Japanese, Bangladeshi and Chinese patients (14–19).
Therefore, there is a significant correlation between TGFBI
mutations and their associated phenotype. However, the exact
mechanisms and pathological roles of TGFBI mutations in the
development of LCD remain largely unknown, and the correlation
between phenotype and genotype of TGFBI in the Chinese
population has not been extensively investigated.
In the present study, a Chinese family with LCDI was
investigated to determine the mutation underlying this disease, and
to characterize the clinical features and phenotype-genotype
correlation of this family.
Materials and methods
Study participants
The current study was approved by the Institutional
Review Boards of Sichuan Academy of Medical Sciences and Sichuan
Provincial People's Hospital, University of Electronic Science and
Technology of China (Chengdu, China). Written informed consent was
obtained from all subjects prior to the study. All procedures in
the present study adhered to the tenets of the Declaration of
Helsinki. Five affected and 11 unaffected individuals from a
Chinese family (Fig. 1A) were enrolled
in the study after obtaining informed consent. Slit-lamp
examination was performed for all participating individuals in this
family to determine whether they were affected or unaffected with
corneal dystrophies and to determine the disease phenotype. Three
individuals in the family (II:2, III:1 and III:4) were evaluated by
laser scanning in vivo confocal microscopy (Heidelberg
Retina Tomograph II, Rostock Corneal Module; Heidelberg Engineering
GmbH, Heidelberg, Germany). Detailed clinical histories of all the
participating subjects, such as the age of onset, initial signs and
symptoms, progression of disease, other ocular therapeutic
procedures, and so on, were obtained. A total of 400 unrelated
healthy control subjects were recruited from the Hospital of
University of Electronic Science and Technology of China. These
control subjects had no medical history of associated visual
disorders.
DNA extraction
All genomic DNA was extracted from 2 ml peripheral
blood, using a blood DNA extraction kit (QIAamp DNA Blood Midi kit,
cat. no. 51106; Qiagen GmbH, Hilden, Germany) according to the
manufacturer's protocol. DNA samples were stored at −20°C until
used. DNA integrity was evaluated by 1% agarose gel
electrophoresis. Briefly, 5 µl of DNA with 1 µl loading buffer were
loaded into 1% agarose gel and run at 120 V for 30 min in an
electrophoresis apparatus (Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
Mutation screening
The coding sequences of TGFBI (NM_000358.2)
was amplified by polymerase chain reaction (PCR) with 35 cycles (30
sec at 95°C for initial denaturation, 30 sec for annealing at
different temperatures and 30 sec at 72°C for extension), using a
MyCycler thermocycler (Bio-Rad Laboratories, Inc.). Sequencing
primers forthe flanking sequence of each exon were designedusing
Primer 5.0 software (Premier Biosoft International, Palo Alto, CA,
USA). Amplified PCR products were purified with spin columns
(QIAquick PCR Purification kit, cat. no. 28104; Qiagen, Inc.,
Valencia, CA, USA) and sequenced directly (BigDye Terminators
Sequencing kit (cat. no. 4337455; Applied Biosystems; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) in two directions using an
automated genetic analysis system (model 3130; Applied Biosystems;
Thermo Fisher Scientific, Inc.). Multiple sequence alignment ofthe
human TGFBI protein was performed along with other TGFBI proteins
across different species to assessfor the conservation of residues.
The possible damaging effects of this mutation on the structure and
function of TGFBI were predicted using Sorting Intolerant From
Tolerant (SIFT; http://sift.jcvi.org) and PolyPhen-2
(http://genetics.bwh.harvard.edu/pph2/).
Results
Clinical data
A family including 16 members from the Sichuan
province of China was recruited in the current study (Fig. 1A). Ophthalmic examinations identified
five affected individuals (II:2, II:5, II:7, III:4 and III:5) among
the family members. The affected subjects with LCDI exhibited
similar clinical features, except that patient III:5 presented with
mild symptom. The proband, patient II:2 in this family, was
initially diagnosed as LCDI in each eye when he visited the
Hospital of University of Electronic Science and Technology of
China at the age of 43 years. The patient had experienced bilateral
foreign body sensation and visual defects since the age of 26, and
progressive deterioration in visual acuity bilateral over the past
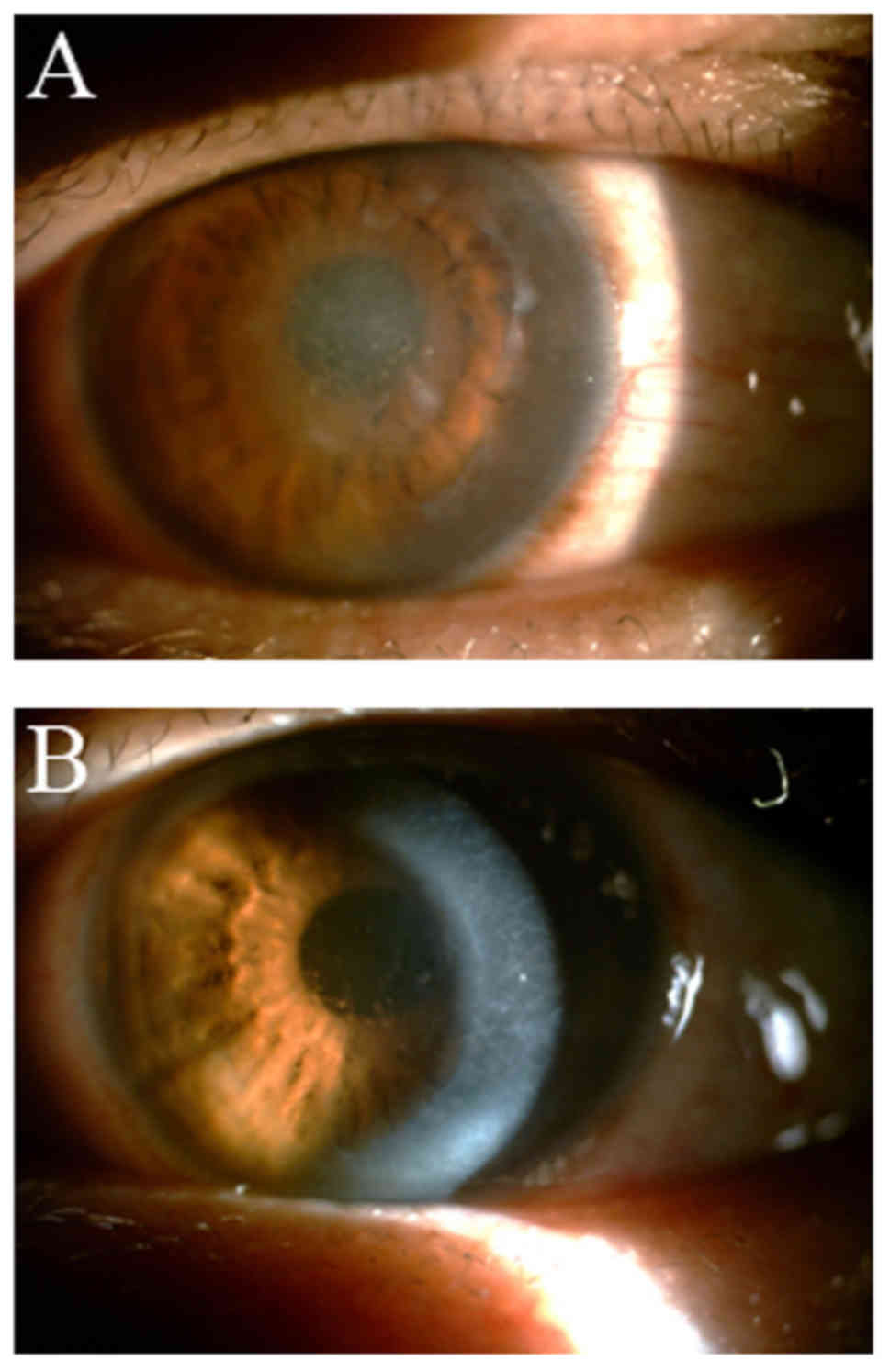
10 years (Table I). Slit-lamp
examination demonstrated various distinct linear deposits in the
superficial stroma of the central corneal accompanied by epithelial
erosions (Fig. 2). The brother and
sister of the proband (II:5 and II:7) were also found to be
affected with LCDI. They began to exhibit the initial symptom with
a mild foreign body sensation at approximately 21–25 years old and
exhibited obvious visual impairment.
 | Table I.Clinical data of family members with
LCDI. |
Table I.
Clinical data of family members with
LCDI.
| Patient | Sex/age | Age at onset
(years) | Duration (years) | Surgical treatment
(patient's age when performed) |
|---|
| II:2 | Male/43 | 26 | 20 | NK |
| II:5 | Male/41 | 24 | 18 | PKP
(37-year-old) |
| II:7 | Female/38 | 23 | 16 | PKP
(35-year-old) |
| III:4 | Female/19 | 19 | 1 | NK |
| III:5 | Male/15 | 24 | 5 | NK |
The nieces of the proband (III:4 and III:5) were 19
and 15 years old, respectively. They complained of bilateral
blurred vision, recurring ocular pain, photophobia and tearing. The
two patients had multiple linear deposits and a few confluent
opacities in the anterior stroma of the central cornea. An obvious
visual defect was detected in III:4, but not in III:5.
Mutation screening of TGFBI in the
family with LCD
Sequencing analysis of the TGFBI gene
revealed a heterozygous mutation, c.370C>T (p.R124C), which was
located at nucleotide 370 in the coding sequence of exon 4
(Fig. 1B). This missense mutation was
present in the five affected patients (II:2, II:5, II:7, III:4 and
III:5). This mutation was not observed in the unaffected family
members and 400 normal control subjects. Comparative amino acid
sequence alignment of other TGFBI proteins across different species
revealed that the mutation occurred at highly conserved positions
(Fig. 1C). SIFT and PolyPhen-2
predicted this mutation to be damaging, and it may result in a
substitution from arginine to cysteine at position 124.
Confocal microscopy examination
In order to determine whether the detected mutation
in the family is associated with LCDI, in vivo confocal
microscopy was performed to investigate the difference in corneal
structure between healthy and affected members. Laser scanning
in vivo confocal microscopy was performed on the proband
(II:2), his affected niece (III:4) and one of the healthy
individuals (III:1) in the family. The basal epithelial cells of
patients were characterized by irregular hyperreflective dots,
compared with that of healthy individuals (Fig. 3A-C). In the lamina elastic anterior
(Bowman's membrane), numerous nerve fibers were clearly detected in
the healthy individual, whereas, focal depositions of reflective
materials and decreased nerve fibers were identified in the
patients (Fig. 3D-F). In the
superficial one-third of the stroma, the number of cell nuclei was
largely reduced, and collagenous fibers were thinner and fewer in
the patients than in the healthy individuals (Fig. 3G-I). In the deeper one-third of the
stroma, there were coarser nerve fibers across the layer detected
in the patients compared with those in the healthy individuals
(Fig. 3J-L). The two patients and the
healthy individual exhibited a normal posterior elastic lamina
(Descemet's membrane) and endothelium cells, except that the
endothelial cells in the patients were visualized faintly, although
they appeared to be normal (data not shown).
Discussion
LCDI is an autosomal, dominantly inherited corneal
amyloidosis associated with severe visual defects, and mutations in
the TGFBI gene are the main causes of LCDI (7). In the present study, a heterozygous
mutation, c.370C>T (p.R124C)was detected in TGFBI and the
phenotypic characteristics in a Chinese family affected with LCDI
were described. TGFBI mutations have been reported to be
closely involved with at least five types of inherited corneal
dystrophy. Among those mutations reported, R124 in TGFBI
appeared to be a ‘hot-spot’ point mutation detected in different
types of corneal dystrophy in various ethnic groups worldwide
(7). R124 in exon 4 of TGFBI is
conserved among numerous different species (Fig. 1C), indicating that this residue is an
important functional and structural site of the protein.
Furthermore, it is important to investigate the mutation underlying
this disease and to characterize the clinical features and
phenotype-genotype correlations within this family.
In the present study, five affected individuals
among the 16 family members were diagnosed with LCDI, although
there is already a study of phenotypic variability with the R124C
mutation in LCDI pedigrees (17). LCD
is generally divided into three subtypes and is classified
predominantly by clinical and pathological findings. The underlying
mechanism of the phenotypic variability for corneal dystrophy with
the same mutation and even within the same family requires further
investigation. Therefore, it is necessary to identify the specific
mutation in patients to assess a final diagnosis due to genetic
heterogeneity. In the current family, the affected members with the
R124C mutation exhibited similar clinical features of LCDI. They
all had prominent delicate linear opacities that tended to
predominantly be in the superficial corneal stroma accompanied with
epithelial erosions, although patient III:5 exhibited mild
symptoms. Molecular genetic analysis, which is important in
establishing the classification of corneal dystrophy, may reveal
reliable clinical diagnostic criteria and may improve the accuracy
of the clinical diagnosis. The patient, III:5, who carried the same
R124C mutation as the other four patients, was thus diagnosed as
LCDI although she did not exhibit typical clinical features of
LCDI. It is also possible that she was younger and may develop
corneal dystrophy in the future, indicating that genetic analysis
may be used during prenatal and postnatal DNA diagnosis for
clinical work.
In order to confirm the clinical features of patient
III:4, she was recruited for examination by in vivo confocal
microscopy. The images indicated that multiple corneal layers,
including the basal epithelial cells, stroma cells and Bowman's
membrane, were affected in the two patients (the proband, II:2 and
III:4) of this family (Fig. 3). The
proband seemed to exhibit stronger irregular hyperreflective
materials compared with patient III:4 in the basal epithelial cells
and Bowman's membrane (Fig. 3A-C). It
is likely that patient III:4 was markedly younger than the proband.
Patient III:4 was found to have milder symptom than the proband,
indicating that the worse the visual acuity is, the more likely
that the morphology of corneal layers was affected. During confocal
microscopy and slit-lamp examination of the affected members, their
corneal epithelia were particularly fragile and corneal epithelial
erosions were identified. They also developed photophobia and
lacrimation by microscopic examination. Observations of the
abnormal morphology of Bowman's membrane in the patients (Fig. 3D-F) identified that the density of
nerve fibers was significantly decreased and even disappeared; it
is likely that the damaged Bowman's membrane cannot provide normal
nutrition for corneal nerve growth, thus leading to corneal
deposits. If this corneal dystrophy was due to lack of nutrition,
the disease maybe treated with neurotrophic factors. The TGFBI
protein is presumably involved in cell adhesion and migration of
various cell types, including epithelial cells, fibroblasts,
endothelial cells, and vascular smooth muscle cells (20). Therefore, patients with the
TGFBI R124C mutation may exhibit relaxation of the
connection between the corneal epithelial cells and the entire
epithelial layer, leading to the abnormity of Bowman stromal
corneal dystrophy in this family. Furthermore, nerve fibers may not
grow into the Bowman's layer and the corneal epithelial cells lack
nerve nutrition, resulting in exfoliation of the corneal epithelium
(Fig. 2).
In conclusion, a mutation in the TGFBI gene
was identified in a Chinese family with LCDI. The current results
characterized the clinical features of LCDI and revealed a clear
genotype-associated phenotype in this family, which may contribute
to further understanding the role of this TGFBI mutation in
the development of LCDI. However, the composition of the affected
corneal layer in patients remains unknown and thus its specific
underlying mechanism requires further investigation. Future
functional work is required to confirm the role of TGFBI and
the underlying mechanisms in the disease.
Acknowledgements
The authors would like to thank the family and the
healthy volunteers for their participation in the study. The study
was supported by grants from the Natural Science Foundation of
China (grant nos. 81371048, 81570848, 81170882 and 81570888) and
the Department of Science and Technology of Sichuan Province (grant
nos. 2015JZ0004, 2015HH0031 and 2014JZ0004).
References
|
1
|
Klintworth GK: The molecular genetics of
the corneal dystrophies - current status. Front Biosci. 8:687–713.
2003. View Article : Google Scholar
|
|
2
|
Sacchetti M, Macchi I, Tiezzi A, La Cava
M, Massaro-Giordano G and Lambiase A: Pathophysiology of corneal
dystrophies: From cellular genetic alteration to clinical findings.
J Cell Physiol. 231:261–269. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weiss JS, Møller HU, Lisch W, Kinoshita S,
Aldave AJ, Belin MW, Kivelä T, Busin M, Munier FL, Seitz B, et al:
The IC3D classification of the corneal dystrophies. Cornea. 27
Suppl 2:S1–S83. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schorderet D: Corneal dystrophies:
Overview and summary. Prog Mol Biol Transl Sci. 134:73–78. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Klintworth GK: Advances in the molecular
genetics of corneal dystrophies. Am J Ophthalmol. 128:747–754.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lisch W and Seitz B: Lattice corneal
dystrophy type 1: An epithelial or stromal entity? Cornea.
33:1109–1112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lakshminarayanan R, Chaurasia SS,
Anandalakshmi V, Chai SM, Murugan E, Vithana EN, Beuerman RW and
Mehta JS: Clinical and genetic aspects of the TGFBI-associated
corneal dystrophies. Ocul Surf. 12:234–251. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Munier FL, Frueh BE, Othenin-Girard P,
Uffer S, Cousin P, Wang MX, Héon E, Black GC, Blasi MA, Balestrazzi
E, et al: BIGH3 mutation spectrum in corneal dystrophies. Invest
Ophthalmol Vis Sci. 43:949–954. 2002.PubMed/NCBI
|
|
9
|
Dudakova L, Palos M, Jirsova K, Skalicka
P, Dundr P and Liskova P: Novel TGFBI mutation p.(Leu558Arg) in a
lattice corneal dystrophy patient. Ophthalmic Genet. 37:473–474.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chae H, Kim M, Kim Y, Kim J, Kwon A, Choi
H, Park J, Jang W, Lee YS, Park SH and Kim MS: Mutational spectrum
of Korean patients with corneal dystrophy. Clin Genet. 89:678–689.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cai J, Zhu L, Zha Y and Kang Q: TGFBI gene
mutation analysis in Chinese families with corneal dystrophies.
Genet Test Mol Biomarkers. 20:388–392. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ann LB, Abbouda A, Frausto RF, Huseynli S,
Gupta K, Alió JL and Aldave AJ: Variant lattice corneal dystrophy
associated with compound heterozygous mutations in the TGFBI gene.
Br J Ophthalmol. 101:509–513. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Costagliola C, Romano V, Cifariello F,
Aceto F and Porcellini A: Lattice corneal dystrophy: A report of
two cases in twin sisters due to 3 mutations (T1620C, C1416T,
A1924G) in the TGFBI (BIGH3) gene. Clin Ter. 165:e73–e75.
2014.PubMed/NCBI
|
|
14
|
Hellenbroich Y, Tzivras G, Neppert B,
Schwinger E and Zühlke C: R124C mutation of the betaIGH3 gene leads
to remarkable phenotypic variability in a Greek four-generation
family with lattice corneal dystrophy type 1. Ophthalmologica.
215:444–447. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yoshida S, Yoshida A, Nakao S, Emori A,
Nakamura T, Fujisawa K, Kumano Y and Ishibashi T: Lattice corneal
dystrophy type I without typical lattice lines: Role of mutational
analysis. Am J Ophthalmol. 137:586–588. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
El-Ashry MF, El-Aziz Abd MM, Ficker LA,
Hardcastle AJ, Bhattacharya SS and Ebenezer ND: BIGH3 mutation in a
Bangladeshi family with a variable phenotype of LCDI. Eye (Lond).
18:723–728. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu Z, Wang YQ, Gong QH and Xie LX: An
R124C mutation in TGFBI caused lattice corneal dystrophy type I
with a variable phenotype in three Chinese families. Mol Vis.
14:1234–1239. 2008.PubMed/NCBI
|
|
18
|
Courtney DG, Poulsen ET, Kennedy S, Moore
JE, Atkinson SD, Maurizi E, Nesbit MA, Moore CB and Enghild JJ:
Protein composition of TGFBI-R124C- and TGFBI-R555W-associated
aggregates suggests multiple mechanisms leading to lattice and
granular corneal dystrophy. Invest Ophthalmol Vis Sci.
56:4653–4661. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang QN, Zhao YW, Guo LH, Yan NH, Liu XY
and Cai SP: Arg124Cys mutation of the TGFBI gene in a Chinese
pedigree of Reis-Bücklers corneal dystrophy. Int J Ophthalmol.
4:235–238. 2011.PubMed/NCBI
|
|
20
|
Runager K, Enghild JJ and Klintworth GK:
Focus on molecules: Transforming growth factor beta induced protein
(TGFBIp). Exp Eye Res. 87:298–299. 2008. View Article : Google Scholar : PubMed/NCBI
|