Introduction
Numerous studies have reported that the central and
peripheral sympathetic nervous systems serve important roles in
bone remodeling and bone fracture healing (1–4).
Particularly, bone-forming osteoblasts and bone-resorbing
osteoclasts are established to express α- and β-adrenergic
receptors (α- and β-ARs) (5–7). In osteoblastogenesis, it has been
documented that α1-ARs promote cell proliferation through the
suppression of potassium channels (8). Additionally, α1B-AR signaling may
stimulate bone formation through the promotion of proliferation via
upregulation of CCAAT/enhancer-binding protein δ in osteoblasts
(9). Furthermore, it has been
reported that leptin binding to hypothalamic receptors contributed
to the regulation of bone homeostasis via β2-ARs (1), and that these β2-ARs inhibited
cyclic-adenosine monophosphate (c-AMP)-responsive element-binding
protein phosphorylation, leading to a decrease in osteoblast
proliferation (10). α1-AR agonist
but not β2-AR agonist may also induce fracture callus contraction
via promotion of osteogenesis (11).
In osteoclastogenesis, it has been documented that
an agonist to β-AR (isoprenaline) could promote bone-resorbing
activity in human osteoclast-like cells (6). Furthermore, β2-ARs have been reported to
stimulate osteoclastogenesis via reactive oxygen species generation
(12). A key feature of ARs in bone
remodeling is their ability to mediate interactions of osteoblasts
with osteoclasts, since activation of α1- and β-ARs induces
expression of receptor activator of nuclear factor κB (NF-κB)
ligand (RANKL) in osteoblasts, resulting in RANKL-driven promotion
of osteoclastogenesis (13–15). To the best of our knowledge, however,
little is known of the role of agonists to α-ARs in the development
of osteoclast precursors.
α2-ARs, the prime focus in the present study, belong
to the G-protein-coupled receptor (GPCR) family. There are three
α2-AR subtypes (α2A, α2B and α2C), which are established to
regulate various physiological functions via suppression of
adenylyl cyclase and reduction of c-AMP (16–21). For
instance, α2-ARs on presynaptic membranes may inhibit
norepinephrine secretion from sympathetic nerves (16). It has also been reported that α2A-AR
serves a principal role in the hypotensive response (17), and that it is a primary mediator of
sedative, analgesic and anesthetic-sparing responses (18). Furthermore, α2A-AR on pancreatic
β-cells has been documented to inhibit insulin secretion (19). Although α2-ARs have been established
to serve various roles in homeostasis, little is understood of
their direct involvement in osteoclastogenesis.
In the current study, RAW264.7 pre-osteoclast cells
and primary bone marrow cells were used to evaluate the role of
α2-ARs in osteoclastogenesis. In the presence and absence of α2-AR
agonists (guanabenz, clonidine and xylazine) and α2-AR antagonists
(yohimbine and idazoxan), these cells were cultured in an
osteoclast differentiation medium. Real-time quantitative
polymerase chain reaction (qPCR) and tartrate-resistant acid
phosphatase (TRAP) staining, as well as western blot analysis, were
then conducted to determine the effects of these agonists and
antagonists in osteoclastogenesis.
Materials and methods
Animals
To harvest bone marrow cells, C57BL/6J mice were
purchased from Chubu Kagaku Shizai Co., Ltd. (Nagoya, Japan). A
total of 35 female mice (8–10 weeks old) were used in the current
study. The mice were housed under a 12-h light/dark cycle, and
water and food were provided ad libitum. The protocols for
animal experiments were approved by the Aichi-Gakuin University
Animal Research Committee (Nagoya, Japan).
Cell culture
After the female mice were scarified by cervical
dislocation, mouse bone marrow cells were isolated from long bones
(femur and tibia). For isolation of the bone marrow cells, the
distal and proximal ends of the long bone were removed. Using a
needle (25G), the bone marrow cavity was flushed out with
phosphate-buffered saline. The buffer was then filtered with a cell
strainer (100 µm; BD Falcon™; BD Biosciences, Durham, NC, USA) and
the filtered solution consisting of bone marrow derived cells was
used (22). The mouse bone marrow
cells as well as murine RAW264.7 pre-osteoclast cells obtained from
American Type Culture Collection (Manassas, VA, USA) were cultured
in α-Minimum Essential Media containing 10% fetal bovine serum and
antibiotics (100 U/ml penicillin, 100 µg/ml streptomycin; Wako Pure
Chemical Industries, Ltd., Osaka, Japan). The cells were maintained
at 37°C with 5% CO2 in a humidified incubator.
In vitro osteoclast formation and TRAP
staining
Mouse bone marrow cells were plated at densities of
1.2×105 and 1.0×106 cells into 12-well and
60-mm dishes, respectively, and cultured with 10 ng/ml macrophage
colony-stimulating factor (M-CSF; PeproTech, Inc., Rocky Hill, NJ,
USA) at 37°C for 3 days. The surface-attached cells were used as
osteoclast precursors (22). These
cells were cultured with 10 ng/ml M-CSF and 50 ng/ml RANKL
(PeproTech, Inc.). A total of 5–20 µM guanabenz (R&D Systems,
Inc., Minneapolis, MN, USA) or 10–20 µM xylazine (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) was applied at the same time point
as RANKL; 10–20 µM clonidine (Sigma-Aldrich; Merck KGaA) was
administered with RANKL or 1 day after RANKL administration. After
a 60-h treatment with RANKL at 37°C, cells were fixed in 10%
formalin neutral buffer solution at room temperature and stained
with TRAP for 1 h at 37°C. The number of TRAP-positive cells
containing three or more nuclei was determined. All positive cells
in each well were counted using a light microscope (magnification,
×100; Zeiss AG, Oberkochen, Germany).
RAW264.7 cells were plated at 1.0×105
cells into 60-mm dishes and cultured with 25 ng/ml RANKL in the
presence or absence of 5–20 µM guanabenz, 10–20 µM clonidine or
10–20 µM xylazine with or without 10–20 µM yohimbine or 10–20 µM
idazoxan (Sigma-Aldrich; Merck KGaA) at 37°C for 2–4 days for qPCR
analysis.
Reverse transcription-qPCR
Mouse bone marrow cells and RAW264.7 cells were
treated with RANKL and α2 agonists/antagonists at 37°C for 2–4 days
prior to qPCR analysis. Total RNA was extracted using an RNeasy
Plus Mini kit (Qiagen Sciences, Inc., Gaithersburg, MD, USA).
Reverse transcription was conducted with a High-Capacity cDNA
Reverse Transcription kit (Applied Biosystems; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), and real-time qPCR was
performed using a Takara Thermal Cycler Dice Real Time System III
(Takara Bio, Inc., Otsu, Japan) with Thunderbird SYBR qPCR mix
(Toyobo Life Science, Osaka, Japan). The PCR cycling conditions
were 95°C for 10 min (pre-denaturation), 40 cycles at 95°C for 15
sec (denaturation) and 60°C for 1 min (extension). The mRNA levels
of α2A-, α2B-, and α2C-ARs, nuclear factor of activated T-cells,
cytoplasmic 1 (NFATc1), TRAP and cathepsin K were evaluated with
the PCR primers listed in Table I.
The expression of GAPDH was used as the internal control. The PCR
results were interpreted using the 2−ΔΔCq method
(23).
 | Table I.Real-time polymerase chain reaction
primers used in the present study. |
Table I.
Real-time polymerase chain reaction
primers used in the present study.
|
| Primer sequence
(5′-3′) |
|---|
|
|
|
|---|
| Target | Forward | Reverse |
|---|
| α2A-AR |
GGTGTGTTGGTTTCCGTTCT |
CGGAAGTCGTGGTTGAAGAT |
| α2B-AR |
TCGGAGAGGCTAATGGACAC |
TCTTCAGCTCCCTTCTCTGC |
| α2C-AR (ref.
7) |
CATGGGCGTGTTCGTACTGT |
CAGGCCTCACGGCAGATG |
| Cathepsin K |
CAGCTTCCCCAAGATGTGAT |
AGCACCAACGAGAGGAGAAA |
| NFATc1 |
GGTGCTGTCTGGCCATAACT |
GCGGAAAGGTGGTATCTCAA |
| TRAP |
TCCTGGCTCAAAAAGCAGTT |
ACATAGCCCACACCGTTCTC |
| GAPDH |
TGCACCACCAACTGCTTAG |
GGATGCAGGGATGATGTTC |
Western blot analysis
RAW264.7 cells were lysed in 1X
radioimmunoprecipitation assay buffer containing protease
inhibitors (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and
phosphatase inhibitors (Merck KGaA). Isolated proteins were
quantified using a Pierce bicinchoninic acid protein assay kit
(Thermo Fisher Scientific, Inc.). A total of 10 µg protein per lane
was fractioned using 10% SDS gels and electro-transferred to
Immobilon-P membranes (EMD Millipore, Billerica, MA, USA). The
membranes were blocked with 1% nonfat dry milk at 4°C for overnight
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The membranes were
then incubated for 1 h at room temperature with primary antibodies
followed by a 45-min incubation at room temperature with goat
anti-rabbit (2,000-fold dilution) or anti-mouse (2,000-fold
dilution) immunoglobulin G conjugated with horseradish peroxidase
(cat. nos. 7074 and 7076, respectively; Cell Signaling Technology,
Inc., Danvers, MA, USA). The primary antibodies used were against
eukaryotic translation initiation factor 2α (eIF2α; 1,000-fold
dilution; cat. no. 9722; Cell Signaling Technology, Inc.),
phosphorylated (p)-eIF2α (1,000-fold dilution; cat. no. PA1-26686;
Thermo Fisher Scientific, Inc.) and β-actin (10,000-fold dilution;
cat. no. A5441; Sigma-Aldrich; Merck KGaA). Protein levels were
assayed using a SuperSignal West Femto Maximum Sensitivity
Substrate (Thermo Fisher Scientific, Inc.). To determine band
intensities, images were scanned with a luminescent image analyzer
(LAS-3000; Fujifilm, Tokyo, Japan) and quantified using Image J
v1.48 (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Statistical analyses were performed using Microsoft
Excel for Mac 2011 (version 14.6.9; Microsoft Corporation, Redmond,
WA, USA). Data were expressed as the mean ± standard deviation of
three to five independent experiments. Statistical significance was
evaluated using Student's t-test at P<0.05.
Results
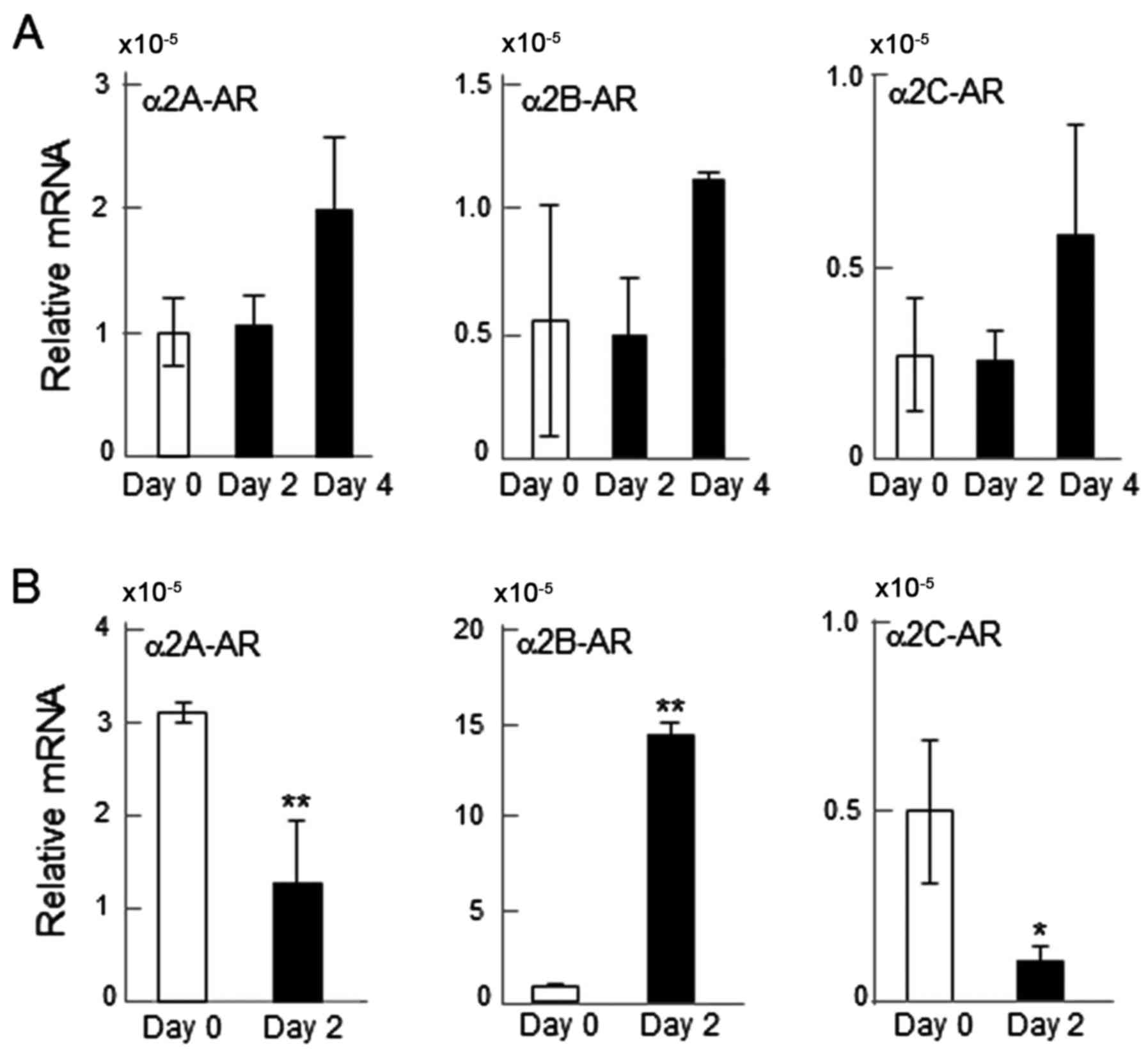
mRNA expression of α2-ARs
The mRNA levels of α2A-, α2B- and α2C-ARs were
determined. All three of the α2-ARs were detectable (Fig. 1). However, the responses to RANKL
differed between RAW264.7 and primary bone marrow cells. RANKL
administration on days 0, 2 and 4 did not significantly alter the
mRNA levels of α2-ARs in RAW264.7 cells (Fig. 1A); while it significantly altered
their mRNA levels in primary bone marrow cells when detected after
2 days. Specifically, the mRNA expression of α2A- and α2C-ARs was
significantly downregulated by RANKL administration (P<0.01 and
P<0.05, respectively), while that of α2B-AR was upregulated
(P<0.01; Fig. 1B).
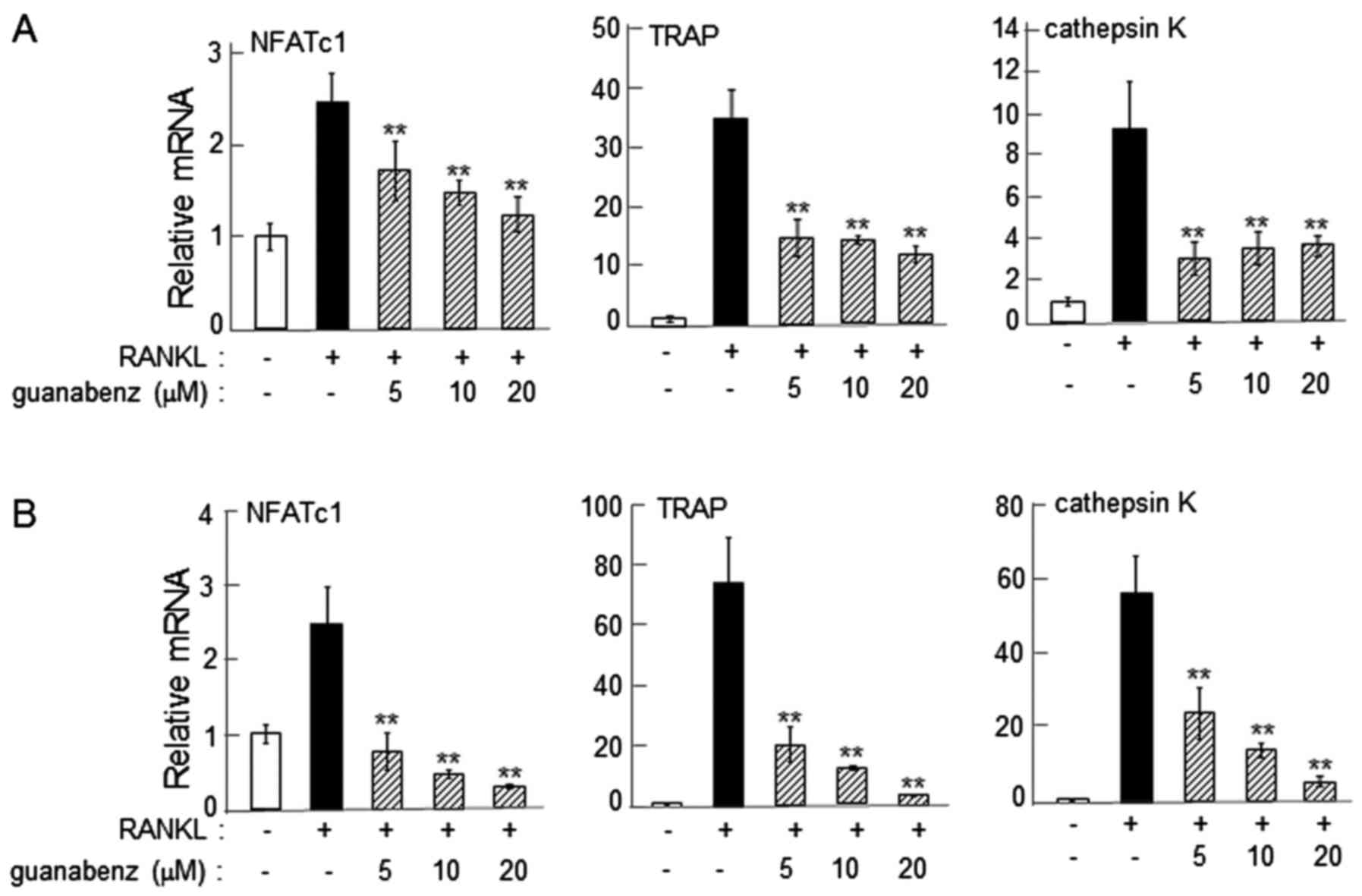
α2-AR agonist-driven reduction in the
expression of osteoclast genes
On day 2 following the administration of RANKL, the
mRNA levels of NFATc1, TRAP and cathepsin K were significantly
reduced by 5–20 µM guanabenz in RAW264.7 and primary bone marrow
cells (P<0.01; Fig. 2).
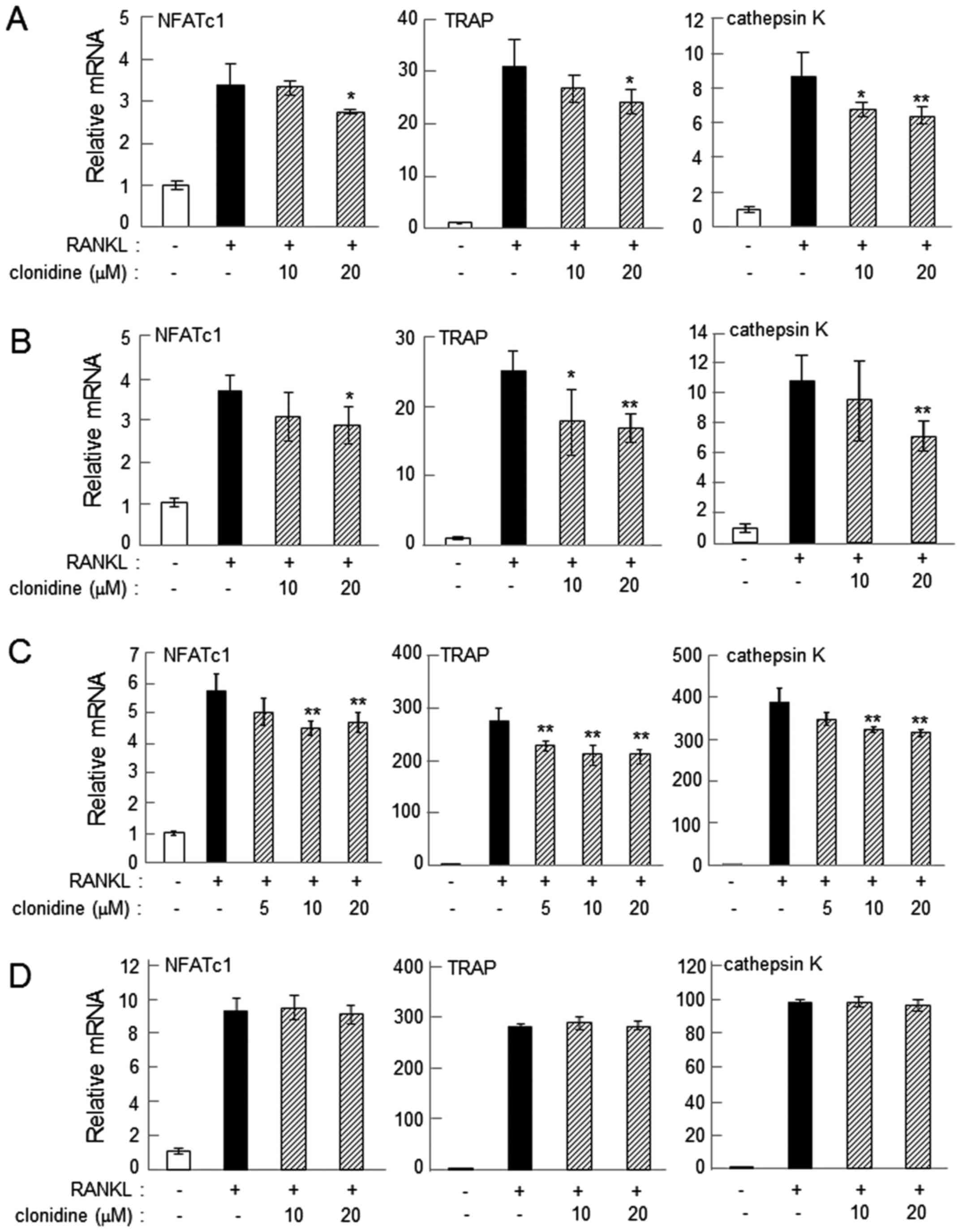
Administration of 20 µM clonidine consistently suppressed
RANKL-induced upregulation of NFATc1, TRAP and cathepsin K on days
2 and 4 in RAW264.7 cells (P<0.05; Fig. 3A and B). In primary bone marrow cells,
administration of 10–20 µM clonidine suppressed the mRNA levels of
the osteoclast genes when clonidine was applied alongside RANKL
(P<0.01; Fig. 3C). However, when
clonidine was administered 1 day after the administration of RANKL,
it did not alter the mRNA levels of NFATc1, TRAP or cathepsin K
(Fig. 3D). The mRNA levels of these
osteoclast genes were also consistently downregulated by 20 µM
xylazine in RAW264.7 and primary bone marrow cells on day 2 after
administration of RANKL (P<0.05; Fig.
4).
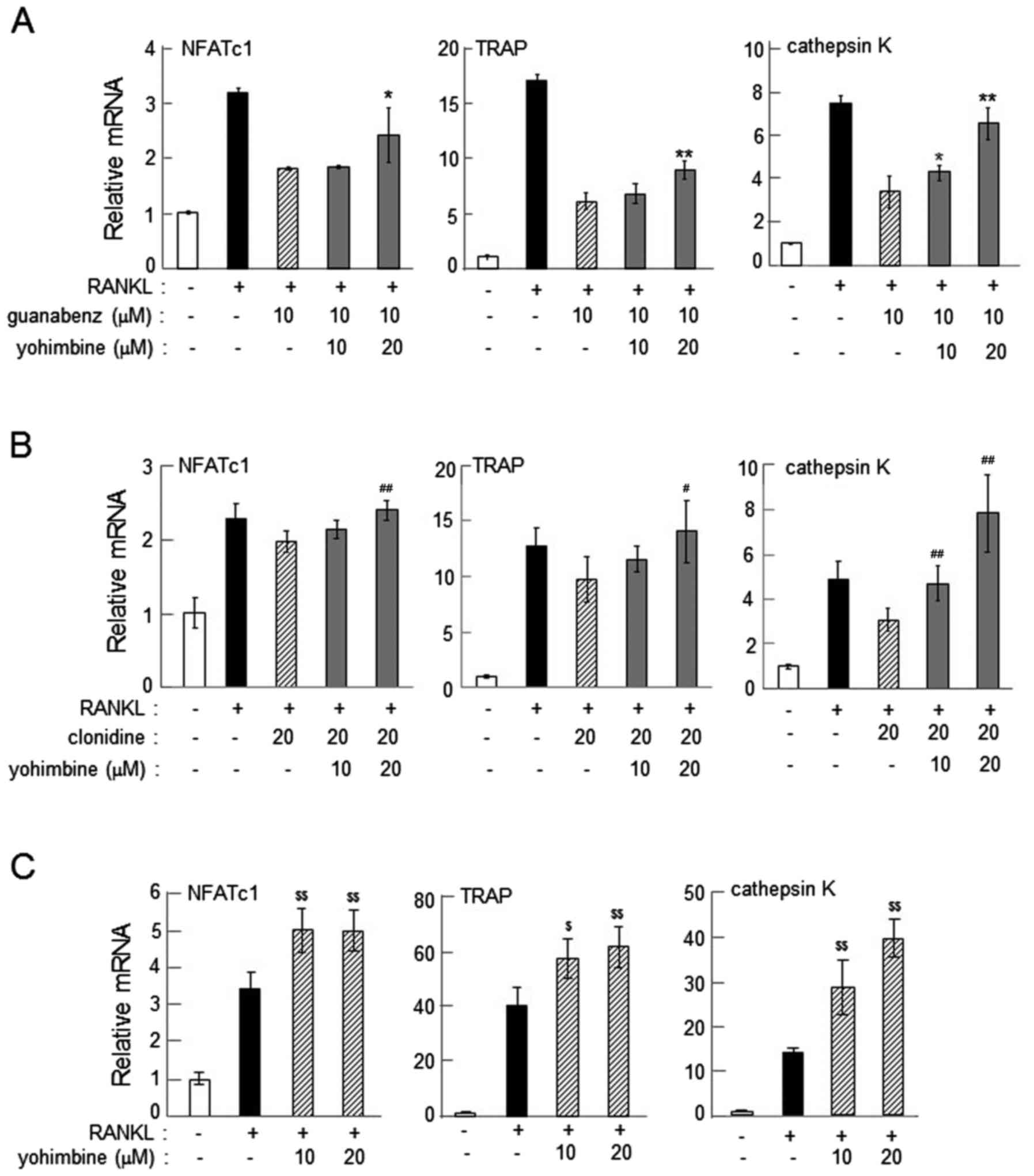
Suppression of α2-AR agonist-driven
reduction of osteoclast gene expression by yohimbine or
idazoxan
The reduction in the mRNA levels of NFATc1, TRAP and
cathepsin K in response to guanabenz and clonidine was consistently
suppressed by 20 µM yohimbine (P<0.05; Fig. 5A and B) or 20 µM idazoxan (P<0.05;
Fig. 6). This result indicates that
α2-AR antagonists inhibit the action of α2-AR agonists, and also
supports the notion that α2-ARs may be involved in regulation of
osteoclast gene expression. Of note, administration of yohimbine
alone upregulated the expression of the osteoclast genes at
concentrations of 10 (P<0.05) and 20 (P<0.01) µM (Fig. 5C).
Inhibitory effects of α2-AR agonist on
osteoclastogenesis
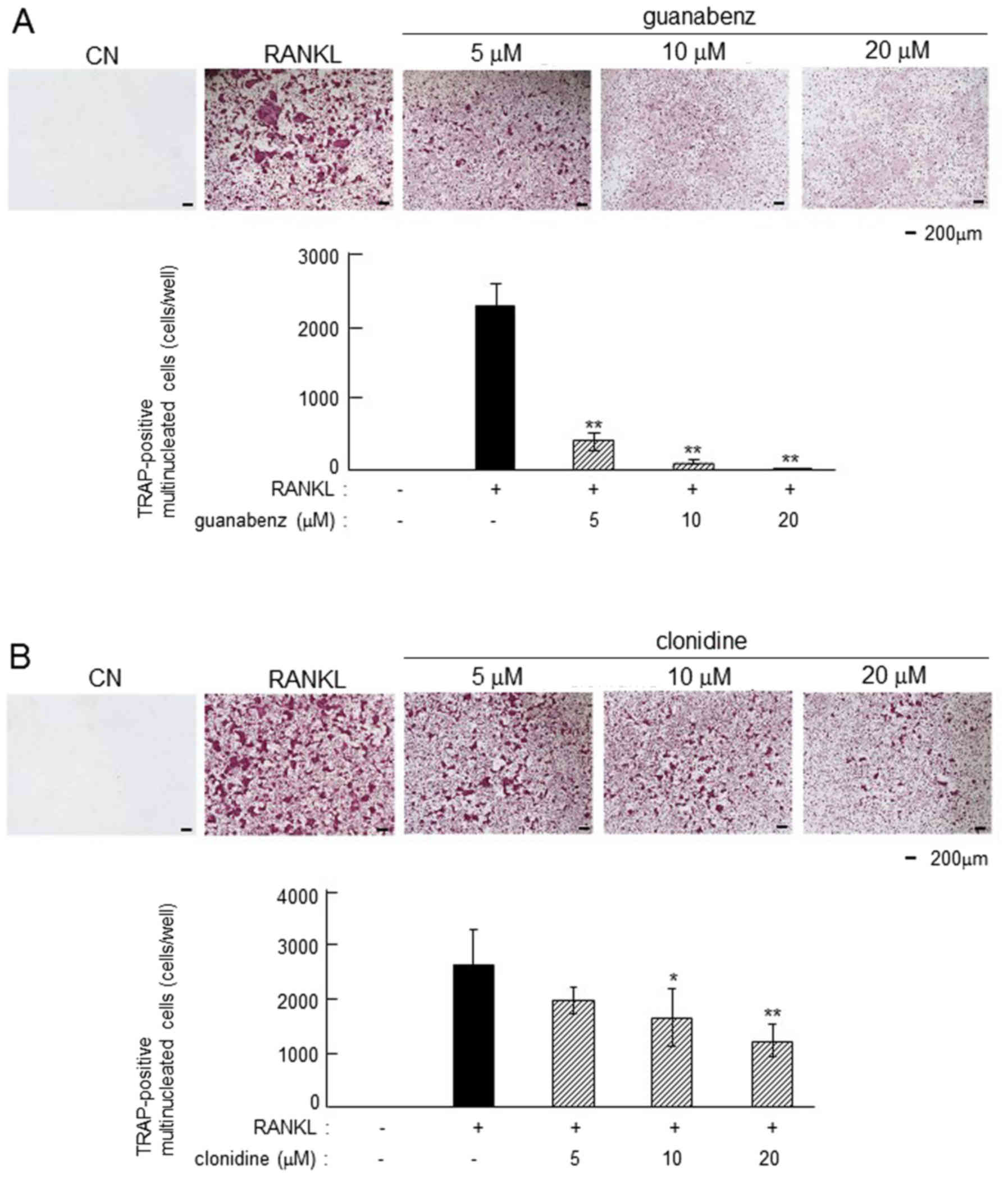
In RANKL-induced primary bone marrow cells,
guanabenz and clonidine suppressed osteoclastogenesis in a
dose-dependent manner (Fig. 7). The
number of TRAP-positive multi-nucleated osteoclasts was
significantly reduced by 5, 10 and 20 µM guanabenz (P<0.01) and
10 and 20 µM clonidine (P<0.05 and P<0.01, respectively).
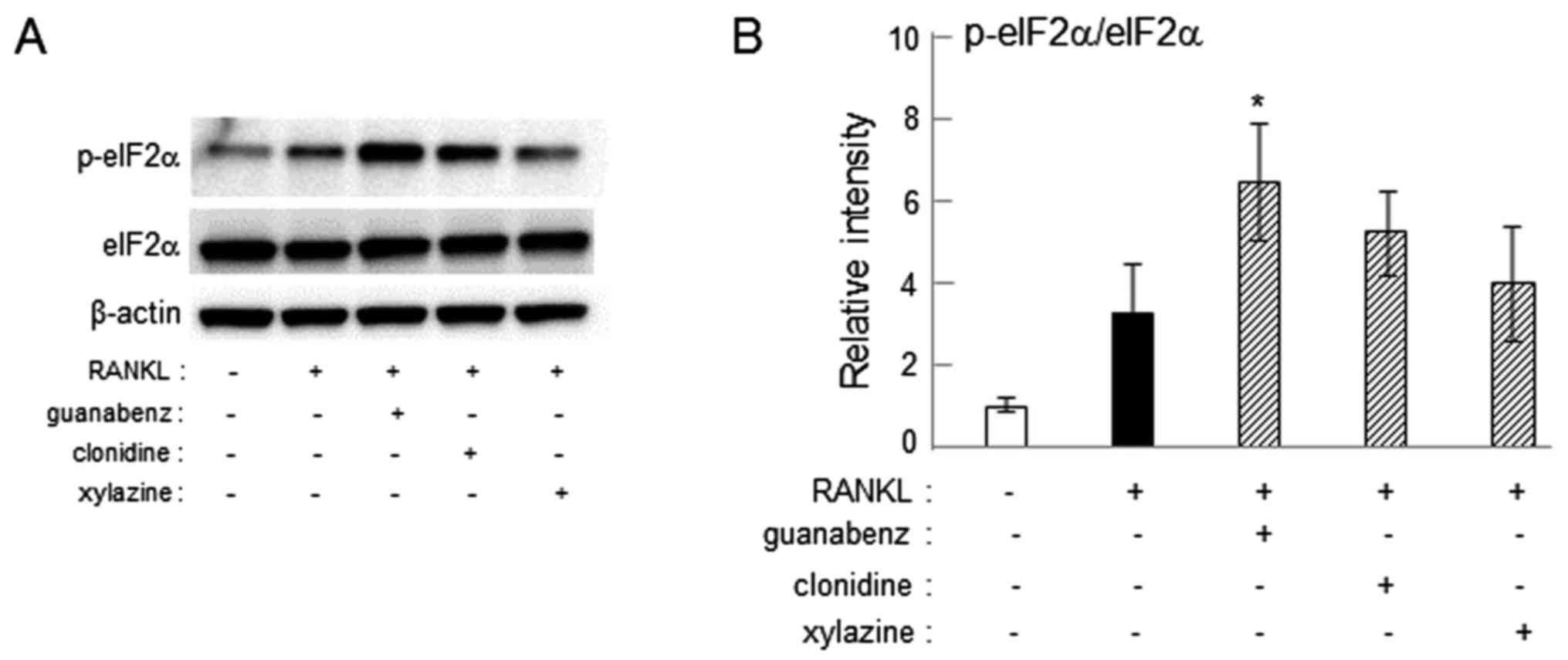
Increase in eIF2α phosphorylation by
guanabenz
Guanabenz is established to suppress
osteoclastogenesis by inhibiting dephosphorylation of eIF2α
(24–26). To determine whether clonidine and
xylazine also inhibit dephosphorylation of eIF2α, the level of
p-eIF2α was assessed in RAW264.7 cells. Western blot analysis
demonstrated that administration of 20 µM guanabenz increased the
phosphorylation of eIF2α (P<0.05), while treatment with 20 µM
clonidine or xylazine did not significantly affect the
phosphorylation level (Fig. 8).
Discussion
The current study demonstrated that three chemical
agents, guanabenz, clonidine and xylazine, which serve as α2-AR
agonists, suppressed the mRNA expression of three osteoclast genes
(NFATc1, TRAP and cathepsin K) in RAW264.7 and primary bone marrow
cells, and reduced the number of TRAP-positive multi-nucleated
osteoclasts in mouse bone marrow cells. Consistent with the
observed involvement of α2-ARs in response to guanabenz and
clonidine, administration of yohimbine and idazoxan, as α2-AR
antagonists, suppressed the α2-AR agonist-induced reduction in the
mRNA levels of the target genes. Compared with clonidine and
xylazine, the results also indicated that the greater inhibitory
effect of guanabenz in osteoclastogenesis may be associated with a
guanabenz-driven elevation in p-eIF2α. Furthermore, the findings
suggest that the responses to agents including clonidine may differ
depending on the administration window during
osteoclastogenesis.
Since yohimbine and idazoxan are established to
serve as α2-AR antagonists (27), it
was determined whether yohimbine and idazoxan could suppress the
effect of α2-AR agonists including guanabenz and clonidine. The
results demonstrated that administration of yohimbine or idazoxan
suppressed α2-AR agonist-driven reduction in the expression of
osteoclast genes. This indicates that the selected antagonists may
block binding of agonists to α2-ARs. Notably, administration of
yohimbine alone increased mRNA expression of the osteoclast genes.
It has been reported that certain ionotropic receptors, including
the γ-aminobutyric acid A receptors, as well as GPCRs, including
adrenergic, histamine and adenosine receptors, are constitutively
activated even in the absence of agonists, and this constitutive
activity may be inhibited by so-called inverse agonists (28–32). For
instance, adenosine A1 receptor is constitutively activated in
osteoclast precursors and rolofylline, a receptor antagonist, has
been reported to inhibit osteoclast differentiation as an inverse
agonist (32). Yohimbine may also
serve as an inverse agonist of α2-ARs, resulting in increased
expression of osteoclast genes.
Although guanabenz, clonidine and xylazine are α2-AR
agonists, there are differences in specificity among these agents.
Guanabenz is known to inhibit dephosphorylation of eIF2α and
attenuate endoplasmic reticulum stress, leading to downregulation
of osteoclast genes and attenuation of osteoclastogenesis (24–26).
Consistent with the action of guanabenz, western blot analysis
revealed that administration of guanabenz to RAW264.7 cells
elevated the level of p-eIF2α, while administration of clonidine or
xylazine did not significantly alter the phosphorylation level.
This result indicates that guanabenz serves as an inhibitor of
eIF2α dephosphorylation, as well as an α2-AR agonist, and induces
stronger suppression of osteoclast genes and attenuation of
osteoclastogenesis compared with clonidine and xylazine.
The action of clonidine and guanabenz via α2-ARs is
possibly mediated by c-AMP, since RANKL has been reported to
increase the level of c-AMP in osteoclast precursors (33). Elevation of c-AMP may activate
exchange protein directly activated by c-AMP (34,35), which
has been documented to promote osteoclast differentiation via
nuclear translocation of NF-κB (33).
Furthermore, activation of an adenylyl cyclase followed by
elevation of c-AMP upregulated c-Fos, which is established to
promote osteoclast development (36,37). Since
α2-ARs are known to suppress adenylyl cyclase and reduce c-AMP
(21), and clonidine and guanabenz
have been reported to reduce c-Fos expression (25,38), it is
possible that the α2 agonists in the current study reduce
RANKL-induced c-AMP, resulting in the suppression of
osteoclastogenesis.
While clonidine served as an inhibitor of
osteoclastogenesis in the current study, its effect during the
course of osteoclastogenesis is not completely understood. A
previous report identified that clonidine increased the number of
TRAP-positive osteoclasts in mouse bone marrow cells, and that its
administration did not alter the number of TRAP-positive
osteoclasts in α2A and α2C double knockout mice (7). It has also been reported that the number
of TRAP-positive osteoclasts was not affected by clonidine in
cluster of differentiation 14+ osteoclast precursors
(39). A major difference among these
studies appears to be the timing of clonidine administration. In
the current study, clonidine was applied on day 0 together with
RANKL or 1 day after administration of RANKL, while it was
administered on days 2 and 11 in the previous reports (7,39). The
present study revealed that when clonidine was applied alongside
RANKL in primary bone marrow cells, it suppressed the mRNA levels
of NFATc1, TRAP and cathepsin K; however, when clonidine was
administered 1 day after the administration of RANKL, it did not
alter mRNA levels. Therefore, as the mRNA levels of α2A- and
α2C-ARs in primary bone marrow cells were downregulated on day 2,
it is possible that the efficacy of clonidine as an inhibitor may
depend on the timing of its administration as well as the
expression profiles of α2A and α2C receptors.
While it was demonstrated in the current study that
α2-ARs on osteoclast precursors suppressed osteoclastogenesis,
pre-clinical studies in laboratory animals are necessary prior to
clinical studies and application in patients. As the current study
was an in vitro analysis, animal studies using conditional
knockout mice or other appropriate models are recommended to verify
the present findings. Nonetheless, the results indicated that
α2-ARs may be involved in the regulation of osteoclastogenesis in
RAW264.7 and primary bone marrow cells in vitro.
Acknowledgements
Not applicable.
Funding
The current study was supported in part by the
Grants-in-Aid for Scientific Research project of the Ministry of
Education, Culture, Sports, Science and Technology, Japan (grant
no. 17K11657, awarded to KaH) and by the National Institutes of
Health, Bethesda, MD, USA (grant no. NIH R01AR52144, awarded to
HY).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
KoH, KaH, HY, HK, KT, KI, DK, TH, KM, SG and AT
designed the research. KoH, KaH, AC, HM and SY performed the
experiments and analyzed the data. KaH wrote the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The protocols for animal experiments were approved
by the Aichi-Gakuin University Animal Research Committee (Nagoya,
Japan).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Karsenty G: Convergence between bone and
energy homeostases: Leptin regulation of bone mass. Cell Metab.
4:341–348. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Togari A, Arai M and Kondo A: The role of
the sympathetic nervous system in controlling bone metabolism.
Expert Opin Ther Targets. 9:931–940. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Elefteriou F, Campbell P and Ma Y: Control
of bone remodeling by the peripheral sympathetic nervous system.
Calcif Tissue Int. 94:140–151. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Niedermair T, Kuhn V, Doranehgard F,
Stange R, Wieskötter B, Beckmann J, Salmen P, Springorum H-R,
Straub RH, Zimmer A, et al: Absence of substance P and the
sympathetic nervous system impact on bone structure and chondrocyte
differentiation in an adult model of endochondral ossification.
Matrix Biol. 38:22–35. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Togari A: Adrenergic regulation of bone
metabolism: Possible involvement of sympathetic innervation of
osteoblastic and osteoclastic cells. Microsc Res Tech. 58:77–84.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arai M, Nagasawa T, Koshihara Y, Yamamoto
S and Togari A: Effects of beta-adrenergic agonists on
bone-resorbing activity in human osteoclast-like cells. Biochim
Biophys Acta. 1640:137–142. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fonseca TL, Jorgetti V, Costa CC, Capelo
LP, Covarrubias AE, Moulatlet AC, Teixeira MB, Hesse E, Morethson
P, Beber EH, et al: Double disruption of α2A- and α2C-adrenoceptors
results in sympathetic hyperactivity and high-bone-mass phenotype.
J Bone Miner Res. 26:591–603. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kodama D and Togari A: Noradrenaline
stimulates cell proliferation by suppressing potassium channels via
G(i/o) -protein-coupled α(1B) -adrenoceptors in human osteoblasts.
Br J Pharmacol. 168:1230–1239. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tanaka K, Hirai T, Kodama D, Kondo H,
Hamamura K and Togari A: α1B-Adrenoceptor signalling regulates bone
formation through the up-regulation of CCAAT/enhancer-binding
protein δ expression in osteoblasts. Br J Pharmacol. 173:1058–1069.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kajimura D, Hinoi E, Ferron M, Kode A,
Riley KJ, Zhou B, Guo XE and Karsenty G: Genetic determination of
the cellular basis of the sympathetic regulation of bone mass
accrual. J Exp Med. 208:841–851. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McDonald SJ, Dooley PC, McDonald AC,
Djouma E, Schuijers JA, Ward AR and Grills BL: α(1) adrenergic
receptor agonist, phenylephrine, actively contracts early rat rib
fracture callus ex vivo. J Orthop Res. 29:740–745. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kondo H, Takeuchi S and Togari A:
β-Adrenergic signaling stimulates osteoclastogenesis via reactive
oxygen species. Am J Physiol Endocrinol Metab. 304:E507–E515. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Takeuchi T, Tsuboi T, Arai M and Togari A:
Adrenergic stimulation of osteoclastogenesis mediated by expression
of osteoclast differentiation factor in MC3T3-E1 osteoblast-like
cells. Biochem Pharmacol. 61:579–586. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nishiura T and Abe K: α1-adrenergic
receptor stimulation induces the expression of receptor activator
of nuclear factor kappaB ligand gene via protein kinase C and
extracellular signal-regulated kinase pathways in MC3T3-E1
osteoblast-like cells. Arch Oral Biol. 52:778–785. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aitken SJ, Landao-Bassonga E, Ralston SH
and Idris AI: Beta2-adrenoreceptor ligands regulate osteoclast
differentiation in vitro by direct and indirect mechanisms. Arch
Biochem Biophys. 482:96–103. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hein L, Altman JD and Kobilka BK: Two
functionally distinct α2-adrenergic receptors regulate sympathetic
neurotransmission. Nature. 402:181–184. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
MacMillan LB, Hein L, Smith MS, Piascik MT
and Limbird LE: Central hypotensive effects of the
alpha2a-adrenergic receptor subtype. Science. 273:801–803. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lakhlani PP, MacMillan LB, Guo TZ, McCool
BA, Lovinger DM, Maze M and Limbird LE: Substitution of a mutant
α2a-adrenergic receptor via ‘hit and run’ gene targeting reveals
the role of this subtype in sedative, analgesic, and
anesthetic-sparing responses in vivo. Proc Natl Acad Sci USA.
94:9950–9955. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fagerholm V, Haaparanta M and Scheinin M:
α2-adrenoceptor regulation of blood glucose homeostasis. Basic Clin
Pharmacol Toxicol. 108:365–370. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Albarrán-Juárez J, Gilsbach R, Piekorz RP,
Pexa K, Beetz N, Schneider J, Nürnberg B, Birnbaumer L and Hein L:
Modulation of α2-adrenoceptor functions by heterotrimeric Galphai
protein isoforms. J Pharmacol Exp Ther. 331:35–44. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Storch U, Straub J, Erdogmus S, Gudermann
T, Mederos Y and Schnitzler M: Dynamic monitoring of
Gi/o-protein-mediated decreases of intracellular cAMP by FRET-based
Epac sensors. Pflugers Arch. 469:725–737. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hamamura K, Chen A, Nishimura A, Tanjung
N, Sudo A and Yokota H: Predicting and validating the pathway of
Wnt3a-driven suppression of osteoclastogenesis. Cell Signal.
26:2358–2369. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hamamura K, Tanjung N and Yokota H:
Suppression of osteoclastogenesis through phosphorylation of
eukaryotic translation initiation factor 2 alpha. J Bone Miner
Metab. 31:618–628. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hamamura K, Chen A, Tanjung N, Takigawa S,
Sudo A and Yokota H: In vitro and in silico analysis of an
inhibitory mechanism of osteoclastogenesis by salubrinal and
guanabenz. Cell Signal. 27:353–362. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hamamura K, Tanjung N, Chen A, Yokota H
and Togari A: Suppression of osteoclastogenesis via upregulation of
Zfyve21 and Ddit4 by salubrinal and guanabenz. Oral Therap
Pharmacol. 35:127–135. 2016.
|
|
27
|
Wade SM, Lan K, Moore DJ and Neubig RR:
Inverse agonist activity at the alpha(2A)-adrenergic receptor. Mol
Pharmacol. 59:532–542. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Strange PG: Mechanisms of inverse agonism
at G-protein-coupled receptors. Trends Pharmacol Sci. 23:89–95.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Milligan G: Constitutive activity and
inverse agonists of G protein-coupled receptors: A current
perspective. Mol Pharmacol. 64:1271–1276. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Soudijn W, van Wijngaarden I and Ijzerman
AP: Structure-activity relationships of inverse agonists for
G-protein-coupled receptors. Med Res Rev. 25:398–426. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cotecchia S: Constitutive activity and
inverse agonism at the α1adrenoceptors. Biochem Pharmacol.
73:1076–1083. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
He W, Wilder T and Cronstein BN:
Rolofylline, an adenosine A1 receptor antagonist, inhibits
osteoclast differentiation as an inverse agonist. Br J Pharmacol.
170:1167–1176. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mediero A, Perez-Aso and Cronstein BN:
Activation of EPAC1/2 is essential for osteoclast formation by
modulating NFκB nuclear translocation and actin cytoskeleton
rearrangements. FASEB J. 28:4901–4913. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
de Rooij J, Zwartkruis FJ, Verheijen MH,
Cool RH, Nijman SM, Wittinghofer A and Bos JL: Epac is a Rap1
guanine-nucleotide-exchange factor directly activated by cyclic
AMP. Nature. 396:474–477. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ferrero JJ, Alvarez AM, Ramírez-Franco J,
Godino MC, Bartolomé-Martín D, Aguado C, Torres M, Luján R, Ciruela
F and Sánchez-Prieto J: β-Adrenergic receptors activate exchange
protein directly activated by cAMP (Epac), translocate Munc13-1,
and enhance the Rab3A-RIM1α interaction to potentiate glutamate
release at cerebrocortical nerve terminals. J Biol Chem.
288:31370–31385. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Aerts I, Grobben B, Van Ostade X and
Slegers H: Cyclic AMP-dependent down regulation of ecto-nucleotide
pyrophosphatase/phosphodiesterase 1 (NPP1) in rat C6 glioma. Eur J
Pharmacol. 654:1–9. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Inda C, Bonfiglio JJ, Dos Santos Claro PA,
Senin SA, Armando NG, Deussing JM and Silberstein S: cAMP-dependent
cell differentiation triggered by activated CRHR1 in hippocampal
neuronal cells. Sci Rep. 7:19442017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
El-Mas MM and Abdel-Rahman AA: Clonidine
diminishes c-jun gene expression in the cardiovascular sensitive
areas of the rat brainstem. Brain Res. 856:245–249. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Limonard EJ, Schoenmaker T, de Vries TJ,
Tanck MW, Heijboer AC, Endert E, Fliers E, Everts V and Bisschop
PH: Clonidine increases bone resorption in humans. Osteoporos Int.
27:1063–1071. 2016. View Article : Google Scholar : PubMed/NCBI
|