Introduction
Lung cancer is the second most commonly diagnosed
cancer, with an estimated 2.2 million new cases (11.4% of total
newly diagnosed cancer cases) annually. This disease remains the
leading cause of cancer-associated death, with an estimated 1.8
million deaths (18% of all cancer-associated deaths) per year
(1). Non-small-cell lung cancer
(NSCLC) accounts for 85% of all cases of lung cancer and is linked
to a poor prognosis. Acquired resistance is a major obstacle in
treatment (2-4).
Deregulated PI3K/AKT signaling is associated with tumorigenesis,
tumor progression and drug resistance in NSCLC (5,6). Based
on oncogenic evidence and possible druggability of its components,
PI3K/AKT/mTOR signaling has been explored for development of
anticancer therapy with limited success, mainly because of
intrinsic and acquired resistance and toxicity of PI3K inhibitor
therapy (6,7).
Serum and glucocorticoid regulated kinase 1 (SGK1),
which belongs to the family of AGC serine threonine kinases, is
activated by serum, steroids and cytokines. Therefore, this enzyme
serves as a critical regulator of the transmission of cell survival
signals pertaining to steroids and other growth factors (8). Following phosphorylation and
activation, similar to AKT, SGK1 is rapidly translocated to the
nucleus, where it phosphorylates numerous transcription factors, as
well as antiapoptotic and cell cycle-related proteins, thereby
resulting in cell survival and proliferation of multiple cancer
types including NSCLC (9-11).
The catalytic domain of SGK1 is 54% homologous to AKT and both
kinases share the same phosphorylation consensus motif (RXRXXS/T);
hence, they phosphorylate similar downstream targets (12). However, unlike AKT, the cellular
functions of SGK1 and its physiological targets are largely
uncharacterized.
Studies have shown that SGK1 is activated via
PI3K-independent mechanisms and sustains mTORC1 activity in the
presence of phosphorylated (p-) AKT after PI3K inhibitor treatment
(12,13). Thus, SGK1 could be a promising
target for PI3K-resistant NSCLC tumors either as a standalone
therapy or in combination with PI3K inhibitors. The present study
aimed to investigate the potential role of SGK1 in PI3K inhibitor
resistance in NSCLC and to elucidate molecular mechanism.
Materials and methods
Cell lines, culture conditions and
reagents
A549, NCI-H460, NCI-H441 and NCI-H358 cell lines
were purchased from American Type Culture Collection and grown in
RPMI-1640 (cat. no. #R8758, Sigma-Aldrich; Merck KGaA) supplemented
with 10% fetal bovine serum (cat. no. #13140071, Gibco; Thermo
Fisher Scientific, Inc.). All cell lines were maintained at 37˚C
with 5% CO2 in a cell culture incubator. PI3K inhibitor
BYL719 was obtained from MedChemExpress (cat. no. HY-15244) and
SGK1 inhibitor GSK650394 from Selleck Chemicals (cat. no.
S7209).
Colony formation assay (CFA)
Cells were seeded in a 48-well plate at a density of
500 cells/well and allowed to adhere overnight at 37˚C. Cells were
treated with BYL719 (10 µM) and GSK650394 (both 10 µM) alone or in
combination at 37˚C in CO2 incubator (5%
CO2). 0.1% DMSO treated cells were used as a vehicle
control. Vacuole formation was observed under a light microscope
with 20x magnification (EVOS XL Core system). After 7 days, cells
were stained with crystal violet (0.5% crystal violet, 1% methanol
and 1% formaldehyde in water) at 25˚C for 10 min. Colonies were
destained at 25˚C for 30 min with 10% acetic acid. Absorbance was
read at 592 nm on the Synergy Neo2™ reader (Agilent
Technologies, Inc.) and inhibition of colony formation relative to
the untreated control was determined.
Antiproliferation assay
All cell lines were seeded at a cell density of 250
cells/well in white 96-well plates. Cells were treated with BYL719
and/or GSK650394 for 7 days in a cell culture incubator (37˚C, 5%
CO2). Cell proliferation was measured using
CellTiterGlo® (Promega Corporation; cat. no. #G7570)
reagent as per the manufacturer's instructions.
Cell cycle analysis
NCI-H460 cells were seeded at a cell density of
0.1x106 cells/well in six-well plates. Cells were
treated with BYL719 (3 µM) and GSK650394 (1 and 3 µM) alone or in
combination for 16 h in a cell culture incubator (37˚C, 5%
CO2). 0.1% DMSO treated cells were used as a vehicle
control. Cells were harvested via trypsinization, washed with PBS
and fixed in 70% ethanol at 2-8˚C for 30 min. Cells were stained
with 50 µg/ml propidium iodide solution (containing 1% Triton X-100
and 0.1 mg/ml RNAse). The stained cells were washed with PBS and
analyzed using BD FACS Canto-II Flow Cytometer (BD
FACSDiva™ Software v9.0, both BD Biosciences).
Western blotting
Cells were lysed in 1X cell lysis buffer (cat. no.
#9803, Cell Signaling Technology, Inc.) containing 1X protease
(cat. no. #P8340) and phosphatase inhibitor cocktails (cat. no.
#P5726, both Sigma-Aldrich; Merck KGaA). Protein estimation was
performed using BCA method. SDS-PAGE (10%) and immunoblotting
(nitrocellulose membrane) were performed with 25 µg sample/lane.
The membrane was blocked with blocking buffer [5% bovine serum
albumin (MP Bio; cat. no. 0882045-CF) in Tris-buffered saline with
0.1% Tween-20 (TBST) for 1 h at 25˚C]. Incubation with primary
(1:1,000, overnight at 2-8˚C) and secondary (1:5,000 dilution, 2 h
at 25˚C) antibody solutions was performed with washing of the
membrane with TBST in between. The membrane was then washed in TBST
and developed with Super Signal™ West Femto (cat. no.
#34094, Thermo Fisher Scientific, Inc.) substrate on Biorad
Chemidoc™. For proteosomal inhibition, the cells were
treated with 10 µM MG-132 (cat. no. #M7449, Sigma-Aldrich; Merck
KGaA) for 4 h at 37˚C prior to harvesting. The rest of the protocol
was as aforementioned. 0.1%-DMSO treated cells were used as a
vehicle control. All primary and secondary antibodies are listed in
Table SI. Densitometry analysis
was performed with the ImageJ software (version 1.53t, National
Institutes of Health). The ratio of p- to total protein or total
protein to β-actin was calculated for the estimation of
fold-change.
Synergy score calculation
Bliss, Loewe, ZIP (Zero interaction potency) and HSA
(highest single agent) synergy scores were calculated with the
Synergy Finder software v3.0 (synergyfinder.org).
Statistical analysis
Data are presented as the mean ± SEM of three
independent experiments. Half-maximal inhibitory concentration
(IC50) values were generated using non-linear regression
analysis. Statistical analysis using one-way ANOVA followed by post
hoc test (Sídak's multiple comparisons test) was performed with
GraphPad Prism version 9.0 software (GraphPad Software, Inc.;
Dotmatics). P<0.05 was considered to indicate a statistically
significant difference.
Results
SGK1 protein is variably expressed in
NSCLC cell lines
SGK1 protein expression in NSCLC cell lines was
evaluated. The protein was variably expressed in the cell lines,
with the highest expression in A549, NCI-H460 and NCI-H1299 cells.
NCI-H441 cells showed the lowest expression (Fig. S1A). Furthermore, BYL719 (a specific
inhibitor of PI3Ka) exhibited lower antiproliferation activity in
cells with higher SGK1 protein expression than in those with lower
expression (Fig. S1B). These
results indicated that high SGK1 levels contributed to the
resistance of NSCLC cell lines to PI3K inhibition.
Inhibition of SGK1 sensitizes NSCLC
cell lines to PI3K inhibition
To determine the role of SGK1 in PI3K inhibitor
resistance, the combined potential of SGK1 and PI3K inhibitors in
NSCLC cell lines (A549 and NCI-H460) that showed high SGK1
expression was evaluated. CFA and cell proliferation assay were
performed with the PI3Kα inhibitor BYL719 and/or SGK1 inhibitor
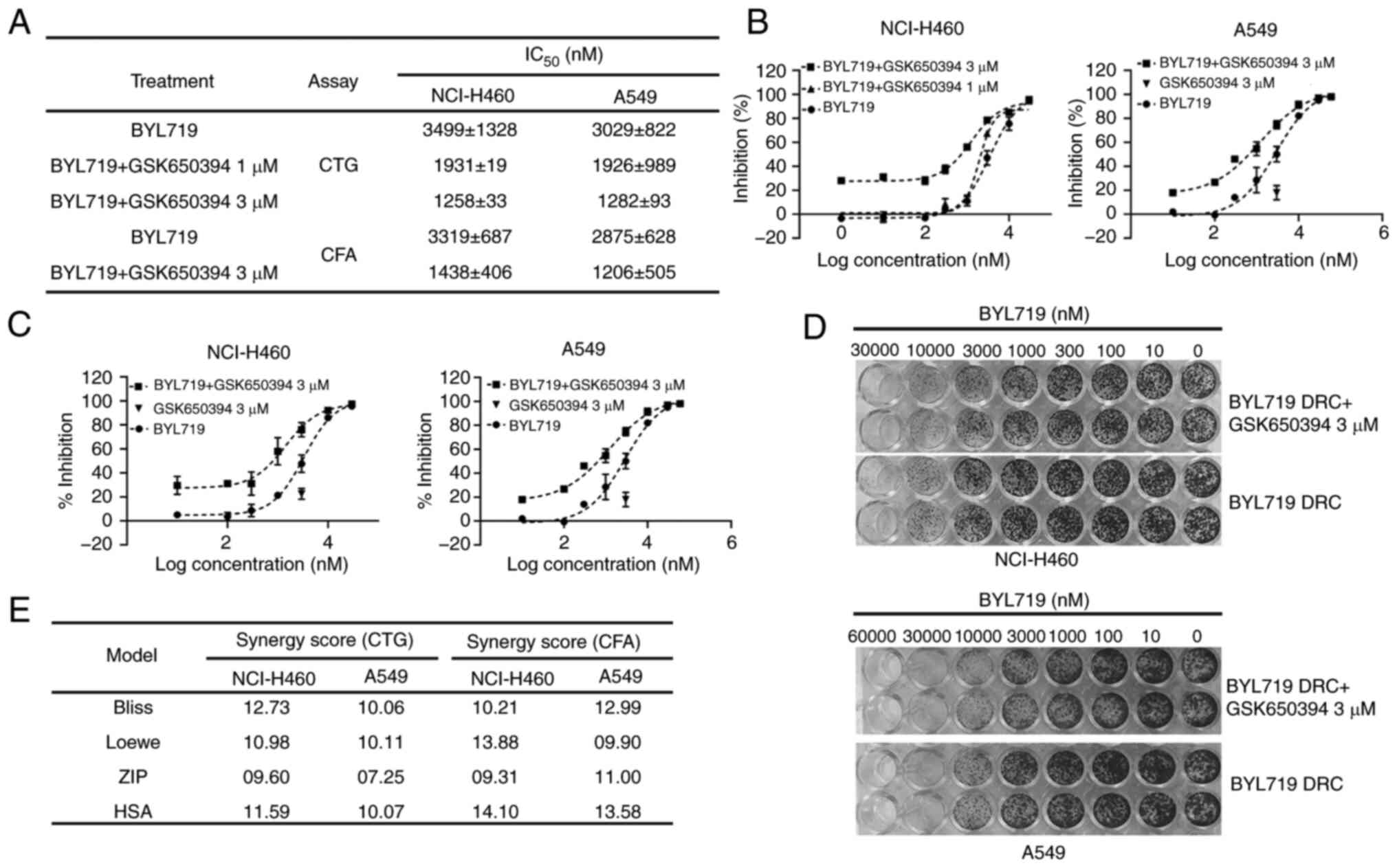
GSK650394. In both cell proliferation and CFA, BYL719-alone
displayed notable antitumor activity in NCI-H460 and A549 cells
(Fig. 1A). Despite the minimal
effect on cell proliferation when used as a single agent, treatment
with 1 and 3 µM GSK650394 sensitized both BYL719-resistant NSCLC
cell lines (NCI-H460 and A549) to PI3K inhibitors (Fig. 1A and B). In both cell lines, >2.5-fold
decrease in IC50 of BYL719 was observed in the presence
of 3 µM GSK650394 compared with BYL719 alone (Fig. 1A). Similarly, 3 µM GSK650394 was
sufficient to sensitize both NSCLC cell lines to PI3K inhibitors in
CFA (Fig. 1A, C and D).
In both assays, slight to moderate synergy in antitumor response
was noted with the combination of BYL719 and GSK650394, with a
Bliss synergy score of >10 (Figs.
1E and S1C and D). Other synergy models also confirmed
slight to moderate synergistic activity of combination treatment in
NSCLC cell lines for both assays (Fig.
1E). Based on these findings, it was hypothesized that elevated
SGK1 was responsible for resistance to PI3K inhibitors in NSCLC
cell lines and that inhibiting its activity restored sensitivity to
PI3K inhibition.
Combined inhibition of PI3K and SGK1
results in inhibition of the AKT/mTOR/S6 axis
Reports suggest that inhibition of PI3K completely
abolishes AKT activity but SGK1 maintains mTORC1 activity and
weakens the antitumor activity of PI3K inhibitors in breast cancer
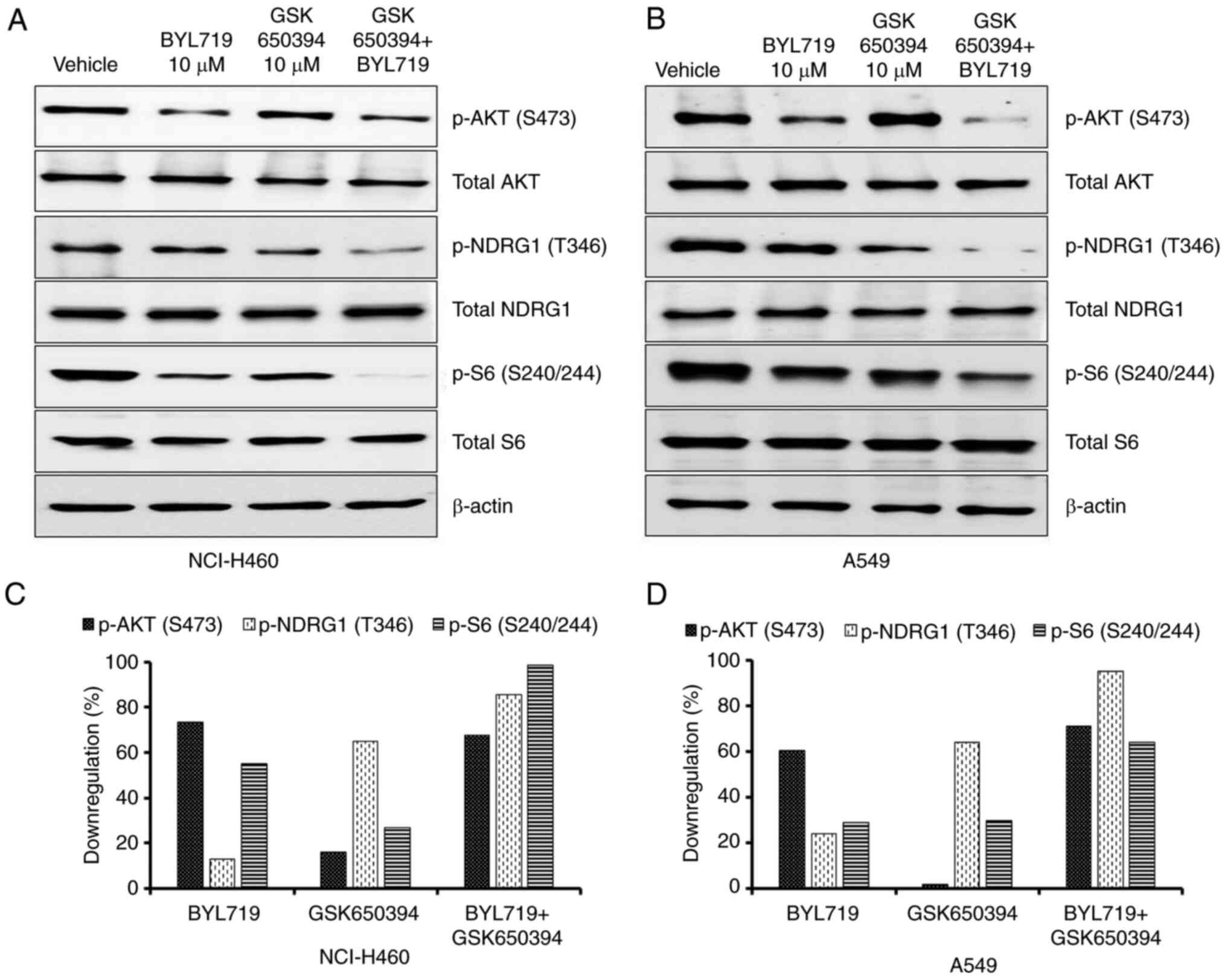
(12,13). NSCLC cell lines were treated with 10
µM BYL719 and/or GSK650394 for 48 h and the effect on mTORC1/S6
pathway activation was evaluated using western blotting. N-Myc
downstream-regulated gene 1 (NDRG1) is specific substrate of SGK1
and phosphorylated by SGK1 at Thr346, which primes NDRG1 for
phosphorylation by Glycogen synthase kinase-3(12). Hence, p-NDRG1(Thr346) levels were
evaluated as marker of SGK1 activity. SGK1 inhibitor (GSK650394)
alone had no effect on phosphorylation of AKT but resulted in
decreased expression of p-NDRG1, indicating GSK650394 inhibited
SGK1 activity (Fig. 2A and B). In NCI-H460 cells, BYL719 treatment
notably suppressed AKT phosphorylation (73%). However, only
moderate inhibition of S6 phosphorylation (55%) was observed with
BYL719 treatment (Fig. 2A and
C). When GSK650394 was combined
with BYL719, p-S6 levels were almost completely abolished (98%),
with similar p-AKT inhibition as in cells treated with BYL719 alone
(Fig. 2A and C). Comparable results were observed in
A549 cells (Fig. 2B and D). These data suggested that in the
absence of AKT phosphorylation, SGK1 maintains mTORC1 signaling and
may contribute to PI3K inhibitor resistance in NSCLC cell
lines.
SGK1 inhibition in combination with
BYL719 significantly enhances the expression of markers for
apoptosis, DNA damage and immunogenicity of NSCLC cell lines
Levels of pro-/antiapoptotic proteins were assessed
to confirm the cell death induced by combination treatment in
NCI-H460 cells. The cells were treated with BYL719 and/or GSK650394
(10 µM) for 48 h and the levels of pro- and antiapoptotic proteins
were estimated using western blot. BYL719 and GSK650394 alone had
no effect on proapoptotic (cleaved PARP1 and caspase 3) or
antiapoptotic markers in NCI-H460 cells (Fig. 3A). However, combination treatment
notably enhanced the expression of cleaved PARP1 and cleaved
caspase 3. In addition, the combination regimen decreased
expression of the antiapoptotic marker Bcl-xl (Fig. 3A). Combination treatment also
notably increased the DNA damage, as suggested by enhanced
expression of p-H2A.X (Fig.
3B).
 | Figure 3Combined inhibition of PI3K and SGK1
activity induces apoptosis, DNA damage and cell cycle arrest in
NCI-H460 cells. (A) Western blot analysis showed enhanced
proapoptotic (cleaved PARP1 and cleaved caspase 3) and decreased
antiapoptotic markers (Bcl-xl). (B) Levels of the DNA damage marker
(p-H2AX) increased following combination treatment. (C) Combination
treatment significantly augmented BYL719-induced G0/G1 phase cell
cycle arrest (**P<0.01, ***P<0.001), as
observed by (D) flow cytometry analysis of PI staining. P2, G0/G1;
P3, S; P4, G2/M; P5, sub-G1 phase. (E) Combination treatment
modulated cyclin D1, CDK4, cyclin E1 and CDK2 levels, as determined
using western blot. SGK1, Serum and Glucocorticoid kinase 1; p-,
phosphorylated; H2AX, Histone 2A X; ns, non-significant. |
Programmed death ligand 1 (PD-L1), also known as
CD274, is predominantly expressed on antigen-presenting and tumor
cells, whereas its receptor PD-1 is chiefly expressed on cytotoxic
T cells (14). The binding of PD-L1
to the PD-1 receptor suppresses the immune response against tumor
cells, which results in tumor immune evasion (15,16).
Glucocorticoids are commonly administered to manage side effects of
chemo- or immunotherapy and exert immunosuppressive and
anti-inflammatory effects via upregulation of SGK1(17). On the other hand, PD-L1 expression
is regulated by glucocorticoids via PI3K/signaling (18). Therefore, it was hypothesized that
SGK1 could be an important player in PD-L1 mediated
immunosuppression. Cells were treated with the SGK1 inhibitor alone
or in combination with PI3K inhibitor and the PD-L1 expression was
assessed using western blot. PD-L1 was highly expressed in the
NSCLC cell line NCI-H460, and its expression was partially
decreased by the treatment with BYL719 or GSK650394 alone. However,
the combination of both inhibitors resulted in inhibition of PD-L1
levels in NCI-H460 cells (Fig.
S2A). These data indicated possible involvement of the
SGK1/PD-L1 axis in immune evasion. Cells treated with GSK650394
alone or in combination showed morphological features of
cytoplasmic vacuole formation, which were not observed in BYL719-
or DMSO-treated cells (Fig.
S2B).
Combined inhibition of SGK1 and PI3K
results in cell cycle arrest in G1/S phase
To determine how combined inhibition of SGK1 and
PI3K inhibits cell viability, flow cytometry was used to examine
cell cycle progression. In NCI-H460 cells, a notable dose-dependent
increase in proportion of cells in G2/M phase was seen after 16 h
treatment with GSK650394. In addition, there was a decrease in
percentage of cells in G0/G1 phase (Figs. 3C and S2C and D). At a concentration of 1 µM, BYL719
resulted in non-significant cell cycle arrest in G0/G1 phase
compared with the vehicle-treated group (Fig. 3C and D). When BYL719 (1 µM) was combined with
GSK360394 (1 and 3 µM), a significant increase in the percentage of
G0/G1 cells was observed compared with the BYL719-alone group (63
to 72 and 76% at 3 and 1 µM GSK650394, respectively; Fig. 3C and D). This increase in G0/G1 phase cells was
in line with the decrease in G2/M phase cells in the combination
group compared with the BYL719-alone group (from 25 to 17 and 11%
at 3 and 1 µM GSK650394, respectively; Fig. S2C). Combining BYL719 with 1 µM
GSK650394 resulted in higher G0/G1 phase arrest compared with
BYL719 + 3 µM GSK950394. This may be due to significantly lower
population of G0/G1 cells in the 3 µM compared with 1 µM GSK650394
group (46 vs. 53%; Figs. 3C and
S2C).
Cell cycle progression is driven by heterodimeric
complexes of cyclins (A, B, D and E) with cyclin-dependent kinases
(CDK1, CDK2, CDK4 and CDK6). Cyclin D/CDK4 and cyclin E1/CDK2 are
the two major cyclin/CDK complexes required for cell cycle entry
and exit from the G1 phase (19).
To confirm that combination of BYL719 and GSK650394 induced G1/G0
cell cycle arrest, modulation of cell cycle arrest markers was
assessed using western blotting. Expression of cyclin D1 with its
binding partner CDK4 was notably decreased in the combination
treatment group compared with BYL719- and GSK650394-alone (Fig. 3E). Similarly, expression of cyclin
E1 with its binding partner CDK2 was also decreased by combination
treatment (Fig. 3E). Increased
phosphorylation of histone H3 at the serine 10 residue and cyclin
B1 levels confirmed G2/M phase arrest following GSK650394-alone
(Fig. S2D). Furthermore, p-H3
(S10) and cyclin B1 levels were reduced in the combination group
owing to the decreased proportion of cells in the G2/M phase
compared with GSK650394-alone. These findings confirmed the G0/G1
phase arrest in the combination group (Fig. S2D). Western blot data for cell
cycle marker analysis were consistent with the flow cytometry
results, demonstrating cell cycle arrest in the G1/S phase
following the combination treatment.
SGK1 inhibition in combination with
BYL719 inhibits MEK/ERK signaling:
Both AKT and SGK1 are activated by numerous
extracellular stimuli and have a range of downstream effectors.
MAPK/ERK and PI3K/AKT pathways typically act together in
tumorigenesis and tumor growth. Following PI3K stimulation, AKT
directly phosphorylates Raf at S259 and activates the RAF/MEK/ERK
cascade (20). Similarly, SGK1
phosphorylates ERK2 at the serine 29 residue, which activates
MEK/ERK signaling by enhancing the interaction between MEK1/2 and
ERK1/2 complexes (21). When NSCLC
cell lines were treated with BYL719 or GSK650394 alone, both
inhibitors showed little to no effect on the phosphorylation levels
of ERK and its downstream targets p-p90RSK and p-S6 in NCI-H460
cells (Fig. 4A and C). When cells were treated with a
combination of BYL719 and GSK650394, marked inhibition of pERK, as
well as inhibition of pP90RSK and p-S6, was observed (Fig. 4A and C). Similarly, inhibition of pERK and its
downstream targets was seen in A549 cells (Fig. 4B and D). These data confirmed the involvement of
the MEK/ERK pathway in resistance to PI3K inhibition and indicated
that it was overcome, at least partly, by the simultaneous
inhibition of SGK1 activity.
SGK1 inhibition abolishes
β-catenin-induced resistance to PI3K inhibition
The nuclear accumulation of β-catenin following
prolonged exposure to PI3K and AKT inhibitors induces resistance to
FOXO3a-mediated apoptosis in cancer cells. However, this resistance
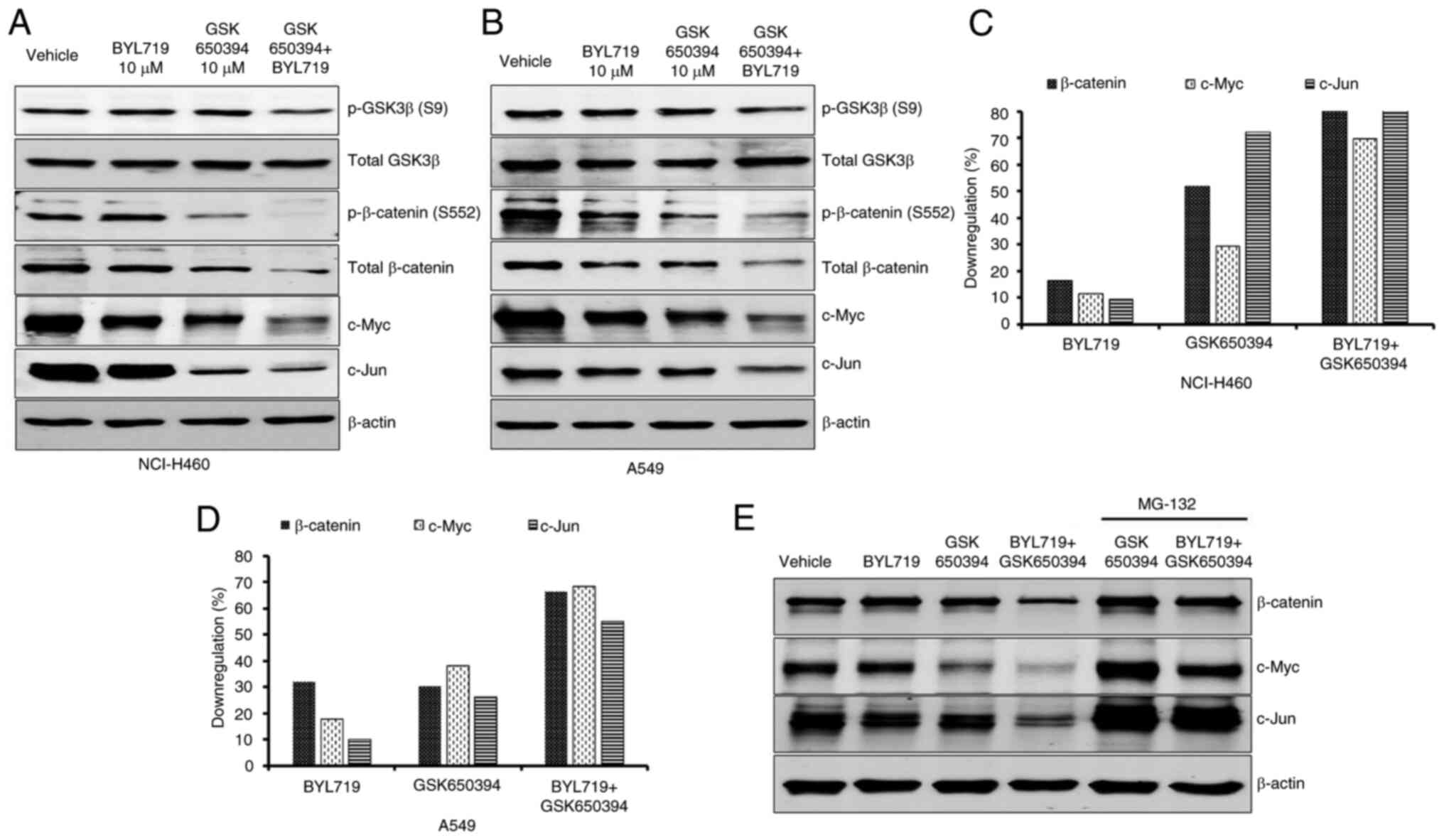
is overcome by the inhibition of Wnt β-catenin signaling (22). Therefore, the effect of the
combination of PI3K and SGK1 inhibitors on modulation of β-catenin
signaling was evaluated. BYL719- and GSK650394-alone had minimal
effect on S9 phosphorylation of GSK3β. Combination treatment
resulted in inhibition of GSK3β S9 phosphorylation in both NCI-H460
and A549 cells (Fig. 5A and
B). Similarly, levels of p- and
total β-catenin and its downstream targets c-Jun and c-Myc were
notably decreased following combination treatment in NCI-H460
(Fig. 5A and C) and A549 (Fig. 5B and D) cells. Decreased c-Myc levels occurred
due to decreased p-ERK signaling following combination treatment.
Unlike p-GSK3β S9 levels, GSK650394 alone showed marked inhibition
of β-catenin and c-Jun, indicating involvement of the
GSK3β-independent pathway (Fig. 5A
and B). To establish SGK1-mediated
degradation of β-catenin and its target genes c-Myc and c-Jun in
the combination regimen, the proteasomal inhibitor MG-132 was added
with GSK650394 alone and in combination with BYL719 in NCI-H460
cells. MG-132 treatment rescued β-catenin, c-Myc and c-Jun levels
following both GSK650394-alone and combination treatment. This
demonstrated that SGK1 inhibition alone and in combination with
BYL719 induced proteasomal degradation of β-catenin and its target
proteins (Fig. 5E). Collectively,
the present data indicated that reduced β-catenin accumulation by
SGK1 inhibition serves a key role in sensitizing NSCLC cell lines
to PI3K inhibitor treatment.
Discussion
Numerous PI3K inhibitors are currently being
clinically evaluated for treating patients with NSCLC: BYL719
(Alpelisib), a selective PI3Kα inhibitor, was approved for treating
advanced breast cancer in 2019 but showed no efficacy in NSCLC
trials (23,24). Considering the importance of PI3K
signaling in development and progression of NSCLC, PI3K inhibitors
have potential for treatment via strategies such as drug
combination and patient stratification. Here, SGK1 was expressed at
different levels in differing NSCLC cell lines; those expressing
higher SGK1 protein were more resistant to the PI3K inhibitor than
those with lower expression. Subsequently, whether inhibition of
elevated SGK1 activity sensitized the NSCLC cell lines to PI3K
inhibition was tested. A selective and potent SGK1 inhibitor,
GSK650394, with >30-fold selectivity against AKT and other
related kinases and >60-fold selectivity for SGK1 over PDK1 was
used (25). The combined inhibition
of SGK1 and PI3K resulted in synergistic anticancer activity in
BYL719-resistant NSCLC cell lines. These finding suggest that
inhibition of elevated SGK1 might restore sensitivity to PI3K
inhibition in NSCLC cells, however, validation studies
overexpressing or downregulating SGK1 in the presence and absence
of PI3K inhibitors are required. In addition, the increased
anticancer activity was associated with inhibition of mTORC1/S6
signaling as well as increased apoptosis following combination
treatment in NSCLC cell lines.
In breast cancer models, the combined inhibition of
PI3K/AKT and SGK1 is reported to exert notable anticancer effects
in vitro and in vivo; to the best of our knowledge,
however, the underlying molecular mechanisms have not been
investigated (12,13). Here, the combination treatment
notably increased cell cycle arrest in the G1/S phase. These
results were validated by decreased levels of CDK4, cyclin D1, CDK2
and cyclin E1 in the combination group. Along with mTORC1
signaling, the present study also unraveled the contributions of
two crucial oncogenic pathways in SGK1-mediated PI3K inhibitor
resistance in NSCLC cell lines. Extensive crosstalk occurs between
Ras/ERK and PI3K/AKT pathways and they compensate each other,
resulting in drug resistance. Co-inhibition of these pathways
results in higher anticancer activity but causes serious adverse
effects like hyperglycemia, dermatitis and pneumonitis (26). The combination of GSK650394 and
BYL719 facilitated notably higher inhibition of p-ERK and its
downstream targets p-P90RSK and p-S6. Similarly, elevated nuclear
β-catenin levels confer resistance to PI3K/AKT inhibitor-induced
apoptosis in cancer cells (22).
Here, SGK1 inhibition caused a marked decrease in p-β-catenin S553
and total β-catenin levels, which were further reduced by
combination treatment in both cell lines. Moreover, the combination
treatment decreased levels of β-catenin target proteins c-Myc and
c-Jun. SGK1 inhibition alone resulted in a moderate decrease in
β-catenin, c-Myc and c-Jun, but not p-GSK3β, levels. This suggested
p-GSK3β-independent modulation of β-catenin levels by SGK1. This
mechanism should be further investigated to devise better
therapeutic strategies. Similarly, SGK1 overexpression
significantly promotes cell migration and invasion in multiple
types of cancer (8,9). Further studies are needed to determine
these roles of SGK1. Combination of SGK1 and PI3K inhibitor induced
vacuole formation in NCI-H460 cells, which were likely autophagic
vacuoles. However, these results should be confirmed by levels of
autophagic markers.
In conclusion, the present data demonstrated that
elevated tumor SGK1 levels may predict resistance to PI3K/AKT
inhibitors in patients with NSCLC. Further investigation is
required to address the limitations of the present study, such as
lack of evaluation of drug combination in normal cells to test
in vitro toxicity and validation using overexpression or
knockdown of SGK1 in in vitro and in vivo models.
Such studies may aid clinical trials evaluating the therapeutic
potential of PI3K/AKT inhibitors for treating NSCLC. The present
findings also demonstrated that increased anticancer activity is
driven by the simultaneous inhibition of multiple signaling
cascades, such as RAS/MEK/ERK and Wnt/β-catenin, in addition to
mTORC1 signaling. This highlights the need for further studies to
identify the specific pathways and downstream mechanisms controlled
by SGK1 and their role in PI3K/AKT inhibitor resistance.
Supplementary Material
Expression of SGK1 protein in NSCLC
cell lines and its impact on PI3K inhibitor sensitivity. (A) Total
SGK1 expression in a panel of NSCLC cell lines. (B) Anticancer
IC50 for BTL719 in NSCLC cell lines with high and low
SGK1 expression. Bliss synergy score for BTL719 and GSK650394
combination in (C) antiproliferation and (D) colony formation
assay. SGK1, Serum and Glucocorticoid Kinase 1; NSCLC, Non-small
cell lung cancer; IC50, Inhibitory concentration
50%.
Combination treatment increases
immunogenicity of non-small cell lung cancer cell lines and cell
cycle arrest in G1 phase. (A) PD-L1 expression, (B) vacuole
formation (arrows indicate vacuoles, 20X magnification). (C) cell
cycle distribution and (D) expression of G2/M phase markers
following BYL719 and/or GSK650394 treatment in NCI-H460 cells.
Antibodies for western blotting.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Lupin Ltd. (Reg No.
219000006).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RK conceived the study, performed experiments,
analyzed data and wrote and reviewed the manuscript. CS conceived
the study and performed experiments. AB conceived the study and
reviewed the manuscript. MV conceived the study, analyzed data and
reviewed the manuscript. KN and MB conceived the study, analyzed
data and reviewed the manuscript. RK and CS confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Meador CB and Hata AN: Acquired resistance
to targeted therapies in NSCLC: Updates and evolving insights.
Pharmacol Ther. 210(107522)2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang M, Herbst RS and Boshoff C: Toward
personalized treatment approaches for non-small-cell lung cancer.
Nat Med. 27:1345–1356. 2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Travis WD, Brambilla E, Burke AP, Marx A
and Nicholson AG: Introduction to The 2015 World Health
Organization classification of tumors of the lung, pleura, thymus,
and heart. J Thorac Oncol. 10:1240–1242. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Scheffler M, Bos M, Gardizi M, König K,
Michels S, Fassunke J, Heydt C, Künstlinger H, Ihle M, Ueckeroth F,
et al: PIK3CA mutations in non-small cell lung cancer (NSCLC):
Genetic heterogeneity, prognostic impact and incidence of prior
malignancies. Oncotarget. 20:1315–1326. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhou X, Wang X, Zhu H, Gu G, Zhan Y, Liu C
and Sun G: PI3K inhibition sensitizes EGFR wild-type NSCLC cell
lines to erlotinib chemotherapy. Exp Ther Med. 21(9)2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hanker AB, Kaklamani V and Arteaga CL:
Challenges for the clinical development of PI3K inhibitors:
Strategies to improve their impact in solid tumors. Cancer Discov.
9:482–491. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Basnet R, Gong GQ, Li C and Wang MW: Serum
and glucocorticoid inducible protein kinases (SGKs): A potential
target for cancer intervention. Acta Pharm Sin B. 8:767–771.
2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Talarico C, Dattilo V, D'Antona L, Menniti
M, Bianco C, Ortuso F, Alcaro S, Schenone S, Perrotti N and Amato
R: SGK1, the new player in the game of resistance: Chemo-radio
molecular target and strategy for inhibition. Cell Physiol Biochem.
39:1863–1876. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zhang Z, Xu Q, Song C, Mi B, Zhang H, Kang
H, Liu H, Sun Y, Wang J, Lei Z, et al: Serum- and
glucocorticoid-inducible kinase 1 is essential for
osteoclastogenesis and promotes breast cancer bone metastasis. Mol
Cancer Ther. 19:650–660. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lee LYW, Woolley C, Starkey T, Biswas S,
Mirshahi T, Bardella C, Segditsas S, Irshad S and Tomlinson I:
Serum- and glucocorticoid-induced kinase Sgk1 directly promotes the
differentiation of colorectal cancer cells and restrains
metastasis. Clin Cancer Res. 25:629–640. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Toska E, Castel P, Chhangawala S,
Arruabarrena-Aristorena A, Chan C, Hristidis VC, Cocco E, Sallaku
M, Xu G, Park J, et al: PI3K inhibition activates SGK1 via a
feedback loop to promote chromatin-based regulation of ER-dependent
gene expression. Cell Rep. 27:294–306.e5. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Castel P, Ellis H, Bago R, Toska E, Razavi
P, Carmona FJ, Kannan S, Verma CS, Dickler M, Chandarlapaty S, et
al: PDK1-SGK1 signaling sustains AKT-independent mTORC1 activation
and confers resistance to PI3Kα inhibition. Cancer Cell.
30:229–242. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Parra ER, Villalobos P, Mino B and
Rodriguez-Canales J: Comparison of different antibody clones for
immunohistochemistry detection of programmed cell death ligand 1
(PD-L1) on non-small cell lung carcinoma. Appl Immunohistochem Mol
Morphol. 26:83–93. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Juneja VR, McGuire KA, Manguso RT, LaFleur
MW, Collins N, Haining WN, Freeman GJ and Sharpe AH: PD-L1 on tumor
cells is sufficient for immune evasion in immunogenic tumors and
inhibits CD8 T cell cytotoxicity. J Exp Med. 214:895–904.
2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Dantoing E, Piton N, Salaün M, Thiberville
L and Guisier F: Anti-PD1/PD-L1 immunotherapy for non-small cell
lung cancer with actionable oncogenic driver mutations. Int J Mol
Sci. 22(6288)2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Adorisio S, Cannarile L, Delfino DV and
Ayroldi E: Glucocorticoid and PD-1 cross-talk: Does the immune
system become confused? Cells. 10(2333)2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yang L, Huang F, Mei J, Wang X, Zhang Q,
Wang H, Xi M and You Z: Posttranscriptional control of PD-L1
expression by 17β-estradiol via PI3K/Akt signaling pathway in
ERα-positive cancer cell lines. Int J Gynecol Cancer. 27:196–205.
2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hydbring P, Malumbres M and Sicinski P:
Non-canonical functions of cell cycle cyclins and cyclin-dependent
kinases. Nat Rev Mol Cell Biol. 17:280–292. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
McCubrey JA, Steelman LS, Franklin RA,
Abrams SL, Chappell WH, Wong EWT, Lehmann BD, Terrian DM, Basecke
J, Stivala F, et al: Targeting the RAF/MEK/ERK, PI3K/AKT and p53
pathways in hematopoietic drug resistance. Adv Enzyme Regul.
47:64–103. 2007.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Won M, Park KA, Byun HS, Kim YR, Choi BL,
Hong JH, Park J, Seok JH, Lee YH, Cho CH, et al: Protein kinase
SGK1 enhances MEK/ERK complex formation through the phosphorylation
of ERK2: Implication for the positive regulatory role of SGK1 on
the ERK function during liver regeneration. J Hepatol. 51:67–76.
2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Tenbaum SP, Ordóñez-Morán P, Puig I,
Chicote I, Arqués O, Landolfi S, Fernández Y, Herance JR, Gispert
JD, Mendizabal L, et al: β-catenin confers resistance to PI3K and
AKT inhibitors and subverts FOXO3a to promote metastasis in colon
cancer. Nat Med. 18:892–901. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Jiang L, Zhang J, Xu Y, Xu H and Wang M:
Treating non-small cell lung cancer by targeting the PI3K signaling
pathway. Chin Med J (Engl). 135:1272–1284. 2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Mishra R, Patel H, Alanazi S, Kilroy MK
and Garrett JT: PI3K inhibitors in cancer: Clinical implications
and adverse effects. Int J Mol Sci. 22(3464)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sherk AB, Frigo DE, Schnackenberg CG, Bray
JD, Laping NJ, Trizna W, Hammond M, Patterson JR, Thompson SK,
Kazmin D, et al: Development of a small-molecule serum- and
glucocorticoid-regulated kinase-1 antagonist and its evaluation as
a prostate cancer therapeutic. Cancer Res. 68:7475–7483.
2008.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Tolcher AW, Peng W and Calvo E: Rational
approaches for combination therapy strategies targeting the MAP
kinase pathway in solid tumors. Mol Cancer Ther. 17:3–16.
2018.PubMed/NCBI View Article : Google Scholar
|