Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive
neurodegenerative disease that affects nerve cells in the brain and
spinal cord. The hallmark of ALS is progressive muscle weakness,
accompanied by muscle atrophy, fasciculations, muscle cramps and
stiffness and slowness of movements. The main features of ALS
include muscle twitches in the arm, leg, shoulder or tongue, muscle
cramps, spasticity, muscle weakness affecting the limbs or the
neck, slurred and nasal speech, as well as chewing or swallowing
difficulties. As the disease evolves, the symptoms may include
difficulty breathing, inability to stand or walk independently,
weight loss, depression and anxiety (1).
In the evolution of ALS, motor deficits appear at
the level of the upper and lower limbs and the respiratory muscles;
phonation and swallowing are also affected (1). In conclusion, this degenerative
disease is characterized by both central and peripheral motor
neuron injuries. The incidence of ALS at the global level is
unknown. In Europe, 2.2 per 100.000 individuals are diagnosed with
ALS every year; in the United States, the rate of ALS is >1.87
per 100.000 individuals per year (2).
The treatment of ALS is complex and
multidisciplinary. The only medication proven to influence the
evolution course is rilusole (3).
Other treatments, such as respiratory care and nutrition
management, can increase the quality of life for patients with ALS
(1).
There is no biological marker for ALS. The diagnosis
can be established using clinical examination and electrodiagnostic
tests. Identifying biological, clinical, neurophysiological and
genetic biomarkers of the disease still remains a challenge
(4,5). The clinician can use the updated El
Escorial diagnostic criteria (EEC) and Awajishima criteria
(6,7). According to the EEC, the diagnosis of
ALS requires the presence of A-criteria and the absence of
B-criteria as follows:
• A1: Degeneration of the lower motor neurons,
proved by clinical, electrophysiological or neuropathological exami
nation;
• A2: Degeneration of the upper motor neurons,
proved by clinical examination;
• A3: Progressive dissemination beyond typical nerve
supply areas.
• B1: Electrophysiological or neuropathological
findings typical for other diseases that could explain the
degeneration of the upper and lower motor neurons;
• B2: Findings of imaging studies that can explain
the clinical symptoms. The Awaji criteria are less rigid and are
based on the presence of fasciculation potentials in a typical
clinical context of ALS.
The current study presented the case of a
68-year-old woman without any medical history, who accessed the
ambulatory service of the Medical Rehabilitation Clinic of the
Emergency County Hospital of Craiova (Craiova, Romania) for a motor
deficit at the level of the upper and lower limbs, installed 1.5
years before the presentation. The diagnosis of ALS was based on
clinical features and electrodiagnostic studies. Establishing a
correct diagnosis is essential for an appropriate therapeutic
approach and consequently for the patient's prognosis.
Case report
A 68-year-old woman, Caucasian, from a rural
environment with a primary education was hospitalized in a Medical
Rehabilitation Clinic of the Emergency County Hospital of Craiova
(Craiova, Romania) due to dizziness, gait disorders, decreased grip
strength and fasciculations. The symptoms began insidiously 1.5
years before the hospitalization. The patient had not previously
seen a doctor and had not received any medical treatment, and there
was no family history of ALS.
The clinical examination showed that the patient was
underweight [body mass index (BMI=18.1 kg/m2)] and was
well-oriented. When examining the cranial nerves, no pathological
elements were highlighted, except for the lingual fasciculations.
Orthostatism was possible, walking was possible with support but
impossible on the toes and tips.
On examination of the hand, the osteoarthritic
aspect was noticed, but also the aspect of the ‘simian hand’, with
hypotrophy of the thenar and hypothenar eminences, retraction of
the palmar aponeurosis and a grasping deficit. These abnormalities
were observed in both hands.
Clinical examination of the lower limbs indicated a
motor deficit in the calf muscles upon muscular testing: The
muscular strength (on a scale ranging from 0 to 5) of the anterior
tibial muscle was 3/4 right/left, the strength of the common
extensor of the toes was 3 bilaterally and the strength of the
hallux extensor muscle was 2/3 right/left (6).
The Babinski response was present bilaterally, while
osteotendinous reflexes in the upper and lower limbs were abolished
(6). Movement coordination was not
affected in the patient and no objective sensitivity disorders were
noted.
Regarding the imaging investigations, the lumbar
spine radiograph showed both degenerative changes and vertebral
osteoporosis, a fact confirmed by the osteodensity examination,
where a T-score of -3.8 was obtained (dual X-ray absortiometry
T-score >-1 means normal; T-score from-1 to -2.5 means
osteopenia; T-score <-2.5 means osteoporosis). These
comorbidities were not typical for ALS (6).
A specialized neurological examination was
requested, following which the diagnosis of neurogenic spinal
atrophy was established and the patient was placed under
observation for amyotrophic lateral sclerosis.
The electroneuromyography (EMG) for the median and
ulnar nerves pointed out the diagnosis of motor chronic
demyelinating neuropathy and a needle EMG was needed for
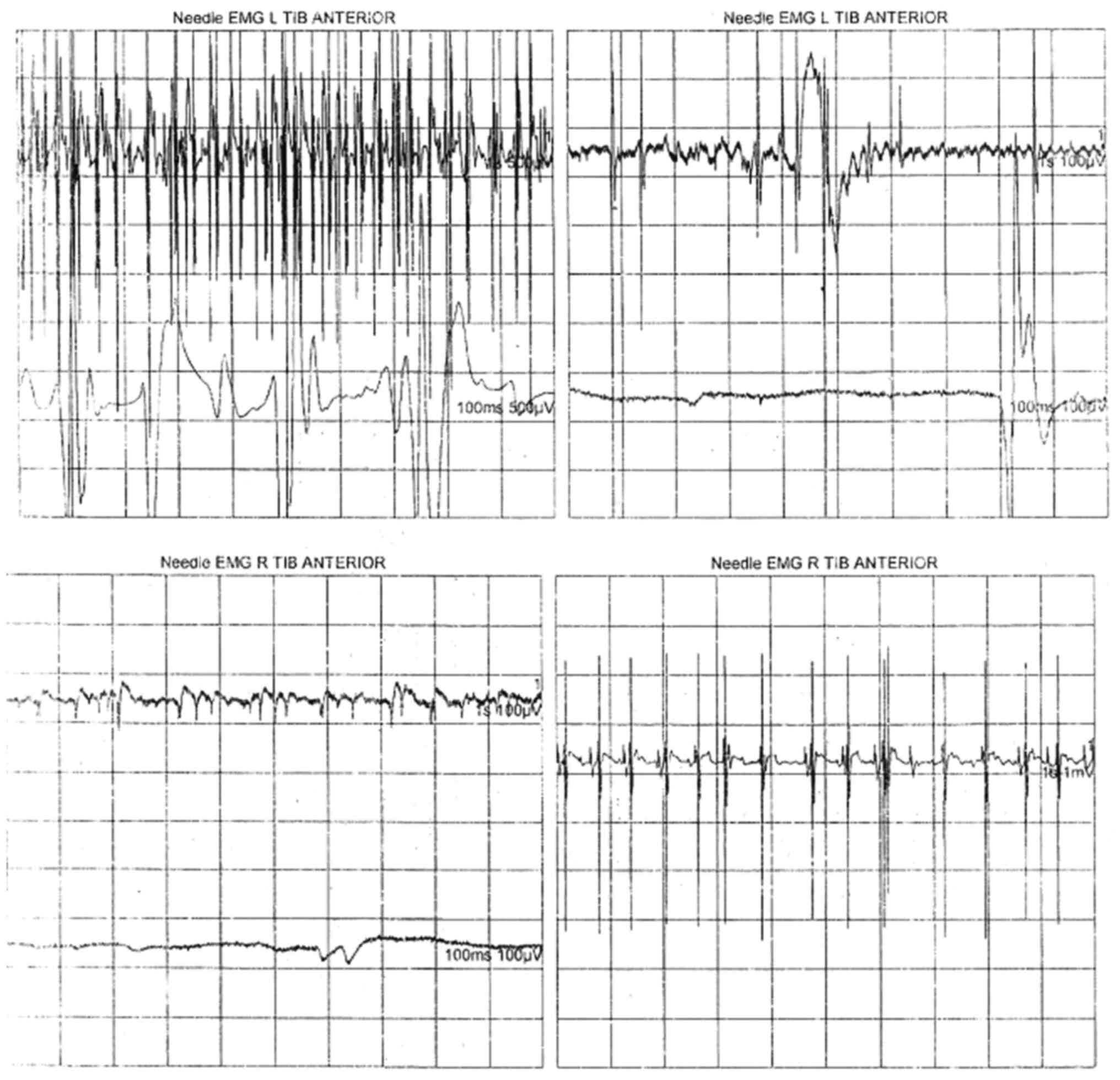
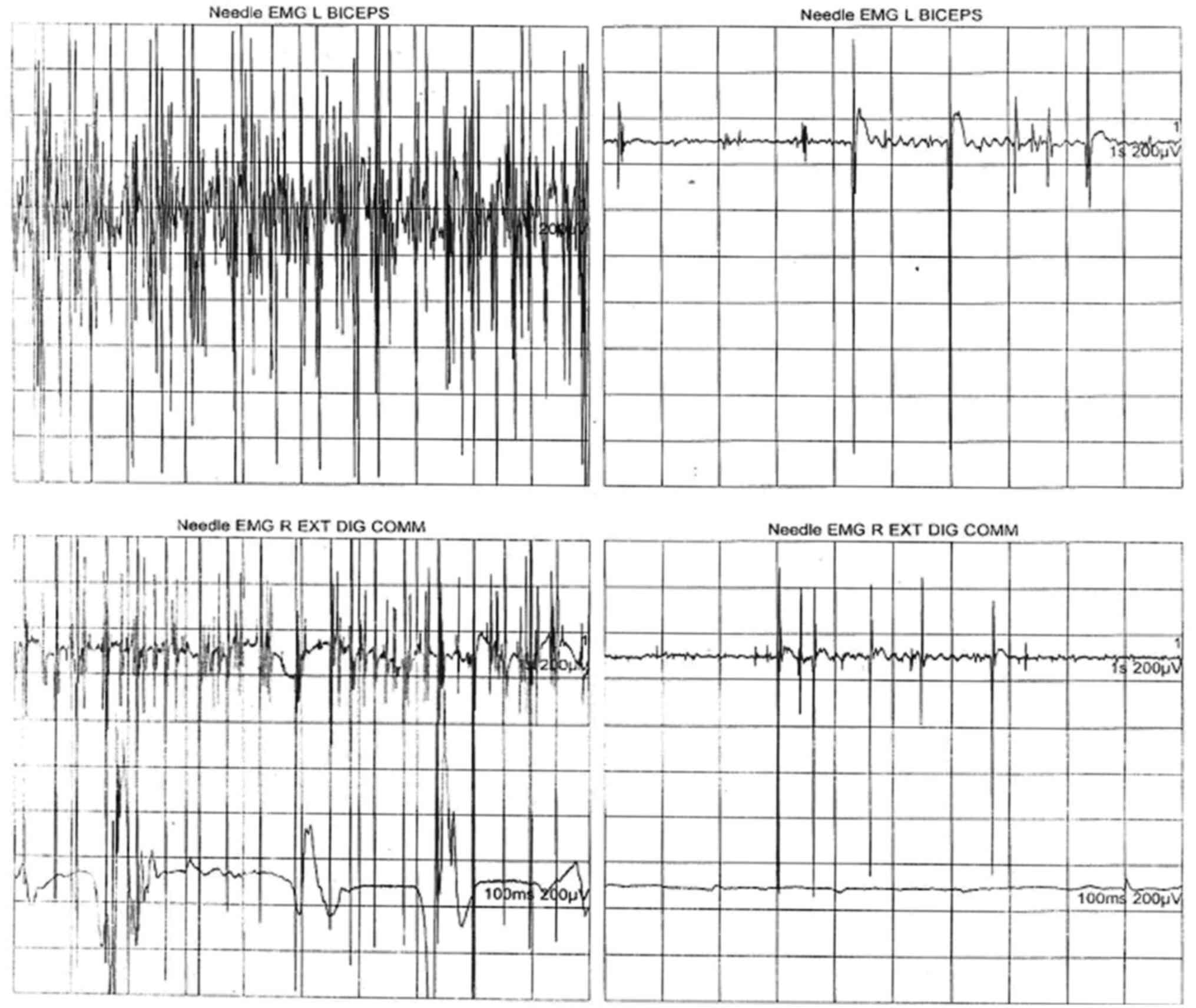
confirmation of ALS. Electromyographic examination performed for
the brachial biceps muscle, common extensor muscle of the fingers
and anterior tibialis muscle revealed an aspect of generalized
chronic active neurogenic lesion. This aspect was characteristic
ofan injury of motor neurons, confirming the diagnosis of ALS. The
recording of the neurogenic pathway was characterized by the
spontaneous presence of fasciculations, fibrillation, positive
sharp waves, stable and instable neurogenic motor unit potentials
with increased duration and amplitude, and a reduced interference
pattern by decreased motor neuron recruitment and activation
(Figs. 1 and 2, Table
I).
 | Table INeedle electroneuromyography
parameters for tibial anterior, biceps and extensor digitorum
communis muscles highlight an aspect of chronic active generalized
neurogenic injury characteristic of motor neuron disease. |
Table I
Needle electroneuromyography
parameters for tibial anterior, biceps and extensor digitorum
communis muscles highlight an aspect of chronic active generalized
neurogenic injury characteristic of motor neuron disease.
| | Spontaneous | MUAP | |
|---|
| Muscle | IA | Fib | PSW | Fasc | H.F. | Amp | Dur | PPP | Engagement
pattern |
|---|
| L. Tibial
anterior | N | 2+ | 1+ | 2+ (slow) | None | Giant | 3+ | 3+ | Reduced |
| R. Tibial
anterior | N | 1+ | 3+ | None | None | Giant | 3+ | 3+ | Discrete |
| L. Biceps | N | 2+ | 1+ | 2+ (slow) | None | 2+ | 3+ | 3+ | Reduced |
| R. Extensor digitorum
communis | N | 1+ | 1+ | 2+ (fast) | None | Giant | 3+ | 3+ | Reduced |
ALS is a neurodegenerative disease that occurs in
motor nerve cells and leads to gradual amyotrophy and muscle
weakness until complete paralysis. The patient of the present study
had weakness in the legs, a decreased ability to hold her balance
and to go up and down the stairs and required assistance with
walking. The patient's arms also showed obvious weakness with arm
lifting difficulties and decreased grip strength of both hands, and
the patient gradually lost the ability to use the hands and arms.
The patient had limb muscle atrophy, muscle bundle tremor and
weight loss. The only characteristic sign of cranial nerve damage
was lingual fasciculations.
No MRI was performed for this patient, while it is
helpful for studying ALS-related changes in the brain or spinal
cord. The patient was administered riluzole 50 mg twice daily.
After the diagnosis of ALS, the patient was referred
to the neurology outpatient clinic to establish a specialized
therapeutic approach. During the 12 days of hospitalization at the
Physical Medicine and Rehabilitation Clinic of the Emergency County
Hospital of Craiova (Craiova, Romania), the patient underwent a
complex kinetic and physical rehabilitation treatment. The
individualized physicotherapy program consisted of light aerobic
exercises and had the objectives of increasing muscular strength,
maintenance of the tone and strength of the unaffected muscles and
improvement of the cardiovascular status. This program included
walking exercises and a cycle ergometer, also helping the patient
fight fatigue and depression. Stretching exercises and mobilization
to increase the amplitude of joint movement also aimed to reduce
spasticity and combat painful muscle contractions. The patient also
responded positively to electrostimulation procedures for lower
limb muscles. The patient was monitored through the neurology
service and underwent recovery treatment ~6 months later.
Discussion
At present, there is no universally accepted
specific diagnostic test for ALS. The diagnosis of the disease is
based in particular on the presence of symptoms, signs and
laboratory tests that show progressive injury to motor neurons,
such as electrodiagnostic tests. Imaging and laboratory tests can
be used to rule out other neurological conditions. The classic
clinical presentation of ALS is characterized by the asymmetric
decrease in muscle strength in the extremities (60-80%) and bulbar
muscles (1-9%), axial onset with fall of the cephalic extremity or
decrease in the strength of the paravertebral extensor muscles,
fasciculations and muscle atrophies (5-7%) (8). Anteflexion of the head due to muscle
strenght decrease.
The lack of diagostic tests with sensitivity and
specificity for ALS is a major obstacle in the early diagnosis of
patients, although in 2000, the diagnostic criteria for this
condition were revised (6,8). The diagnosis of patients with ALS with
a typical presentation of the disease is relatively easy for the
clinician in the presence of signs of upper and lower motoneuron
damage and progressive evolution of the disease. However, for
patients with a slow evolution of the disease, at the onset of the
disease or for those with other concomitant disorders of the
central or peripheral nervous system, the probability of
misdiagnosis with the ‘ALS-mimicking syndromes’ is ~7-8% (9). In such cases, peripheral neuropathies,
myopathies, spinal muscular atrophy and paraplegia should be ruled
out.
Given the complexity of the disease, >40 clinical
randomized controlled trials performed over the last decades failed
to show any influence on disease progression or life expectancy in
patients with ALS (10). In most
European countries, riluzole 50 mg twice daily is the only
medication used for the treatment of patients with ALS due to its
antiglutamatergic effects and it is a disease-modifying drug proven
to prolong life by 3-6 months (11,12).
Muscle spasticity in patients with ALS can be
influenced using baclofen and stretching exercises (13). For the treatment of muscle cramps,
carbamazepine or gabapentin and also magnesium supplements can be
used. Antidepressant medication may also be used for emotional
lability. Nutrition changes and speech therapy can also be useful
for patients with ALS (14).
The success of the treatment of patients with ALS is
ensured by a multidisciplinary collaboration (15). Future therapy may include edaravone
(16,17), a drug that has been approved for the
treatment of ALS in several countries, or masitinib, a tyrosine
kinase inhibitor, drugs used in clinical trials (18). Genetic studies for ALS treatment are
also in progress (19), as well as
stem cell treatments (20,21).
The patient presented in this case study had an
asymmetric decrease in muscle strength at the extremities level, at
the clinical examination, which could suggest the diagnosis of ALS.
When first seen, the patient of the present study had asymmetric
weakness of the extremities that was consistent with ALS, but this
lack of muscle strength in the extremities can also indicate a
demyelinating neurological disease (22). Fasciculations may also be
characteristic of this condition, but may be associated with other
neurological diseases. These symptoms may be significant to clarify
the diagnosis of ALS when they are accompanied by changes in the
motor unit highlighted by the needle EMG examination (7). Electrodiagnostic analysis is essential
in patients with this condition, both to confirm the diagnosis of
ALS and to identify other potentially treatable neurological
disorders.
Patients with ALS frequently exhibit weight loss
that occurs late in the course of the disease. There are multiple
causes of weight loss, which may include decreased muscle strength
in the upper limbs and seizure disorders, dysphagia, dyspnea during
swallowing, chewing problems and a hypermetabolic status (23). The patient presented in the current
study was underweight, with a BMI of 18.1 kg/m2.
According to clinical studies, a BMI <18.51 kg/m2
caused by weight loss in patients with ALS is a negative prognostic
factor and is associated with a decreased survival time (23).
This situation in which the patient with ALS first
presented at the Medical Rehabilitation service is rare, which was
an additional reason why establishing the diagnosis was
challenging, requiring a multidisciplinary evaluation (medical
rehabilitation doctor, neurologist specializing in
electromiography, physioterapists).
In conclusion, as there are no disease-specific
diagnostic tests, diagnosing ALS is a challenge for the clinician.
The neurological clinical examination must be associated with a
series of paraclinical investigations necessary for the
differential diagnosis of other neurological diseases. In this
sense, the importance of electrodiagnostic tests is highlighted,
particularly electromyography, as well as imaging, the most useful
being nuclear magnetic resonance. At times, muscle or nerve biopsy
is required, determination of serum levels of thyroid and
parathyroid hormones, as well as detection of heavymetals in urine.
The average survival time in patients with ALS is between 2 and 5
years after diagnosis, but in numerous cases, it can be
exceeded.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
IRM contributed to the data acquisition. OCR
contributed to manuscript writing and critical revision for
important intellectual content. RP contributed to the
systematization of the data. VP and DD contributed to analysis and
supervision. All authors have read and approved the final version
of the manuscript. IRM and DD checked and confirmed the
authenticity of the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written informed consent was obtained from the
patient for the publication of this case report and any
accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gordon PH: Amyotrophic lateral sclerosis:
An update for 2013: Clinical features, pathophysiology, management
and therapeutic trials. Aging Dis. 4:295–310. 2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kiernan MC, Vucic S, Cheah BC, Turner MR,
Eisen A, Hardiman O, Burell JR and Zoing MC: Amyotrophic lateral
sclerosis. Lancet. 377:942–955. 2011.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Miller RG, Mitchell JD and Moore DH:
Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron
disease (MND). Cochrane Database Syst Rev.
2012(CD001447)2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bowser B, Turner MB and Shefner J:
Biomarkers in amyothrophic lateral sclerosis: Opportunities and
limitations. Nat Rev Neurol. 7:631–638. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Li D, Shen D, Tai H and Cui L:
Neurofilaments in CSF as diagnostic biomarkers in motor neuron
disease: A metaanalysis. Front Aging Neuroscl.
8(290)2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Brooks BR: El Escorial world federation of
neurology criteria for the diagnosis of amyotrophic lateral
sclerosis. Subcommittee on motor neuron diseases/amyotrophic
lateral sclerosis of the world federation of neurology research
group on neuromuscular diseases and the El Escorial ‚clinical
limits of amyotrophic lateral sclerosis’ workshop contributors. J
Neurol Sci. 124 (Suppl):S96–S107. 1994.PubMed/NCBI View Article : Google Scholar
|
|
7
|
de Carvalho M, Dengler R, Eisen A, England
JD, Kaji R, Kimura J, Mills K, Mitsumoto H, Nodera H, Shefner J and
Swash M: Electrodiagnostic criteria for diagnosis of ALS. Clin
Neurophysiol. 119:497–503. 2008.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Talbot K: Motor neuron disease. Postgrad
Med J. 78:513–519. 2002.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Traynor BJ, Codd MB, Corr B, Forde C,
Frost E and Hardiman O: Amyotrophic lateral sclerosis mimic
syndromes: A population-based study. Arch Neurol. 57:109–113.
2000.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Mitsumoto H, Brooks BR and Silani V:
Clinical trials in amyotrophic lateral sclerosis: Why so many
negative trials and how can trials be improved? Lancet Neurol.
13:1127–1138. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
De Sousa EA, Chin RC, Sander HW, Latov N
and Brannigan TH III: Demyelinating findings in typical and
atypical chronic inflammatory demyelinating polyneuropathy:
Sensitivity and specificity. J Clin Neuromuscul Dis. 10:163–164.
2009.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Hinchcliffe M and Smith A: Rilusole: Real
world evidence supports significant extension of median survival
times in patients with amyotrophic lateral sclerosis. Degener
Neurol Neuromuscul Dis. 7:61–70. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Marcu IR, Dop D, Padureanu V, Niculescu
SA, Padureanu R, Niculescu CE and Rogoveanu OC: Non-steroidal
anti-inflammatory drug etoricoxib facilitates the application of
individualized exercise programs in patients with ankylosing
spondylitis. Medicina (Kaunas). 56(270)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Masrori P and Van Damme P: Amyotrophic
lateral sclerosis: a clinical review. Eur J Neurol. 27:1918–1929.
2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
EFNS Task Force on Diagnosis and
Management of Amyotrophic Lateral Sclerosis. Andersen PM, Abrahams
S, Borasio GD, de Carvalho M, Chio A, Van Damme P, Hardiman O,
Kollewe K, Morrison KE, et al: EFNS guidelines on the clinical
management of amyotrophic lateral sclerosis (MALS)-revised report
of an EFNS task force. Eur J Neurol. 19:360–375. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Writing Group; Edaravone (MCI-186) ALS 19
Study Group. Safety and efficacy of edaravone in well defined
patients with amyotrophic lateral sclerosis: A randomised,
double-blind, placebo-controlled trial. Lancet Neurol. 16:505–512.
2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Al Chalabi A, Andersen PM, Chandran S,
Chio A, Corcia P, Couratier P, Danielsson O, de Carvalho M,
Desnuelle C, Grehl T, et al: July 2017 ENCALS statement on
edaravone. Amyotroph Lateral Scler Frontotemporal Degener.
18:471–474. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Mora JS, Genge A, Chio A, Estol CJ,
Chaverri D, Hernández M, Marín S, Mascias J, Rodriguez GE, Povedano
M, et al: Masitinib as an add-on therapy to riluzole in patients
with amyotrophic lateral sclerosis: A randomized clinical trial.
Amyotroph Lateral Scler Frontotemporal Degener. 21:5–14.
2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Jiang J, Zhu Q, Gendron TF, Saberi S,
McAlonis-Downes M, Seelman A, Stauffer JE, Jafar-Nejad P, Drenner
K, Schulte D, et al: Gain of toxicity from ALS/FTD-linked repeat
expansions in C9ORF72 is alleviated by antisense oligonucleotides
targeting GGGGCC-containing RNAs. Neuron. 90:535–550.
2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Mazzini L, Mareschi K, Ferrero I, Vassallo
E, Oliveri G, Nasuelli N, Oggioni GD, Testa L and Fagioli F: Stem
cell treatment in amyotrophic lateral sclerosis. J Neurol Sci.
265:78–83. 2008.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Deda H, Inci MC, Kürekçi AE, Sav A,
Kayihan K, Ozgün E, Ustünsoy GE and Kocabay S: Treatment of
amyotrophic lateral sclerosis patients by autologous bone
marrow-derived hematopoietic stem cell transplantation: A 1-year
follow-up. Cytotherapy. 11:18–25. 2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Desport JC, Preux PM, Magy L, Boirie Y,
Vallat JM, Beaufrere B and Couratier P: Factors correlated with
hypermetabolism in patients with amyotrophic lateral sclerosis. Am
J Clin Nutr. 74:328–334. 2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Desport JC, Preux PM, Truong TC, Vallat
JM, Sautereau D and Couratier P: Nutritional status is a prognostic
factor for survival in ALS patients. Neurology. 53:1059–1063.
1999.PubMed/NCBI View Article : Google Scholar
|