Introduction
Hepatocellular carcinoma (HCC) is the predominant
form of primary liver cancer, as well as the third leading cause of
cancer-associated deaths worldwide (1,2). The
prognosis of advanced HCC is particularly concerning because of
high rates of recurrence and metastasis, which result in a poor
5-year survival rate worldwide (3).
There are still gaps in the understanding of the molecular
mechanisms that trigger HCC and facilitate its development, posing
challenges for effective treatment (4). Consequently, a comprehensive
understanding of these underlying mechanisms is crucial for
advancing therapeutic strategies and improving patient
outcomes.
PAX-interacting protein 1 (PAXIP1) was initially
identified due to its interaction with paired box 2(5) and other transcription factors (TFs)
(6). Previous studies have
highlighted the involvement of PAXIP1 in the DNA damage response
and the regulation of histone modifications (7-10).
Specifically, during the repair of double-stranded DNA breaks,
PAXIP1, in conjunction with p53-binding protein 1 (53BP1), enhances
non-homologous end-joining repair processes (10). Furthermore, PAXIP1 is crucial for
the assembly of the histone methyltransferase complex at a Pax
DNA-binding site, which is a significant aspect of mammalian
development (6,9). PAXIP1 also interacts with the histone
methyltransferase complex, suggesting that it serves a role in
histone methylation and demethylation (7,9,11,12).
As a tandem BRCA1 C-terminal domain protein
(5,13), PAXIP1 is associated with multiple
types of cancer. For instance, PAXIP1 has been shown to function as
a prognostic biomarker in ovarian cancer (14,15).
Additionally, reduced PAXIP1 levels have been observed in patients
diagnosed with breast cancer and an unfavorable prognosis (16). PAXIP1 has been demonstrated to
modulate the cell response in lung cancer (17). Our previous study also demonstrated
that PAXIP1 inhibited cell invasion via modulation of EPH receptor
A2 (EphA2) expression in esophageal squamous cell carcinoma
(18). Despite these findings, the
expression patterns and precise role of PAXIP1 in HCC remain
inadequately explored. Therefore, the protein and mRNA expression
levels, prognostic significance and potential functions of PAXIP1
in HCC were assessed. Using multidimensional analysis and various
public databases, an in-depth examination of the genomic
alterations and functional networks pertaining to the role of
PAXIP1 in HCC was conducted, thereby elucidating its involvement in
tumor immunity.
Materials and methods
Cell culture and small interfering RNA
(siRNA) transfection
The HuH-7 and PLC-PRF-5 human liver cancer cell
lines were obtained from FuHeng Biology. HuH-7 cells were cultured
in DMEM (cat. no. C11995500BT; Gibco; Thermo Fisher Scientific,
Inc.) and PLC-PRF-5 cells were cultured in Minimum Essential Medium
(MEM; cat. no. C11095500BT; Gibco; Thermo Fisher Scientific, Inc.).
Cell media were supplemented with 10% FBS (cat. no. FBS500-S;
Ausgenex Pty, Ltd.) and 1% penicillin/streptomycin (cat. no.
15140122; Gibco; Thermo Fisher Scientific, Inc.). Cells were kept
at 37˚C in a humidity-controlled incubator with 5% CO2
supplementation. All cell lines underwent rigorous verification for
mycoplasma contamination and were confirmed to be free of
contamination. The cells were authenticated using short tandem
repeat analysis.
siRNA transfection was conducted using Lipofectam
ine™ 2000 transfection reagent (cat. no. 52887;
Invitrogen; Thermo Fisher Scientific, Inc.). All siRNA
transfections were performed for 48 h at room temperature. HuH-7
and PLC-PRF-5 cells were cultured in 6-well plates to allow for
adhesion and proliferation overnight. Before the introduction of
the transfection agent, 1 ml serum-free DMEM/MEM was used to
replace the medium. The scrambled control, CCCTC binding factor
(CTCF) and nuclear respiratory factor 1 (NRF1) siRNA molecules were
chemically synthesized by Nanjing GenScript Biotech Co., Ltd. For
6-well plates, 10 µl siRNA (20 µM) was dissolved in 5 µl siRNA
transfection reagent and incubated for 5 min at room temperature.
The aforementioned mixture was then introduced to the cells. After
12 h, 1 ml DMEM/MEM supplemented with 10% FBS was added to each
well. Cells were seeded at a density of 6x105 cells/well
in 6-well culture plates. After 24 h, the cells were transfected
with the siRNA for 48 h and then RNA was extracted. The siRNA
sequences used were as follows: NRF1, 5'-GGAAACUUCGAGCCACGUU-3';
CTCF, 5'-GCGAAAGCAGCAUUCCUAUAU-3'; and control,
5'-UUCUCCGAACGUGUCACGU-3'.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from HCC cells using RNeasy
Kits (cat. no. 74104; Qiagen GmbH) according to the manufacturer's
instructions. cDNA synthesis was conducted with the Primescript
RT-reagent kit (cat. no. RR047A; Takara Bio, Inc.). The temperature
and duration of reverse transcription were: 37˚C for 15 min and
85˚C for 5 sec. qPCR was performed using SYBR Premix Ex Taq (Takara
Biotechnology Co., Ltd.) on an ABI7500 system (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The 2-ΔΔCq method was
utilized for data analysis, using β-actin for normalization
(19,20). The thermocycling conditions were as
follows: 95˚C for 5 min; 40 cycles of 95˚C for 15 sec and 60˚C for
30 sec; 1 cycle of 95˚C for 15 sec, 60˚C for 60 sec and 95˚C for 15
sec. The primer sequences used for qPCR were as follows: NRF1
forward, 5'-CCGGAAGAGGCAACAAACAC-3' and reverse,
5'-CTTGCTGTCCCACACGAGTAGT-3'; CTCF forward,
5'-CATCCAGCATCAGAAGTCACACA-3' and reverse,
5'-GCCTCTCCTGTCTACAAGCGTAA-3'; PAXIP1 forward,
5'-CCAGCTGTACGGACACTGAGG-3' and reverse,
5'-TTGTATGTCCCTGCTGGCTGT-3'; and β-actin forward,
5'-CACTCTTCCAGCCTTCCTTC-3' and reverse,
5'-GTACAGGTCTTTGCGGATGT-3'.
Chromatin
immunoprecipitation-sequencing (ChIP-seq) analysis
ChIP-seq data for PAXIP1, MYB proto-oncogene like 2
(MYBL2) and FOXO1 were retrieved from the ChIP-Atlas database
(http://chip-atlas.org/). The cutoff of broad peak
call was q<1x10-5 (transcription start site ±1 kb).
These datasets (GSE32465 and GSE104247) (21,22)
underwent further analysis using the ChIP-seq pipeline, mainly
using the open-source BEDTools and deepTools software suites.
BEDTools (version 2.29.2) (23) was
employed for the genome arithmetic. The computeMatrix program
within deepTools (version 3.4.3) (24) facilitated the calculation of scores
across genome regions and generated intermediate files for
subsequent visualization with plotHeatmap in deepTools. For genome
annotation, R (version 4.1.0; http://www.r-project.org/) was used to analyze
ChIPseeker (25). Gene Ontology
(GO) analysis was implemented using Database for Annotation,
Visualization and Integrated Discovery (DAVID) functional
annotation tools (https://david.ncifcrf.gov/) (26). Metascape (https://metascape.org/) (27) was also employed to perform DisGeNET
and PaGenBase enrichment analyses for PAXIP1 target genes.
Integrative molecular database of HCC
(HCCDB) analysis
HCCDB (http://lifeome.net/database/hccdb/) is a comprehensive
HCC expression atlas encompassing 15 publicly available HCC gene
expression datasets, which collectively include 3,917 samples
(28). This repository integrates
data from prominent sources such as Gene Expression Omnibus, The
Cancer Genome Atlas (TCGA) Liver HCC Project (TCGA-LIHC) and Liver
Cancer-RIKEN, Japan Project from the International Cancer Genome
Consortium. The HCCDB provides a platform for visualizing outcomes
of various computational analyses, including differential
expression analysis, as well as tissue- and tumor-specific
expression assessments. The 15 HCC datasets were searched with the
keyword ‘PAXIP1’.
Cancer cell line encyclopedia, human
protein atlas (HPA) and cBioPortal database analysis
The mRNA and protein expression profiles were
downloaded from the Cancer Cell Line Encyclopedia (https://depmap.org/portal/interactive/)
and the HPA (https://www.proteinatlas.org/) (29,30).
cBioPortal (https://www.cbioportal.org/), an open-source cancer
genomics data platform, was used to analyze the mutations,
copy-number alterations and gene expression of PAXIP1 in patients
with HCC (31,32). Liver studies were selected and the
keyword ‘PAXIP1’ was searched on the query page of the cBioPortal
website (33-42).
LinkedOmics database analysis
Analysis of PAXIP1 expression in TCGA LIHC cohort
was performed using the LinkedOmics database (http://www.linkedomics.org/) (43). Statistical analysis of PAXIP1
co-expression was conducted using Pearson's correlation
coefficient, with results visualized using volcano plots, heatmaps
and scatter plots. The functional module of the platform
facilitates the examination of GO biological processes, Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathways, kinase-target
enrichment, microRNA (miRNA)-target enrichment and TF-target
enrichment via gene set enrichment analysis (GSEA).
Kaplan-Meier (KM) survival and
nomogram analysis
KM survival analysis and plotting were performed
using the R packages survival and survminer (https://CRAN.R-project.org/package=survminer).
Analysis was performed using default parameters. The area under the
curve (AUC) was analyzed using the R package timeROC (https://CRAN.R-project.org/package=timeROC). Based on
prognostic clinical indicators and the survival analysis of the Cox
regression model, age and pT_stage were entered into the risk
model. The points against each factor were counted, and 1-, 3- and
5-year survival rates were also calculated. The nomogram was
constructed using the rms package (https://CRAN.R-project.org/package=rms) (44). Additionally, the risk score was
calculated as follows: Risk score=0.278 x PAXIP1 + 0.1718 x MYBL2 +
0.0175 x NRF1-0.2226 x FOXO1. Based on the risk score, patients
with HCC were divided into the low-risk group and the high-risk
group using the median risk score as the cutoff (45).
Prediction of TFs of PAXIP1
The human TF (hTF) target database (http://bioinfo.life.hust.edu.cn/hTFtarget#!/)
represents an extensive resource dedicated to the regulation of
hTFs and their respective targets (46). In the present study, this database
was used to predict potential upstream TFs of PAXIP1. These TFs
were then ranked according to their R- and P-value, providing a
systematic evaluation of their potential regulatory roles. Find
Individual Motif Occurrences (v4.10.0; https://meme-suite.org/meme/tools/fimo) was used to
scan both the test and control sets, and then the numbers of
recurrent motifs within the two sets were used to evaluate the
significance of motifs for the TF (t-test with Bonferroni
correction P<0.01) (46).
Tumor immune estimation resource
(TIMER) database
The TIMER database (https://cistrome.shinyapps.io/timer/) was used to
analyze the expression profile of PAXIP1 and immune cell presence
in HCC. For gene expression levels, log2 transformed
transcripts per million values were used.
Analysis of cancer data using the
University of alabama at birmingham cancer data analysis portal
(UALCAN) database and gene expression profiling interactive
analysis (GEPIA)
UALCAN (http://ualcan.path.uab.edu) uses level 3
RNA-sequencing and clinical data from TCGA data of HCC. This
platform facilitates a comprehensive analysis of gene expression,
comparing tumor samples with healthy control tissues and examining
variations across diverse tumor subgroups classified by cancer
stage, tumor grade or other clinicopathological parameters. UALCAN
was used to examine mRNA expression levels according to the online
instructions. GEPIA (http://gepia.cancer-pku.cn/) was used to investigate
MYBL2 and FOXO1 expression in LIHC.
Visualization
Integrative Genomics Viewer (v2.17.0; http://software.broadinstitute.org/software/igv/home)
was adopted to visualize ChIP-seq tracks, while ChIP-seq heat maps
were generated using deepTools (47).
Drug sensitivity analysis
The Genomics of Drug Sensitivity in Cancer (GDSC;
https://www.cancerrxgene.org/) database
was utilized to evaluate the sensitivity of various
chemotherapeutic agents. The pRRophetic package was employed to
estimate the IC50 of these drugs (48).
Statistical analysis
All experiments were performed in triplicate and
repeated three times. All statistical analyses and subsequent
visualization were implemented using R software (version 4.1.0).
The data were assessed for normal distribution using the
Shapiro-Wilk method and for homogeneity of variance using the
Levene method. All two-group comparisons of normally distributed
data were performed using unpaired Student's t test. For multigroup
comparisons, one-way ANOVA with Tukey's post hoc test was used.
Data that were not normally distributed or without homogeneity of
variance were compared using Kruskal-Wallis with Dunn's post hoc
test and Wilcoxon rank sum nonparametric tests. Data are presented
as the mean ± SEM (error bars). All survival analyses were
conducted using KM analysis, the log-rank test and the Cox
proportional hazards model or the two-stage method (49). Pearson's test or Spearman's test was
used to analyze the correlation of two variables. P<0.05 was
considered to indicate a statistically significant difference.
Results
Upregulation of PAXIP1 in HCC
To ascertain the potential involvement of PAXIP1 in
HCC, the transcriptional levels of PAXIP1 across HCC studies
were assessed using HCCDB. Analysis of 11 HCC cohorts from this
database demonstrated that PAXIP1 mRNA was upregulated in
HCC tissues compared with adjacent non-tumor tissues (Fig. 1A). A more granular examination of
TCGA-LIHC samples using the UALCAN database further demonstrated a
marked increase in PAXIP1 expression compared with that in
healthy controls across all tumor grades (Fig. 1B and C). In addition, analysis of subgroups
stratified by sex, age and ethnicity indicated that PAXIP1
expression was higher in patients with HCC compared with healthy
controls (Fig. S1A-C). The
methylation level of the PAXIP1 promoter region varied across
groups based on sex, age and ethnicity (Fig. S1D-F). The mRNA expression matrix
from the Cancer Cell Line Encyclopedia dataset corroborated these
findings, showing that PAXIP1 was highly expressed in HCC cell
lines (Fig. 1D). Furthermore,
immunohistochemical analysis from the HPA database showed an
absence of PAXIP1 protein in adjacent non-tumor liver tissues,
while its expression was elevated in HCC tumor tissues (Fig. 1E and F). Therefore, the aforementioned results
suggested that PAXIP1 expression was upregulated in HCC.
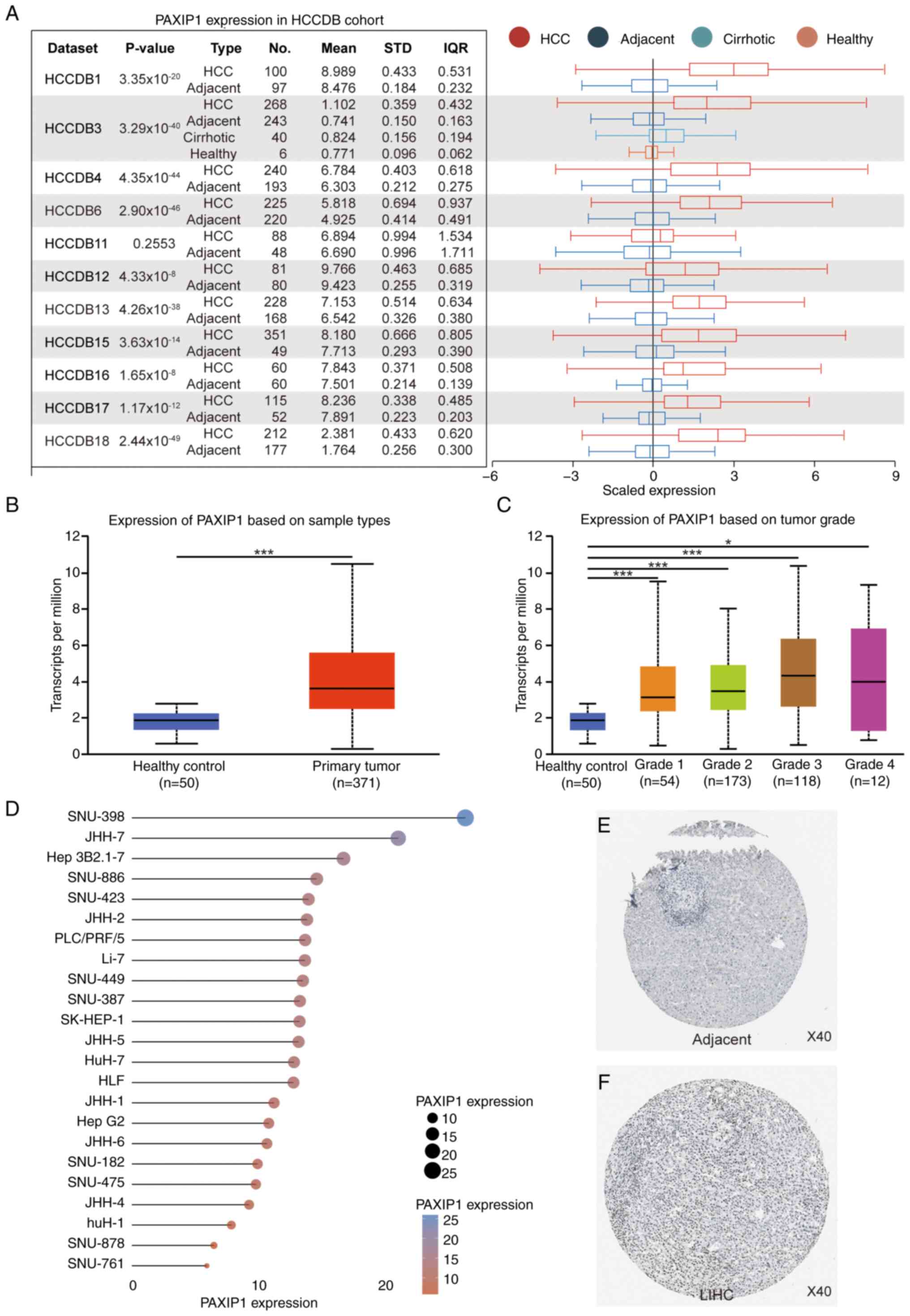 | Figure 1Increased PAXIP1 expression in HCC.
(A) Expression levels of PAXIP1 in tumor tissues vs. adjacent
non-tumor tissues in HCCDB, as analyzed using an unpaired Student's
t-test. For HCCDB3, tumor tissues were compared with adjacent
non-tumor tissues using an unpaired Student's t-test. The sample
sizes (n) were as indicated. Boxplots illustrating the relative
expression levels of PAXIP1 in healthy controls and HCC samples
based on (B) sample types and (C) tumor grade in The Cancer Genome
Atlas. (B) Unpaired two-tailed Student's t-test.
***P<0.001. (C) One-way ANOVA. *P<0.05,
***P<0.001. The sample sizes (n) were as indicated.
(D) Analysis of the expression profile of PAXIP1 mRNA across 23
distinct HCC cell lines based on the Cancer Cell Line Encyclopedia
database. Protein expression levels of PAXIP1 according to
immunohistochemical staining (HPA006694) in (E) adjacent non-tumor
tissues and (F) LIHC tissues from the HPA database. Magnification,
x40. HCC, hepatocellular carcinoma; HCCDB, Integrative Molecular
Database of HCC; HPA, Human Protein Atlas; IQR, interquartile
range; LIHC, liver hepatocellular carcinoma; PAXIP1,
PAX-interacting protein 1; STD, standard deviation. |
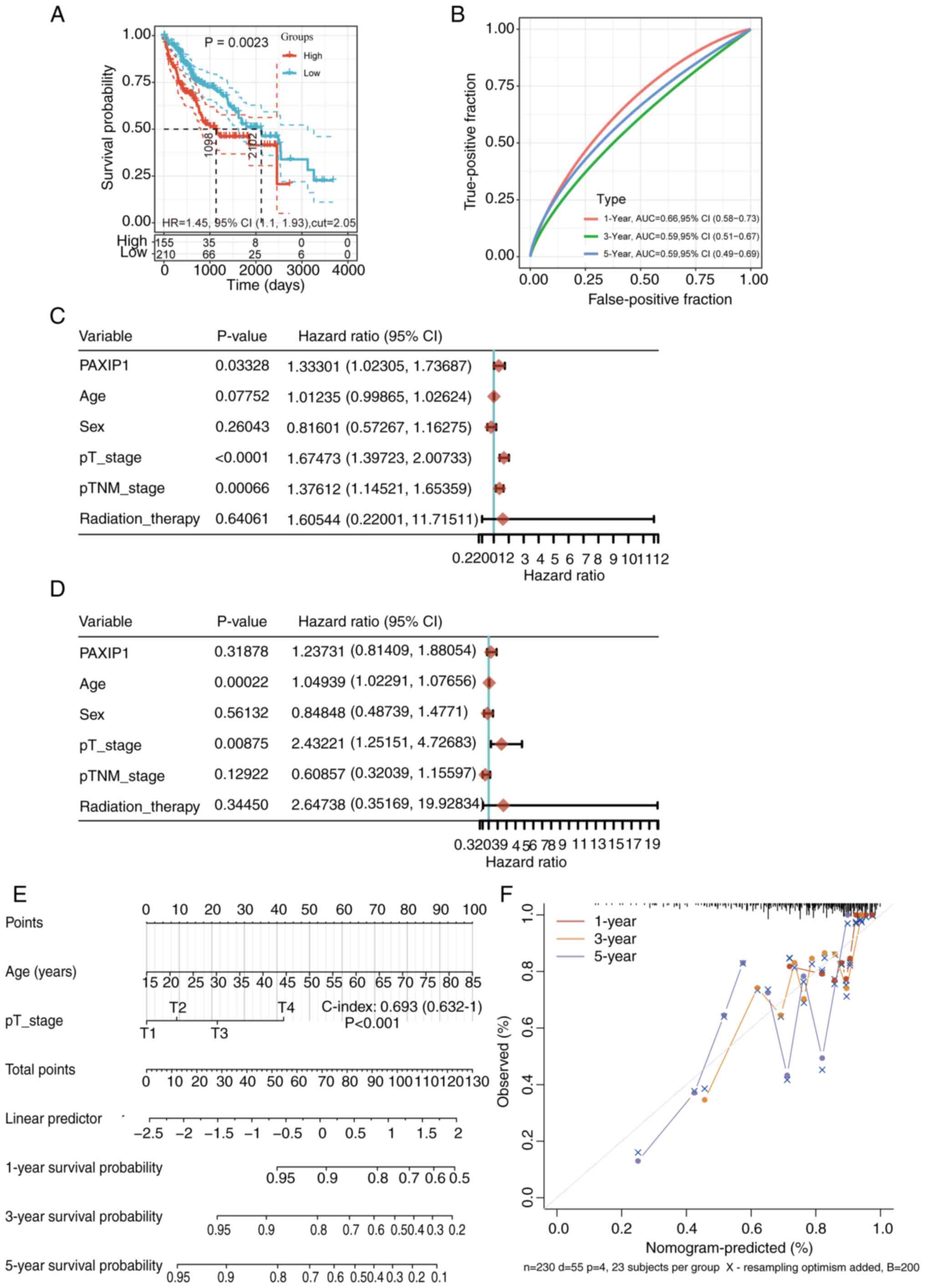
High PAXIP1 expression is associated
with poor prognosis in patients with HCC
To elucidate the relationship between PAXIP1
expression and HCC prognosis, PAXIP1 genomic alterations
were first analyzed in HCC using the cBioPortal website. The
results showed that genomic alterations in PAXIP1 were
present in 1.2% of patients (Fig.
2A). These alterations were diverse in nature (Fig. 2B). Among patients with HCC,
amplification was one of the major types of PAXIP1 copy
number variation (Fig. 2C). The
prognostic significance of PAXIP1 expression was further analyzed
in HCC. Analysis of the overall survival (OS) rate showed that
patients with high PAXIP1 expression had a low survival rate
(Fig. 3A). For the receiver
operating characteristic (ROC) curves, the AUC value range was
0.66-0.59 for 1-, 3- and 5-year prognoses (Fig. 3B). To determine whether PAXIP1 could
be used as an independent prognostic factor, univariate and
multivariate Cox regression analyses were performed. Univariate Cox
regression analysis indicated that PAXIP1 was a significant risk
factor for OS in patients with HCC (Fig. 3C; P=0.03328).
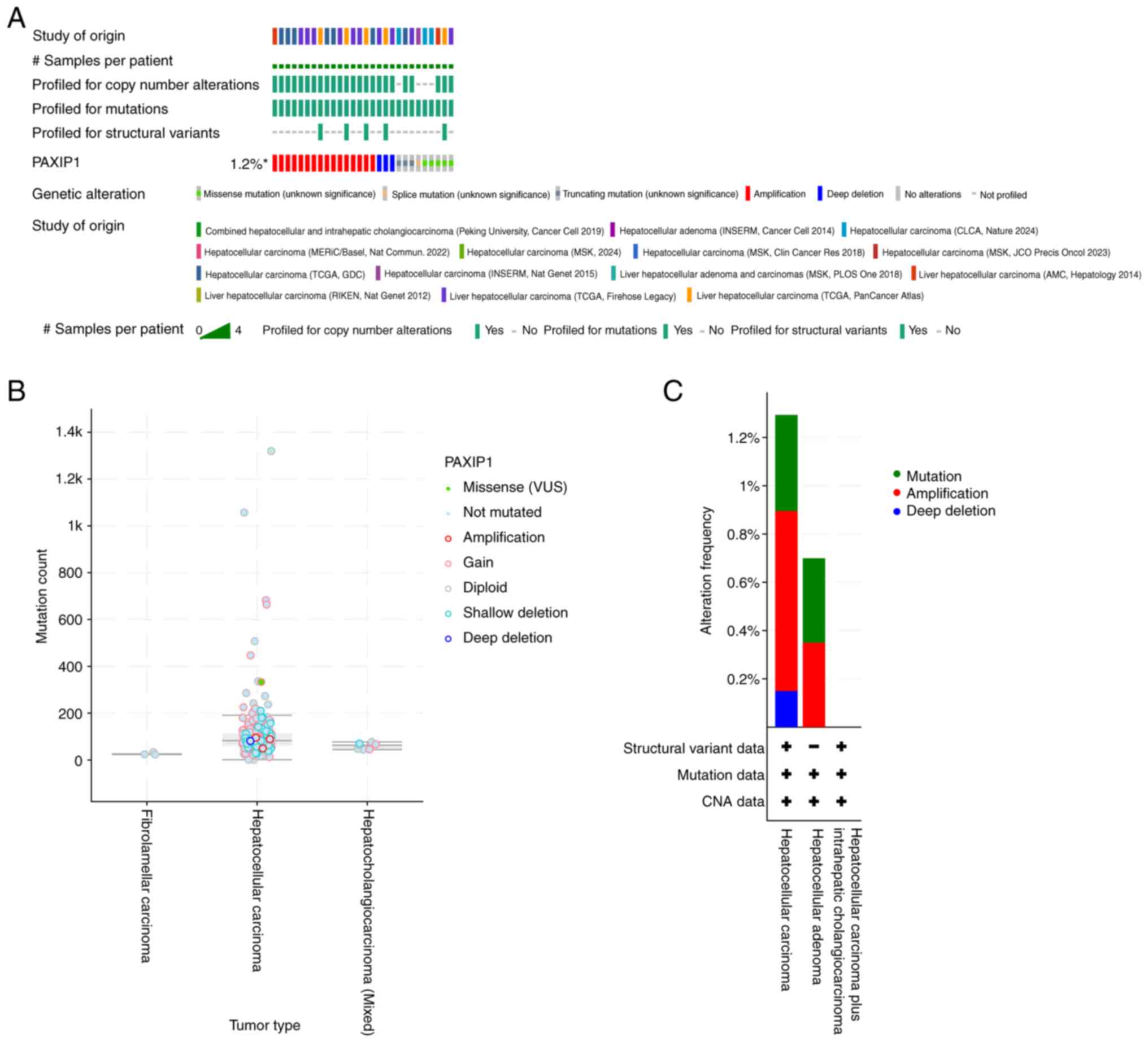 | Figure 2PAXIP1 genomic alterations in
fourteen studies of liver carcinomas analyzed using the cBioPortal
database. (A) OncoPrint analysis of PAXIP1 gene alterations in
cancer cohorts. (B) Diverse alterations of the PAXIP1 gene in
cancer cohorts. (C) Characterization of PAXIP1 Gene Alteration
Types in Cancer Cohorts. INSERM, French National Institute of
Health and Medical Research; CLCA, Chinese Liver Cancer Atlas;
MERiC/Basel, Meric Ataman University of Basel; MSK, Memorial Sloan
Kettering Cancer Center; TCGA, The Cancer Genome Atlas; AMC, Asan
Medical Center; RIKEN, RIKEN Yokohama Japan; N.S., not significant;
PAXIP1, PAX-interacting protein 1; VUS, variants of uncertain
significance. |
However, further multivariate Cox regression
analysis and nomogram results showed that PAXIP1 was not an
independent risk factor for HCC (Fig.
3D-F). Collectively, these findings indicated that a high level
of PAXIP1 may predict a low survival rate of patients with HCC.
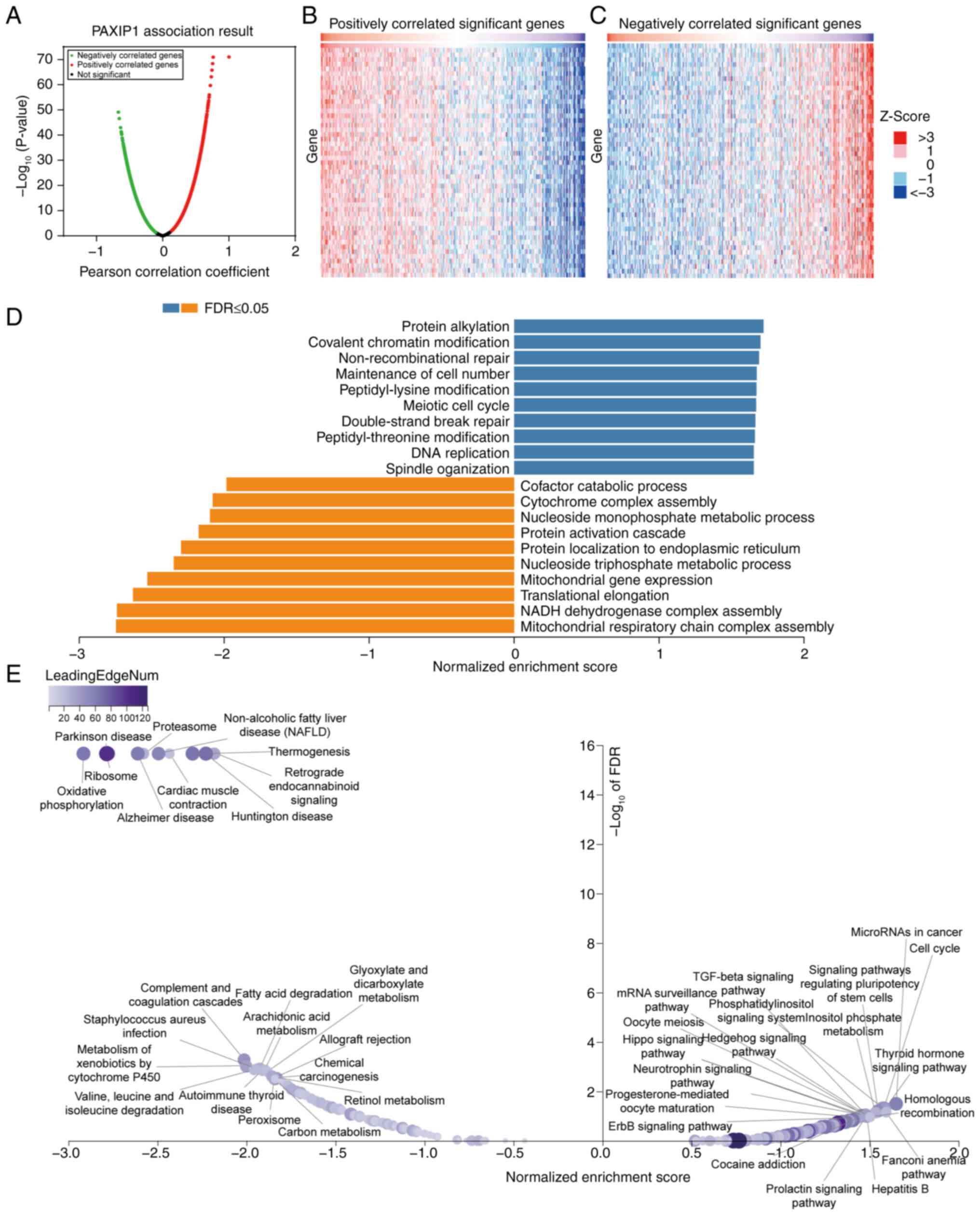
PAXIP1 co-expression networks in
HCC
To investigate the biological significance of PAXIP1
in HCC, PAXIP1 co-expression was examined in an HCC cohort using
the LinkedOmics function module. It was shown that 5,710 genes were
positively correlated with PAXIP1, while 3,317 genes were
negatively correlated with PAXIP1 (false discovery rate <0.01;
Fig. 4A). The heatmap further
illustrates these correlations, highlighting the top 50 most
significant genes, which were positively and negatively correlated
with PAXIP1 expression (Fig. 4B and
C). Subsequent GSEA was conducted
to clarify the principal GO terms associated with PAXIP1
co-expressed genes. The analysis of GO biological process
categories showed that the genes co-expressed with PAXIP1 were
predominantly involved in processes such as ‘Protein alkylation’,
‘Covalent chromatin modification’, ‘Non-recombinational repair’ and
‘Maintenance of cell number’, whereas ‘Mitochondrial respiratory
chain complex assembly’, ‘NADH dehydrogenase complex assembly’,
‘Translational elongation’, ‘mitochondrial gene expression’ and
multiple metabolic processes were found to be downregulated
(Fig. 4D; Table SI). KEGG pathway analysis indicated
notable enrichment in pathways such as ‘MicroRNAs in cancer’, ‘Cell
cycle’, ‘Complement and coagulation cascades’ and ‘Metabolism of
xenobiotics by cytochrome P450’ (Fig.
4E; Table SII). These findings
suggested the extensive impact of PAXIP1 on the global
transcriptome, highlighting its potential role in HCC
pathogenesis.
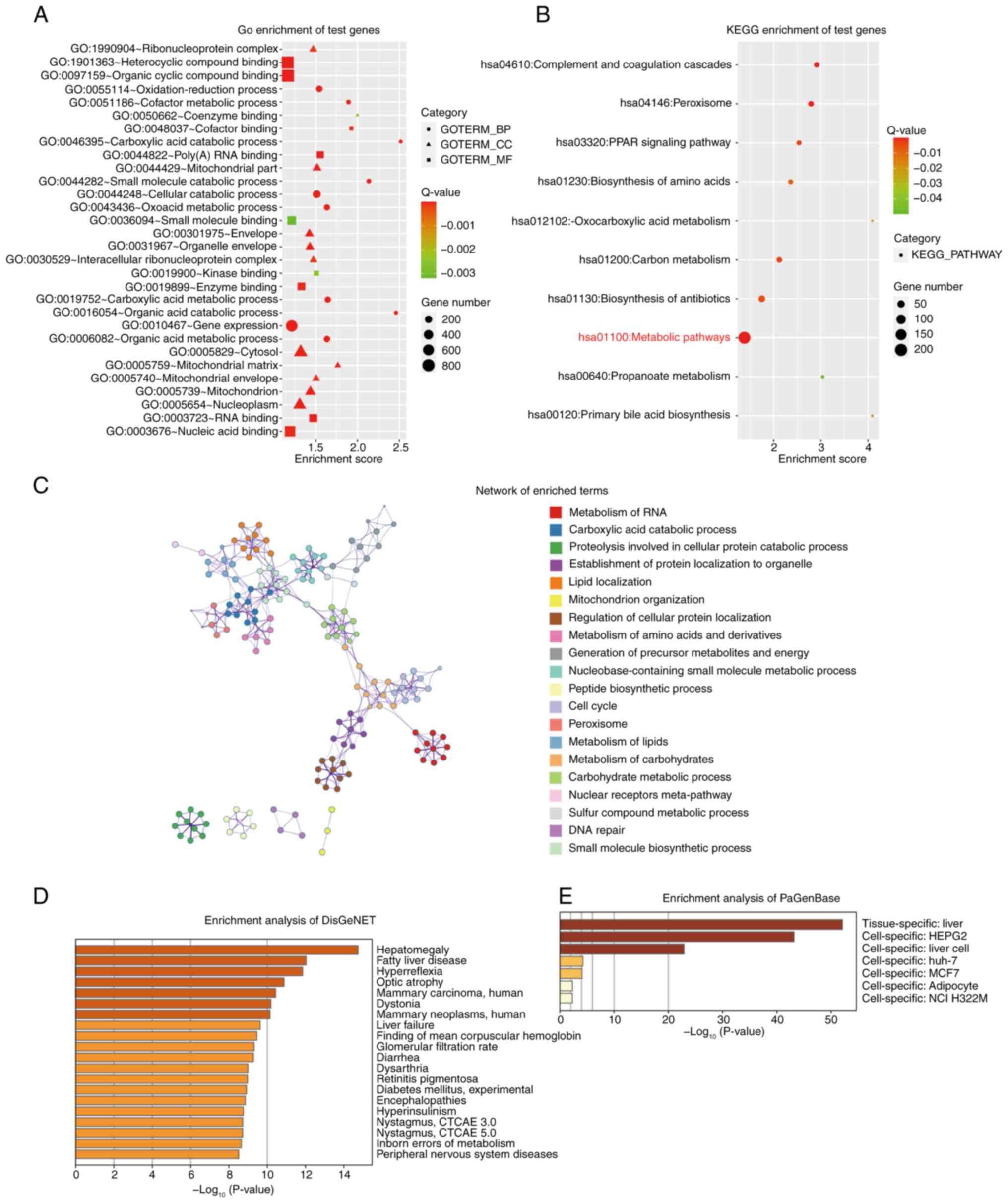
Functional analysis of PAXIP1 target
genes in HCC
PAXIP1 was associated with the survival rate of
patients with HCC. Therefore, a functional analysis of PAXIP1
target genes was performed. Using ChIP-seq data, 2,370 target genes
were identified based on their binding scores (Table SIII). Using DAVID (26,50)
and Metascape (27), functional
annotation of the 2,370 PAXIP1 target genes was performed. The most
enriched terms in the GO (Fig. 5A)
and KEGG (Fig. 5B) analyses are
shown. Notably, metabolism-related pathways or processes showed a
high frequency of occurrence, including ‘Oxocarboxylic acid
metabolism’, ‘Carbon metabolism’ and ‘Propanoate metabolism’,
indicating a potential role of PAXIP1 in the metabolic mechanisms
during HCC tumorigenesis.
The network of enriched terms elucidated the
intricate interactions among the terms with considerable detail
(Fig. 5C). To further understand
the biological functions of the 2,370 PAXIP1 targets, the Metascape
database (https://metascape.org/) was also
employed to perform DisGeNET and PaGenBase enrichment analyses. The
summary of enrichment analysis in DisGeNET and PaGenBase showed
that the differentially expressed genes were mainly enriched in
‘Hepatomegaly’ (Fig. 5D) and
‘liver’ (Fig. 5E). These results
suggested a tissue- or cell-specific role for PAXIP1 and its target
genes in HCC. Therefore, PAXIP1 may be associated with HCC
tumorigenesis via dysregulation of multiple pathways.
Special regions in HCC co-occupied by
PAXIP1, MYBL2 and FOXO1
To understand the involvement of PAXIP1 in HCC and
to expand the investigation to a genomic scale, the binding
features of PAXIP1 in HCC cells were examined. By performing
co-localization analysis of PAXIP1 ChIP-seq data (GSE104247)
derived from HepG2 cells (22), 19
potential genes were identified and a multi-gene summary was
performed using HCCDB. These genes were separated into two groups
(upregulated and downregulated) based on their expression levels in
HCC (Fig. 6A). Analysis using the
GEPIA database revealed a marked increase in MYBL2 expression and a
notable decrease in FOXO1 expression in HCC (Fig. 6B and C). Notably, FOXO1 expression was not
significantly changed when combining TCGA data with Genotype-Tissue
Expression data (Fig. 6C).
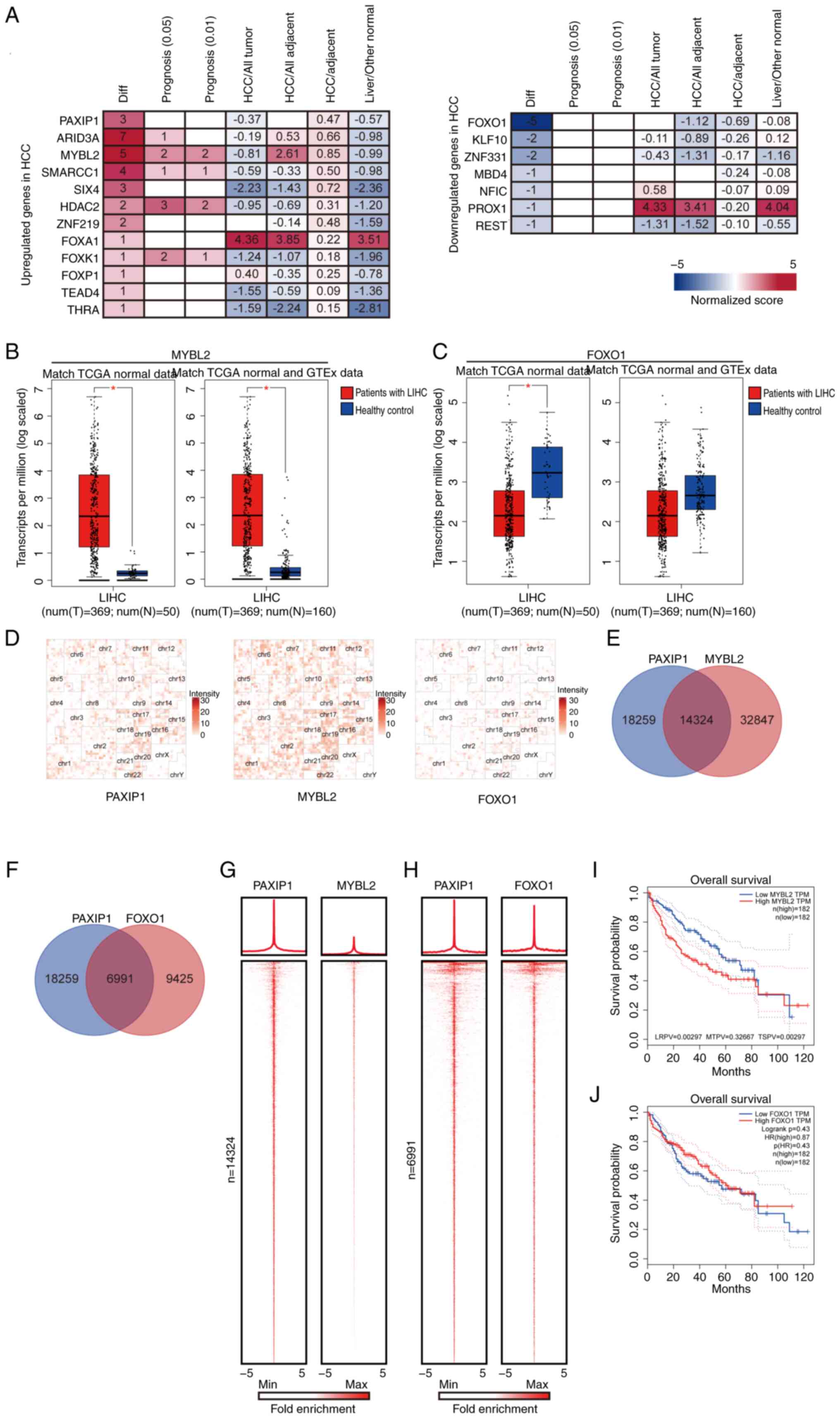 | Figure 6Identification of cofactors of PAXIP1
in HCC. (A) PAXIP1-associated multi-gene summary in the Integrative
Molecular Database of HCC. (B) MYBL2 and (C) FOXO1 expression in
LIHC tissues compared with corresponding TCGA and GTEx tissues from
healthy controls. Unpaired two-tailed Student's t-test.
*P<0.01. The sample sizes (n) were as indicated. (D)
Hilbert curve plots showing the similarity of distribution of
PAXIP1, MYBL2 and FOXO1 in human genomes. (E) Venn diagram showing
the overlapping peaks between PAXIP1 and MYBL2. (F) Venn diagram
showing the overlapping peaks between PAXIP1 and FOXO1. (G) Average
occupancy plots and heatmaps (lower panel) depicting ChIP-seq
enrichment patterns for PAXIP1 and MYBL2 within 5 Kb of the center
of the PAXIP1 peaks at PAXIP1/MYBL2-cobound regions (n=14,324). (H)
Average occupancy plots and heatmaps (lower panel) depicting
ChIP-seq enrichment patterns for PAXIP1 and FOXO1 within 5 Kb of
the center of the PAXIP1 peaks at PAXIP1/FOXO1-cobound regions
(n=6,991). Prognostic value of (I) MYBL2 and (J) FOXO1 in HCC
determined using the Gene Expression Profiling Interactive Analysis
database. The P-value was calculated using the (I) two-stage method
or (J) log-rank test (n=364). ChIP-seq, chromatin
immunoprecipitation-sequencing; GTEx, Genotype-Tissue Expression;
HCC, hepatocellular carcinoma; HR, hazard ratio; LIHC, liver
hepatocellular carcinoma; LRPV, P-value of the log-rank test; MTPV,
P-value of the suggested stage-II test; MYBL2, MYB proto-oncogene
like 2; N, healthy controls; PAXIP1, PAX-interacting protein 1; T,
tumor; TCGA, The Cancer Genome Atlas; TPM, transcripts per million;
TSPV, P-value of the two-stage test (TSPV <0.05 represent a
statistically significant difference); Diff, number of
differentially expressed datasets. |
The genomic distribution of PAXIP1, MYBL2 (GSE32465)
and FOXO1 (GSE104247) was then analyzed using data from previous
studies (21,22). To visualize the overlap of PAXIP1,
MYBL2 and FOXO1 binding sites, chromosomal folding was represented
using a Hilbert curve, which preserves the spatial proximity of
linearly adjacent regions (51).
The distributions of PAXIP1, MYBL2 and FOXO1 exhibited similar
patterns at the whole-genome scale (Fig. 6D). It was further revealed that
PAXIP1 shares 14,324 peaks with MYBL2 and 6,991 peaks with FOXO1
(Fig. 6E and F). To further analyze the relationship
between PAXIP1, MYBL2 and FOXO1, the signals of PAXIP1, MYBL2 and
FOXO1 were plotted in descending order in the PAXIP1/MYBL2- and
PAXIP1/FOXO1-cobound regions. PAXIP1 occupancy showed a similar
pattern with MYBL2 and FOXO1 in their co-bound regions (Fig. 6G and H). Survival analysis revealed that in
patients with HCC, elevated MYBL2 expression was associated with
poorer OS, while reduced FOXO1 expression was also associated with
worse OS outcomes. However, only the difference in OS related to
MYBL2 expression was statistically significant, whereas the
difference associated with FOXO1 expression was not significant
(Fig. 6I and J). Taken together, these findings
demonstrated that PAXIP1 functioned as a cofactor for MYBL2 or
FOXO1 in HCC.
Prediction and analysis of potential
TFs of PAXIP1 in HCC
TFs are responsible for regulating gene expression
in HCC development (52). To assess
whether PAXIP1 was modulated by TFs, the upstream TFs of PAXIP1
were first predicted and the top 10 predicted TFs were identified
using the hTF target database (Fig.
7A). Subsequently, survival analysis for all 10 TFs was
performed (Figs. 7B, C and S2).
Among these TFs, CTCF and NRF1 have been demonstrated to be
prognostic markers in liver cancer (53,54),
according to the HPA database. No statistically significant
differences were found; however, there was a tendency that elevated
CTCF and NRF1 expression levels were associated with poorer patient
outcomes in HCC (Fig. 7B and
C). To confirm the role of NRF1 and
CTCF in the regulation of PAXIP1 in HCC, NRF1 and CTCF were knocked
down with targeted siRNA. Decreased expression of NRF1 resulted in
decreased PAXIP1 expression in HuH-7 and PLC-PRF-5 cells; however,
CTCF knockdown did not affect PAXIP1 expression (Fig. 7D). The aforementioned findings
suggested that NRF1 may regulate PAXIP1 expression during the
development of liver cancer.
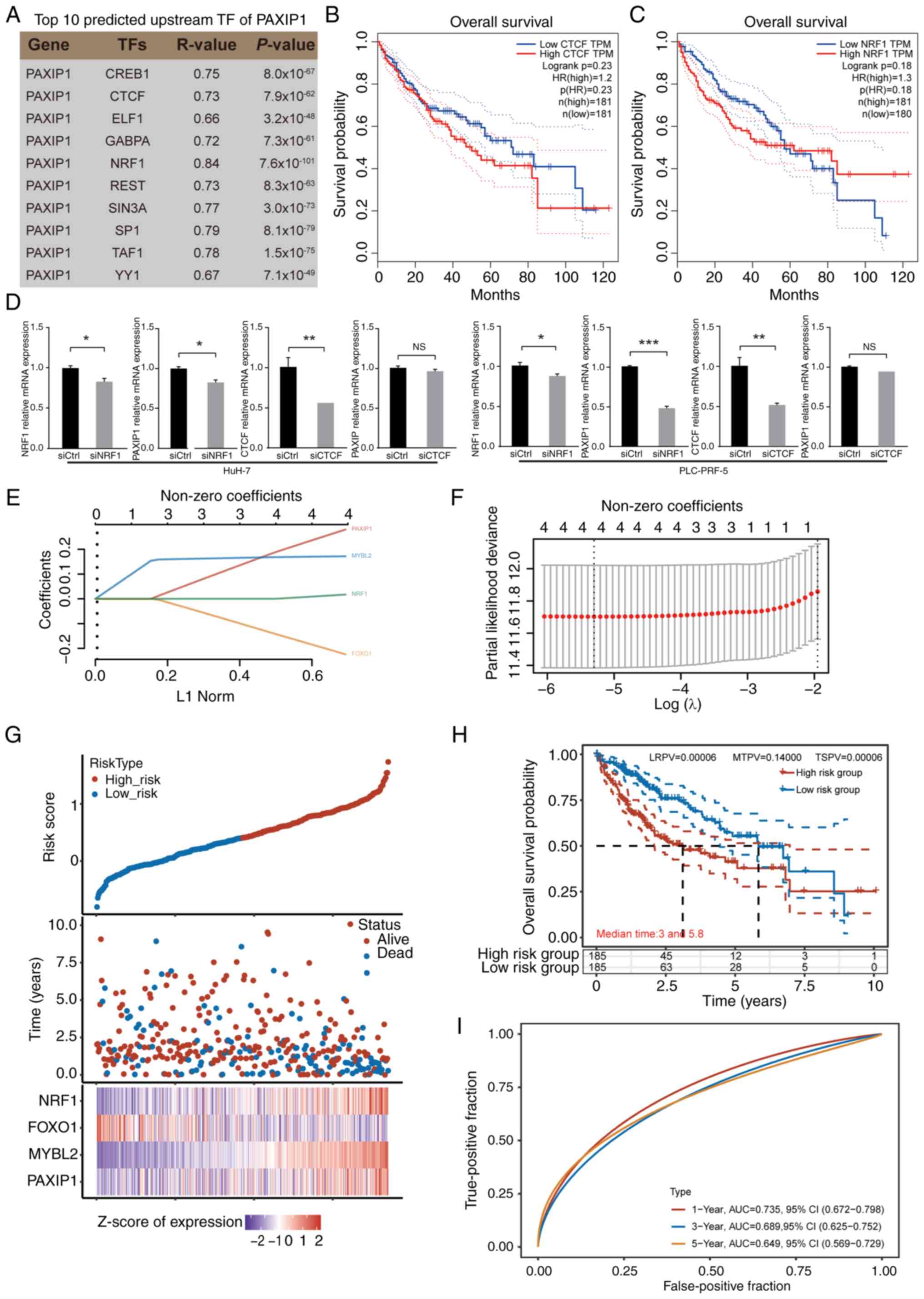 | Figure 7Identification of upstream factors of
PAXIP1 in HCC. (A) List of top 10 predicted upstream TFs of PAXIP1.
The P-value was calculated using Student's t-test. (B) Overall
survival rate of patients with HCC with low and high CTCF
expression. The P-value was calculated using the log-rank test
(n=362). (C) Overall survival rate of patients with HCC with low
and high NRF1 expression. The P-value was calculated using the
log-rank test (n=361). (D) Reverse transcription-quantitative PCR
analysis demonstrated that NRF1 knockdown downregulated PAXIP1
expression, while CTCF knockdown did not change the expression of
PAXIP1 in HCC cells. Mean ± SEM of three independent experiments.
Unpaired two-tailed Student's t-test. *P<0.05,
**P<0.01, ***P<0.001. (E) Coefficients
of PAXIP1, MYBL2, FOXO1 and NRF1 were determined using the lambda
parameter. The x-axis represents the lambda values, while the
y-axis denotes the coefficients of the independent variables. (F)
Relationship between partial likelihood bias and log(λ) plotted
through the application of the least absolute shrinkage and
selection operator Cox regression model. (G) Risk score, survival
time and survival status of selected high-risk and low-risk groups.
The top section displays a scatter plot of risk scores arranged
from low to high, with different colors representing different risk
groups. The middle section shows the scatter plot distribution of
risk scores corresponding to survival time and survival status for
different samples. The bottom section presents a heatmap of the
expression of genes included in the signature. (H) Kaplan-Meier
survival analysis was conducted to assess the risk model derived
from the dataset, and comparisons between different groups were
performed using the two-stage method. (I) Receiver operating
characteristic curve and AUC of PAXIP1-related genes. The data in
(B, C and E-I) were from The Cancer Genome Atlas. AUC, area under
the curve; CTCF, CCCTC binding factor; Ctrl, control; HCC,
hepatocellular carcinoma; HR, hazard ratio; LRPV, P-value of the
log-rank test; MTPV, P-value of the suggested stage-II test; MYBL2,
MYB proto-oncogene like 2; NRF1, nuclear respiratory factor 1; NS,
not significant; PAXIP1, PAX-interacting protein 1; si, small
interfering RNA; TF, transcription factor; TPM, transcripts per
million; TSPV, P-value of the two-stage test. |
Since potential cofactors and regulators of PAXIP1
were identified, the prognostic significance of these genes in HCC
was assessed. Patients with HCC were redivided into the high-risk
(n=185) and low-risk groups (n=185) according to the median risk
score (Fig. 7E-G). Those in the
high-risk group exhibited worse outcomes than their low-risk
counterparts (Fig. 7H). For the ROC
curve, the AUC range was 0.735-0.649 for the 1-, 3- and 5-year
prognoses (Fig. 7I). The results
indicated that the combined expression of four genes, PAXIP1,
MYBL2, FOXO1 and NRF1, could serve as an effective prognostic
marker for HCC.
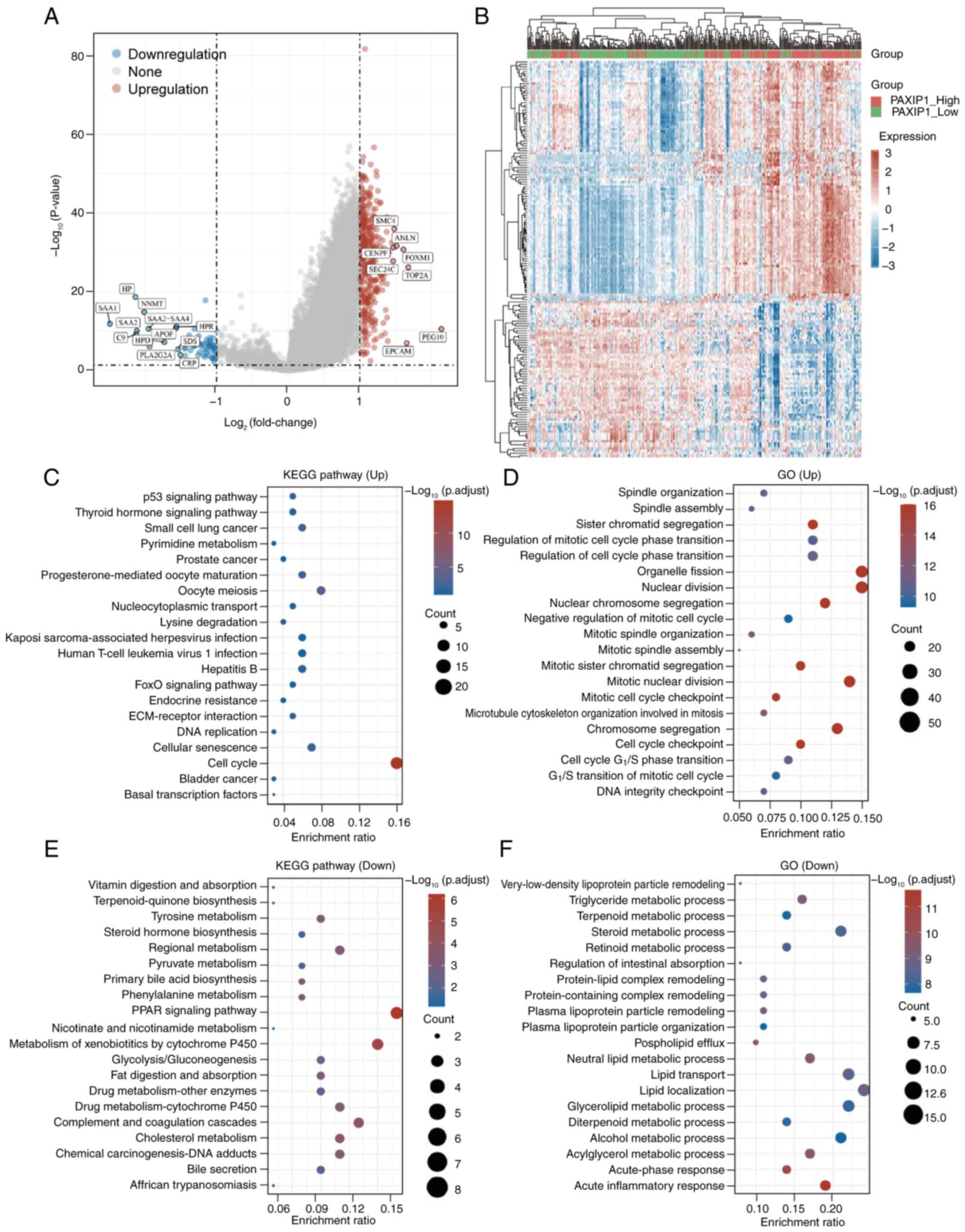
Relationship between PAXIP1 and tumor
immunity in HCC
To elucidate the molecular mechanisms underlying the
role of PAXIP1 in HCC, patients were stratified into two subgroups
based on PAXIP1 expression levels: i) The PAXIP1-high group
(PAXIP1-High; n=186); and ii) the PAXIP1-low group (PAXIP1-Low;
n=185), determined by the median expression value. Differential
expression analysis between these two subgroups was conducted
(Fig. 8A and B). The subsequent GO and KEGG results
revealed that the upregulated genes in the PAXIP1-High group were
predominantly associated with ‘cell cycle’ pathways (Fig. 8C), as well as with processes related
to ‘organelle fission’, ‘nuclear division’ and ‘chromosome
segregation’ (Fig. 8D). Conversely,
the downregulated genes in the PAXIP1-High group were involved in
‘PPAR signaling pathway’, ‘metabolism of xenobiotics by cytochrome
P450’ and ‘complement and coagulation cascades’ (Fig. 8E), and in ‘lipid localization’,
‘glycerolipid metabolic process’ and ‘acute inflammatory response’
(Fig. 8F). These findings indicated
that the differential expression of PAXIP1 target genes may result
in the dysregulation of cell division, immune responses and
metabolic processes.
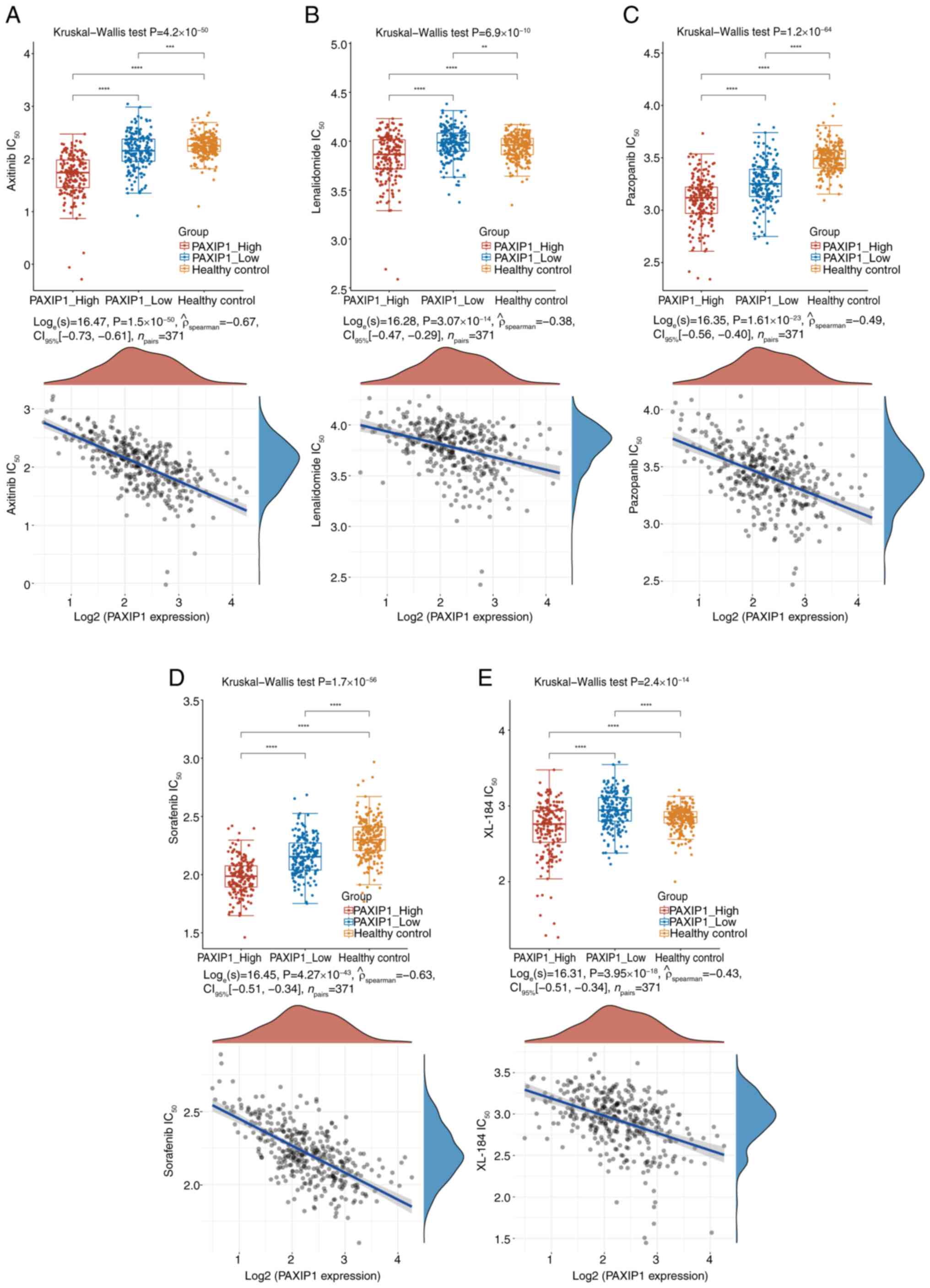
The chemotherapeutic response of each sample was
assessed using the GDSC database. The 50% maximal inhibitory
concentrations for the samples were estimated via ridge regression,
and all the involved parameters were set to their default values.
To eliminate batch effects, combat normalization was applied, and
duplicate gene expression values were summarized by taking their
mean. The results showed that the sensitivities to the
chemotherapeutic drugs axitinib, lenalidomide, pazopanib, sorafenib
and XL-184 (Fig. 9A-E; P<0.001)
were significantly negatively correlated with PAXIP1 expression in
HCC. These analyses indicated that upregulation of PAXIP1
expression may increase sensitivity to the aforementioned
chemotherapy drugs in HCC treatment.
Given the possible oncogenic function of PAXIP1 in
HCC, it is imperative to explore the relationship between PAXIP1
and immune events in HCC. Utilizing the TIMER database, the
correlation between PAXIP1 expression and immune cell infiltration
was evaluated. The analysis revealed a positive association between
PAXIP1 expression and the presence of CD4+ T cells,
neutrophils, macrophages, B cells and myeloid dendritic cells
within HCC tissues (Fig. 10A and
B). Immune checkpoints such as
programmed cell death protein 1 (PD1/PDCD1)/programmed death-ligand
1 (PD-L1/CD274) and cytotoxic T-lymphocyte associated protein 4
(CTLA4) are crucial for the modulation of immune responses and
tumor immune evasion (55). The
current study demonstrated a significant positive correlation
between PAXIP1 expression and the levels of PDCD1, CD274 and CTLA4
(Fig. S3A-C; P<0.05). Further
analysis using the GEPIA database corroborated these findings,
revealing a weak positive correlation between PAXIP1 and PDCD1,
CD274 and CTLA4 expression in HCC (Fig. S3D-F). Therefore, PAXIP1 may
contribute to carcinogenesis in HCC through mechanisms involving
immune cell infiltration and tumor immune escape.
Discussion
HCC is a common and aggressive malignancy found in
several countries. A total of 80-90% of liver cancer cases develop
because of underlying conditions such as hepatitis B/C virus
infection and alcohol-induced liver cirrhosis (56). The exact etiology of liver cancer is
under investigation; however, its development originates from a
complex interplay of genetic and environmental factors (56). The present study highlighted the
significant role of the epigenetic factor PAXIP1 in the
pathogenesis and progression of HCC. The results demonstrated that
PAXIP1 may be a critical node in HCC development, subject to
transcriptional regulation, and may act together with other
cofactors to exert its functions. A model of the NRF1-PAXIP1 axis
in HCC is shown in Fig. 10C. It
was shown that PAXIP1 serves as a prognostic biomarker closely
linked to immune infiltration in HCC. This demonstrates that PAXIP1
may be the key in controlling immune cell infiltration, thereby
highlighting its potential as a valuable prognostic marker for
patients with HCC. Further prospective cohort studies are warranted
to elucidate this association, and further research is necessary to
identify the prognostic significance of PAXIP1 in HCC.
Research on PAXIP1 as an epigenetic factor has
primarily focused on four areas: Developmental biology, DNA damage
repair, immune-related functions and tumor development (18,57-62).
In 2003, Cho et al (13),
using PAXIP1 gene knockout mice, found that these mice exhibited
delayed development, culminating in embryonic lethality around
embryonic day 9.5. Although subsequent analysis revealed that
knockout cells were capable of DNA replication, mitotic division
was decreased (13). A recent study
has also indicated that PAXIP1 is crucial for maintaining mitotic
integrity, with PAXIP1 inactivation leading to increased cell death
during mitotic exit (57). The
results of the present study suggested that PAXIP1 and
co-expression genes were involved in multiple processes related to
cell division in HCC, including DNA replication and covalent
chromatin modifications. PAXIP1 is part of the myeloid/lymphoid or
mixed-lineage leukemia 3 (MLL3)/myeloid/lymphoid or mixed-lineage
leukemia 4 (MLL4)-complex proteins associated with Set1
(COMPASS)-like complex, which serves a critical role in maintaining
DNA modifications and structure (63). These complexes deposit H3K4me1 marks
on enhancers to regulate gene transcription (63). The interaction of PAXIP1 with DNA is
not solely dependent on the COMPASS-like complex; it also interacts
with 53BP1 and participates in DNA damage repair (64).
In the current study, upregulation of PAXIP1 was
associated with poor outcomes in patients with HCC. It has been
demonstrated that multi-gene prognostic models were more effective
and comprehensive than single-gene prognostic models in predicting
cancer outcomes (65).
FOXK1(66), FOXO1(67), histone deacetylase 2(68), MYBL2(69) and SWI/SNF related BAF chromatin
remodeling complex subunit C1(70)
were previously reported to be favorable or unfavorable prognostic
markers in liver cancer, and were associated with PAXIP1 in HCC.
The present results were consistent with the aforementioned
conclusion. In the present study, using PAXIP1, MYBL2, FOXO1 and
NRF1 as a four-gene prognostic marker for HCC had a strong capacity
for predicting prognosis.
Metabolic reprogramming is a defining characteristic
of numerous cancer types, including HCC. Changes in metabolic
processes provide advantages for tumor expansion, tumor growth and
survival by increasing energy production, macromolecular synthesis
and redox equilibrium maintenance (71). In the present study, ChIP-seq data
were analyzed and it was revealed that PAXIP1 binds to genes
associated with metabolism, indicating its potential role in
regulating metabolic processes in HCC development. Deficiency of
UTX, a PAXIP1-interacting protein, in adipocytes leads to metabolic
dysfunction in the liver (72).
Further evidence is needed to clarify whether UTX is required for
PAXIP1 to affect liver cancer metabolism.
Our previous study in Drosophila suggested
that PAXIP1 mediated a molecular switch between histone
modifications, namely H3K4me3/H3K27ac and H3K27me3, in
Trithorax-related or polycomb-occupied regions (59). However, to the best of our
knowledge, the precise mechanism by which PAXIP1 is specifically
recruited to the promoter regions of target genes is yet to be
understood. Typically, PAXIP1 specifically binds to target genes
through interaction with other specific TFs and recruits them to
the promoter region (5). Our
previous study indicated that PAXIP1 interacted with Fosl2 or YY1
to be recruited to the EphA2 promoter region in esophageal squamous
cell carcinoma (18). The present
analysis revealed that PAXIP1, MYBL2 and FOXO1 exhibited similar
genomic distributions, and the expression levels of these factors
were associated with survival rates in HCC except for those of
FOXO1. Therefore, PAXIP1 may be recruited to the promoter regions
of its target genes via MYBL2 or FOXO1 in HCC cells.
A pharmacological screen involving 17 kinases found
that PAXIP1 enhanced sensitivity to AZD1775 in combination with
platinum-based treatment in lung cancer (17). Consistent with a previous study
(17), elevated PAXIP1 expression
was associated with heightened sensitivity of hepatocellular
carcinoma to chemotherapeutic drugs. Further studies are needed to
explore the mechanisms underlying the observed associations.
PAXIP1 serves a crucial yet inadequately understood
role within the immune system. PAXIP1 participates in
immunoglobulin class switching and variable-diversity-joining
rearrangement recombination, which depend on MLL3-MLL4 complex
activity (73). Another study
demonstrated that PAXIP1 regulated thymocyte development in the
thymus (74). In Paxip1 knockout
mice, a marked increase the population of CD4+ and
CD8+ single-positive T cells was observed compared with
that in wild-type mice (74). The
present analysis revealed a positive association between the
expression of PAXIP1 and various immune cell types within the tumor
microenvironment, including CD4+ T cells, neutrophils,
macrophages, B cells and myeloid dendritic cells. Studies have
shown that the infiltration of CD4+ T cells, especially
certain subsets such as regulatory T cells and effector memory T
cells, is associated with the immune response in HCC (75,76).
The results of the present study suggested that PAXIP1 may be
implicated in the tumor immune response, potentially offering novel
insights for HCC treatment. Overall, the results of the present
study revealed a key role of PAXIP1 in HCC development, and
provided a novel index for the clinical diagnosis of HCC.
Supplementary Material
Transcription and promoter methylation
level of PAXIP1 in subgroups of patients with HCC stratified based
on sex, age and ethnicity. Boxplots showing relative PAXIP1
expression in healthy controls and HCC samples based on (A) sex,
(B) age and (C) ethnicity. One way ANOVA. *P<0.05,
**P<0.01, ***P<0.001. The sample sizes
(n) were as indicated. Boxplots showing relative promoter
methylation level of PAXIP1 in healthy controls and HCC samples
based on (D) sex, (E) age and (F) ethnicity. One way ANOVA.
*P<0.05, **P<0.01,
***P<0.001. The sample sizes (n) were as indicated.
HCC, hepatocellular carcinoma; NS, not significant; PAXIP1, PAX
interacting protein 1.
Overall survival rate of patients with
hepatocellular carcinoma with low or high (A) CREB1, (B) ELF1, (C)
GABPA, (D) REST, (E) SIN3A, (F) SP1, (G) TAF1 and (H) YY1
expression. The P value was calculated using the log rank test
(n=362). HR, hazard ratio; TPM, transcripts per million.
PAXIP1 expression is correlated with
CD274, CTLA4 and PDCD1 expression in HCC. (A) Heatmap illustrating
the correlation between PAXIP1 expression and immune checkpoint
genes based on The Cancer Genome Atlas data. Kolmogorov Smirnov and
Wilcoxon rank sum tests. The P value represents the comparison
between the PAXIP1 high group and the PAXIP1 low group.
*P<0.05, ***P<0.001. (B) Spearman's
correlation between HCC tumor purity and PAXIP1 expression. (C)
Spearman correlation analysis of PAXIP1 expression and PDCD1, CD274
and CTLA4 expression in The Cancer Genome Atlas dataset of HCC,
adjusted for tumor purity using Tumor Immune Estimation Resource
2.0. The Spearman correlation coefficient and P value calculated
using Spearman's test are indicated. Correlation of PAXIP1
expression with (D) PDCD1, (E) CD274 and (F) CTLA4 expression in
HCC as assessed using the Gene Expression Profiling Interactive
Analysis database. Pearson correlation coefficient and the P value
calculated using the Pearson test are indicated. CTLA4, cytotoxic T
lymphocyte associated protein 4; HCC, hepatocellular carcinoma;
PAXIP1, PAX interacting protein 1; PDCD1, programmed cell death 1;
TPM, transcripts per million
GO biological process terms enriched
in PAX-interacting protein 1-associated genes in the hepatocellular
carcinoma cohort.
Kyoto Encyclopedia of Genes and
Genomes pathways enriched in PAX-interacting protein 1-associated
genes in the hepatocellular carcinoma cohort.
Potential target genes for
PAXIP1.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by grants from the
Natural Science Foundation of China (grant nos. 31301146, 82203738
and 82102969), Jiangsu Funding Program for Excellent Postdoctoral
Talent (grant no. 2023ZB782), Jiangsu Provincial Medical Key
Discipline Cultivation Unit (grant no. JSDW202233) and Huai'an
Natural Science Research Program (grant nos. HAB202110 and
HAB202101).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
QC, XH and CWZ conceived the study and revised the
manuscript. XH, HX, YLL, FW, XYC and CJ were responsible for data
collection, as well as the subsequent data analysis. QC, XH and CWZ
confirm the authenticity of all the raw data. QC and CWZ wrote the
manuscript. All authors participated in the manuscript development.
All authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Luo Z, Lu L, Tang Q, Wei W, Chen P, Chen
Y, Pu J and Wang J: CircCAMSAP1 promotes hepatocellular carcinoma
progression through miR-1294/GRAMD1A pathway. J Cell Mol Med.
25:3793–3802. 2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
DiStefano JK and Davis B: Diagnostic and
prognostic Potential of AKR1B10 in human hepatocellular carcinoma.
Cancers (Basel). 11(486)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lechner MS, Levitan I and Dressler GR:
PTIP, a novel BRCT domain-containing protein interacts with Pax2
and is associated with active chromatin. Nucleic Acids Res.
28:2741–2751. 2000.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Shimizu K, Bourillot PY, Nielsen SJ, Zorn
AM and Gurdon JB: Swift is a novel BRCT domain coactivator of Smad2
in transforming growth factor beta signaling. Mol Cell Biol.
21:3901–3912. 2001.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Cho YW, Hong T, Hong S, Guo H, Yu H, Kim
D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M and Ge K: PTIP
associates with MLL3- and MLL4-containing histone H3 lysine 4
methyltransferase complex. J Biol Chem. 282:20395–20406.
2007.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Munoz IM, Jowsey PA, Toth R and Rouse J:
Phospho-epitope binding by the BRCT domains of hPTIP controls
multiple aspects of the cellular response to DNA damage. Nucleic
Acids Res. 35:5312–5322. 2007.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Patel SR, Kim D, Levitan I and Dressler
GR: The BRCT-domain containing protein PTIP links PAX2 to a histone
H3, lysine 4 methyltransferase complex. Dev Cell. 13:580–592.
2007.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Escribano-Diaz C and Durocher D: DNA
repair pathway choice-a PTIP of the hat to 53BP1. EMBO Rep.
14:665–666. 2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Issaeva I, Zonis Y, Rozovskaia T, Orlovsky
K, Croce CM, Nakamura T, Mazo A, Eisenbach L and Canaani E:
Knockdown of ALR (MLL2) reveals ALR target genes and leads to
alterations in cell adhesion and growth. Mol Cell Biol.
27:1889–1903. 2007.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Mohan M, Herz HM, Smith ER, Zhang Y,
Jackson J, Washburn MP, Florens L, Eissenberg JC and Shilatifard A:
The COMPASS family of H3K4 methylases in Drosophila. Mol Cell Biol.
31:4310–4318. 2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Cho EA, Prindle MJ and Dressler GR: BRCT
domain-containing protein PTIP is essential for progression through
mitosis. Mol Cell Biol. 23:1666–1673. 2003.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ray Chaudhuri A, Callen E, Ding X, Gogola
E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et
al: Replication fork stability confers chemoresistance in
BRCA-deficient cells. Nature. 535:382–387. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Willis S, Villalobos VM, Gevaert O,
Abramovitz M, Williams C, Sikic BI and Leyland-Jones B: Single gene
prognostic biomarkers in ovarian cancer: A meta-analysis. PLoS One.
11(e0149183)2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
De Gregoriis G, Ramos JA, Fernandes PV,
Vignal GM, Brianese RC, Carraro DM, Monteiro AN, Struchiner CJ,
Suarez-Kurtz G, Vianna-Jorge R and de Carvalho MA: DNA repair genes
PAXIP1 and TP53BP1 expression is associated with breast cancer
prognosis. Cancer Biol Ther. 18:439–449. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Jhuraney A, Woods NT, Wright G, Rix L,
Kinose F, Kroeger JL, Remily-Wood E, Cress WD, Koomen JM, Brantley
SG, et al: PAXIP1 potentiates the combination of WEE1 inhibitor
AZD1775 and platinum agents in lung cancer. Mol Cancer Ther.
15:1669–1681. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Han X, Zhu Y, Shen L, Zhou Y, Pang L, Zhou
W, Gu H, Han K, Yang Y, Jiang C, et al: PTIP inhibits cell invasion
in esophageal squamous cell carcinoma via modulation of EphA2
expression. Front Oncol. 11(629916)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Harland LTG, Simon CS, Senft AD, Costello
I, Greder L, Imaz-Rosshandler I, Göttgens B, Marioni JC, Bikoff EK,
Porcher C, et al: The T-box transcription factor Eomesodermin
governs haemogenic competence of yolk sac mesodermal progenitors.
Nat Cell Biol. 23:61–74. 2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Gertz J, Savic D, Varley KE, Partridge EC,
Safi A, Jain P, Cooper GM, Reddy TE, Crawford GE and Myers RM:
Distinct properties of cell-type-specific and shared transcription
factor binding sites. Mol Cell. 52:25–36. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Partridge EC, Chhetri SB, Prokop JW,
Ramaker RC, Jansen CS, Goh ST, Mackiewicz M, Newberry KM,
Brandsmeier LA, Meadows SK, et al: Occupancy maps of 208
chromatin-associated proteins in one human cell type. Nature.
583:720–728. 2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Quinlan AR and Hall IM: BEDTools: A
flexible suite of utilities for comparing genomic features.
Bioinformatics. 26:841–842. 2010.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ramirez F, Ryan DP, Grüning B, Bhardwaj V,
Kilpert F, Richter AS, Heyne S, Dündar F and Manke T: deepTools2: A
next generation web server for deep-sequencing data analysis.
Nucleic Acids Res. 44 (W1):W160–W165. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yu G, Wang LG and He QY: ChIPseeker: An
R/Bioconductor package for ChIP peak annotation, comparison and
visualization. Bioinformatics. 31:2382–2383. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zhou Y, Zhou B, Pache L, Chang M,
Khodabakhshi AH, Tanaseichuk O, Benner C and Chanda SK: Metascape
provides a biologist-oriented resource for the analysis of
systems-level datasets. Nat Commun. 10(1523)2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Lian Q, Wang S, Zhang G, Wang D, Luo G,
Tang J, Chen L and Gu J: HCCDB: A database of hepatocellular
carcinoma expression atlas. Genomics Proteomics Bioinformatics.
16:269–275. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Riillo C, Polerà N, Di Martino MT, Juli G,
Hokanson CA, Odineca T, Signorelli S, Grillone K, Ascrizzi S,
Mancuso A, et al: A pronectin™ AXL-targeted
first-in-class bispecific T cell engager (pAXLxCD3ε) for ovarian
cancer. J Transl Med. 21(301)2023.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Thul PJ and Lindskog C: The human protein
atlas: A spatial map of the human proteome. Protein Sci.
27:233–244. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
de Bruijn I, Kundra R, Mastrogiacomo B,
Tran TN, Sikina L, Mazor T, Li X, Ochoa A, Zhao G, Lai B, et al:
Analysis and visualization of longitudinal genomic and clinical
data from the AACR project GENIE biopharma collaborative in
cBioPortal. Cancer Res. 83:3861–3867. 2023.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Xue R, Chen L, Zhang C, Fujita M, Li R,
Yan SM, Ong CK, Liao X, Gao Q, Sasagawa S, et al: Genomic and
transcriptomic profiling of combined hepatocellular and
intrahepatic cholangiocarcinoma reveals distinct molecular
subtypes. Cancer Cell. 35:932–947.e8. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Chen L, Zhang C, Xue R, Liu M, Bai J, Bao
J, Wang Y, Jiang N, Li Z, Wang W, et al: Deep whole-genome analysis
of 494 hepatocellular carcinomas. Nature. 627:586–593.
2024.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ng CKY, Dazert E, Boldanova T,
Coto-Llerena M, Nuciforo S, Ercan C, Suslov A, Meier MA, Bock T,
Schmidt A, et al: Integrative proteogenomic characterization of
hepatocellular carcinoma across etiologies and stages. Nat Commun.
13(2436)2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Harding JJ, Nandakumar S, Armenia J,
Khalil DN, Albano M, Ly M, Shia J, Hechtman JF, Kundra R, El Dika
I, et al: Prospective genotyping of hepatocellular carcinoma:
Clinical implications of next-generation sequencing for matching
patients to targeted and immune therapies. Clin Cancer Res.
25:2116–2126. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Cowzer D, White JB, Chou JF, Chen PJ, Kim
TH, Khalil DN, El Dika IH, Columna K, Yaqubie A, Light JS, et al:
Targeted molecular profiling of circulating cell-free DNA in
patients with advanced hepatocellular carcinoma. JCO Precis Oncol.
7(e2300272)2023.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Schulze K, Imbeaud S, Letouzé E,
Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C,
Shinde J, Soysouvanh F, et al: Exome sequencing of hepatocellular
carcinomas identifies new mutational signatures and potential
therapeutic targets. Nat Genet. 47:505–511. 2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zheng J, Sadot E, Vigidal JA, Klimstra DS,
Balachandran VP, Kingham TP, Allen PJ, D'Angelica MI, DeMatteo RP,
Jarnagin WR and Ventura A: Characterization of hepatocellular
adenoma and carcinoma using microRNA profiling and targeted gene
sequencing. PLoS One. 13(e0200776)2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Ahn SM, Jang SJ, Shim JH, Kim D, Hong SM,
Sung CO, Baek D, Haq F, Ansari AA, Lee SY, et al: Genomic portrait
of resectable hepatocellular carcinomas: Implications of RB1 and
FGF19 aberrations for patient stratification. Hepatology.
60:1972–1982. 2014.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Fujimoto A, Totoki Y, Abe T, Boroevich KA,
Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, et al:
Whole-genome sequencing of liver cancers identifies etiological
influences on mutation patterns and recurrent mutations in
chromatin regulators. Nat Genet. 44:760–764. 2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Pilati C, Letouzé E, Nault JC, Imbeaud S,
Boulai A, Calderaro J, Poussin K, Franconi A, Couchy G, Morcrette
G, et al: Genomic profiling of hepatocellular adenomas reveals
recurrent FRK-activating mutations and the mechanisms of malignant
transformation. Cancer Cell. 25:428–441. 2014.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Vasaikar SV, Straub P, Wang J and Zhang B:
LinkedOmics: Analyzing multi-omics data within and across 32 cancer
types. Nucleic Acids Res. 46 (D1):D956–D963. 2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Chen Y, Li B, Wang J, Liu J, Wang Z, Mao
Y, Liu S, Liao X and Chen J: Identification and verification of the
prognostic value of the glutathione S-transferase Mu genes in
gastric cancer. Oncol Lett. 20(100)2020.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Zhang Z, Lin E, Zhuang H, Xie L, Feng X,
Liu J and Yu Y: Construction of a novel gene-based model for
prognosis prediction of clear cell renal cell carcinoma. Cancer
Cell Int. 20(27)2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Zhang Q, Liu W, Zhang HM, Xie GY, Miao YR,
Xia M and Guo AY: hTFtarget: A comprehensive database for
regulations of human transcription factors and their targets.
Genomics Proteomics Bioinformatics. 18:120–128. 2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Thorvaldsdóttir H, Robinson JT and Mesirov
JP: Integrative genomics viewer (IGV): High-performance genomics
data visualization and exploration. Brief Bioinform. 14:178–192.
2013.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Geeleher P, Cox N and Huang RS:
pRRophetic: An R package for prediction of clinical
chemotherapeutic response from tumor gene expression levels. PLoS
One. 9(e107468)2014.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Li H, Han D, Hou Y, Chen H and Chen Z:
Statistical inference methods for two crossing survival curves: A
comparison of methods. PLoS One. 10(e0116774)2015.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Huang da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Anders S: Visualization of genomic data
with the Hilbert curve. Bioinformatics. 25:1231–1235.
2009.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Goodall GJ and Wickramasinghe VO: RNA in
cancer. Nat Rev Cancer. 21:22–36. 2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Shetron SG: Large-scale ecosystem
restoration: Five case studies from the United States. Choice:
Current Reviews for Academic Libraries. 46:715–716. 2008.
|
|
54
|
Wang Y, Guo X, Niu Z, Huang X, Wang B and
Gao L: DeepCBS: Shedding light on the impact of mutations occurring
at CTCF binding sites. Front Genet. 15(1354208)2024.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Srinivasan P, Wu X, Basu M, Rossi C and
Sandler AD: PD-L1 checkpoint inhibition and anti-CTLA-4 whole tumor
cell vaccination counter adaptive immune resistance: A mouse
neuroblastoma model that mimics human disease. PLoS Med.
15(e1002497)2018.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Yang JD, Hainaut P, Gores GJ, Amadou A,
Plymoth A and Roberts LR: A global view of hepatocellular
carcinoma: Trends, risk, prevention and management. Nat Rev
Gastroenterol Hepatol. 16:589–604. 2019.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Zhang F, Wei M, Chen H, Ji L, Nie Y and
Kang J: The genomic stability regulator PTIP is required for proper
chromosome segregation in mitosis. Cell Div. 17(5)2022.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Liu B and Li Z: PTIP-associated protein 1:
More than a component of the MLL3/4 complex. Front Genet.
13(889109)2022.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Cheng Q, Xie H, Zhang XY, Wang MY, Bi CL,
Wang Q, Wang R and Fang M: An essential role for PTIP in mediating
Hox gene regulation along PcG and trxG pathways. FEBS J.
289:6324–6341. 2022.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Xu Y, Zhu D, Yang Q, Su D and Chen YQ:
PTIP deficiency in B lymphocytes reduces subcutaneous fat
deposition in mice. Biochemistry (Mosc). 86:568–576.
2021.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Callen E, Zong D, Wu W, Wong N, Stanlie A,
Ishikawa M, Pavani R, Dumitrache LC, Byrum AK, Mendez-Dorantes C,
et al: 53BP1 Enforces distinct pre- and post-resection blocks on
homologous recombination. Mol Cell. 77:26–38.e7. 2020.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Das P, Veazey KJ, Van HT, Kaushik S, Lin
K, Lu Y, Ishii M, Kikuta J, Ge K, Nussenzweig A and Santos MA:
Histone methylation regulator PTIP is required to maintain normal
and leukemic bone marrow niches. Proc Natl Acad Sci USA.
115:E10137–E10146. 2018.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Cenik BK and Shilatifard A: COMPASS and
SWI/SNF complexes in development and disease. Nat Rev Genet.
22:38–58. 2021.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Wu J, Prindle MJ, Dressler GR and Yu X:
PTIP regulates 53BP1 and SMC1 at the DNA damage sites. J Biol Chem.
284:18078–18084. 2009.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Chang J, Wu H, Wu J, Liu M, Zhang W, Hu Y,
Zhang X, Xu J, Li L, Yu P and Zhu J: Constructing a novel
mitochondrial-related gene signature for evaluating the tumor
immune microenvironment and predicting survival in stomach
adenocarcinoma. J Transl Med. 21(191)2023.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Li P, Yu Z, He L, Zhou D, Xie S, Hou H and
Geng X: Knockdown of FOXK1 inhibited the proliferation, migration
and invasion in hepatocellular carcinoma cells. Biomed
Pharmacother. 92:270–276. 2017.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Yang L, Peng F, Qin J, Zhou H and Wang B:
Downregulation of microRNA-196a inhibits human liver cancer cell
proliferation and invasion by targeting FOXO1. Oncol Rep.
38:2148–2154. 2017.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Noh JH, Chang YG, Kim MG, Jung KH, Kim JK,
Bae HJ, Eun JW, Shen Q, Kim SJ, Kwon SH, et al: MiR-145 functions
as a tumor suppressor by directly targeting histone deacetylase 2
in liver cancer. Cancer Lett. 335:455–462. 2013.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Wei T, Weiler SME, Tóth M, Sticht C, Lutz
T, Thomann S, De La Torre C, Straub B, Merker S, Ruppert T, et al:
YAP-dependent induction of UHMK1 supports nuclear enrichment of the
oncogene MYBL2 and proliferation in liver cancer cells. Oncogene.
38:5541–5550. 2019.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Wang L, Zhao Z, Meyer MB, Saha S, Yu M,
Guo A, Wisinski KB, Huang W, Cai W, Pike JW, et al: CARM1
methylates chromatin remodeling factor BAF155 to enhance tumor
progression and metastasis. Cancer Cell. 25:21–36. 2014.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Todisco S, Convertini P, Iacobazzi V and
Infantino V: TCA cycle rewiring as emerging metabolic signature of
hepatocellular carcinoma. Cancers (Basel). 12(68)2019.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Li F, Wang S, Cui X, Jing J, Yu L, Xue B
and Shi H: Adipocyte utx deficiency promotes high-fat diet-induced
metabolic dysfunction in mice. Cells. 11(181)2022.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Daniel JA, Santos MA, Wang Z, Zang C,
Schwab KR, Jankovic M, Filsuf D, Chen HT, Gazumyan A, Yamane A, et
al: PTIP promotes chromatin changes critical for immunoglobulin
class switch recombination. Science. 329:917–923. 2010.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Callen E, Faryabi RB, Luckey M, Hao B,
Daniel JA, Yang W, Sun HW, Dressler G, Peng W, Chi H, et al: The
DNA damage- and transcription-associated protein paxip1 controls
thymocyte development and emigration. Immunity. 37:971–985.
2012.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Miao Y, Li Z, Feng J, Lei X, Shan J, Qian
C and Li J: The role of CD4+T cells in nonalcoholic
steatohepatitis and hepatocellular carcinoma. Int J Mol Sci.
25(6895)2024.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Zheng X, Jin W, Wang S and Ding H:
Progression on the roles and mechanisms of tumor-infiltrating T
lymphocytes in patients with hepatocellular carcinoma. Front
Immunol. 12(729705)2021.PubMed/NCBI View Article : Google Scholar
|