1. Introduction
One of the major elements of the adaptive immune
response is T cells, which lead to the recognition of antigens and
response to foreign invaders, while simultaneously maintaining
self-tolerance. T cell activation requires an intricate signaling
pathway. It starts with the recognition of antigens by T cell
receptors (TCRs) presented through a major histocompatibility
complex (MHC) on the surface of antigen-presenting cells (APC),
such as macrophages and dendritic cells. Self-tolerance is
preserved by co-stimulatory signals, such as CD28. Finally, a third
signal is initiated by cytokines which play a major role in T cell
differentiation. Any disturbance in this pathway can result in
serious sequelae, such as autoimmune diseases, immunodeficiencies
and malignancies. Recent developments in immunology have focused on
T cell pathways for therapeutic purposes, which has potential.
However, mechanisms underlying T cell anergy and linker of
activated T cells (LAT) phase separation, along with the modulation
of T cells and its use in novel therapeutic approaches, remain
incompletely understood. The aim of the present review was to
provide an overview of T cell activation and regulation along with
the main molecular pathways, with the introduction of novel
mechanisms and newer medical interventions, which would help
further fill the current gaps in research.
2. T cell development
Background
T cell development predominantly takes place during
fetal development and early childhood, shaping the adaptive immune
system for lifelong function (1).
Therefore, the congenital absence of the thymus in DiGeorge
syndrome, due to a microdeletion in chromosome 22, leads to
improper development of the third pharyngeal pouch. Patients with
this syndrome present with deficient cellular immunity or
autoimmune diseases, such as idiopathic thrombocytopenia (2). During puberty, gland shrinkage takes
place. T cell maturation and development diminish without affecting
health, even when the gland is surgically removed (1).
Several immune-deficient mouse strains have been
recognized. Nude and severe combined immunodeficiency (SCID) mice
represent an example of complementary T cell-deficient strains.
Nude mice (they are named so because they lack fur) are naturally
mutated and are bereft of thymus glands (3). Consequently, T cell poverty ensues;
nonetheless, they possess B cells. SCID mice have combined immune
deficiency because they lack the recombination activating gene
(RAG) enzyme that is important for somatic recombination (4). This occurs even though these mice have
a well-developed thymus. The lymphoid progenitors normally thrive
when transferred from nude to SCID mice, since they have an
efficient thymic microenvironment. A more recent study has further
advanced our understanding by showing that murine models play an
essential role in dissecting interactions between thymic stromal
cells and developing thymocytes. The aforementioned interactions
are indispensable for the establishment of the immune scenery along
with T cell selection. Therefore, using these models to further
investigate thymic regeneration would be a viable option aiming to
restore T cell function in human patients (5).
Initial stages of T cells
development
Hematopoietic pluripotent stem cells, which reside
in the bone marrow vascular niche, give rise to lymphoid
progenitors and myeloid progenitors. The myeloid progenitors
develop into the cells of an innate immune response. The lymphoid
progenitors give rise to T and B lymphocyte precursors, which are
adaptive immune components. B cell precursors stay in the bone
marrow and pursue their development there. Concomitantly, the T
cell precursors evolve in the thymus, reaching it through the blood
from the bone marrow (6). Since T
cell development takes place in the thymus, it is considered a
primary lymphoid organ mimicking the bone marrow.
The process of T cell migration from bone marrow to
the thymus is influenced by chemotactic agents, including thymosin,
thymopoietin and thymic factors (7). T cell precursors enter the thymus
through the high endothelial venule. Once they are inside, they
migrate to reach the subcapsular area, where they proliferate and
increase in number. As they descend from the cortical region to the
medulla, they undergo several maturation steps to become mature
naïve T lymphocytes. Developing T cells that fail the selection
processes die by apoptosis (programmed cell death). The process of
T cell development lasts ~3 weeks, after which they become mature
naïve T-lymphocytes. The maturation stages of T cells can be
recognized by the markers expressed on their surface.
Initially, T cells are devoid of CD4 and CD8 markers
of their future lineage. Therefore, they are named double negative
cells (DN). The DN stage is further divided into four substages:
DN1, DN2, DN3 and DN4, demarcated by CD44 and CD25 expression. The
former is an adhesion molecule, while the latter is an IL-2
receptor. At DN3, the pre-TCR is formed by the pairing of a
functional rearranged β chain with pre-α chain. The vast majority
of TCRs in postnatal life are composed of α and β subunits, whereas
γ/δ TCR is predominant in fetal T cell linage (8). TCR receptor assembly commences with
V(D)J recombination at the TCR β gene locus (9). Thereupon, the rearranged functional β
chain binds to the pre-α chain, forming pre-TCR. Next, pre-TCR and
CD3 engender vital signals for survival, proliferation and
cessation of β chain rearrangement. In the bargain, these signals
drive DN cells to become double positive (DP).
At the end of this process, mature TCR recognizes
manifold antigens. This diversity of immune repertoire arises from
variable diversity joining V(D)J recombination of the β subunit,
and VJ recombination only of the α subunit (cut and paste
rearrangement) for the TCR genes. V(D)J starts with DNA breakage
engendered by RAG1/RAG2 at recombination signal sequences (10).
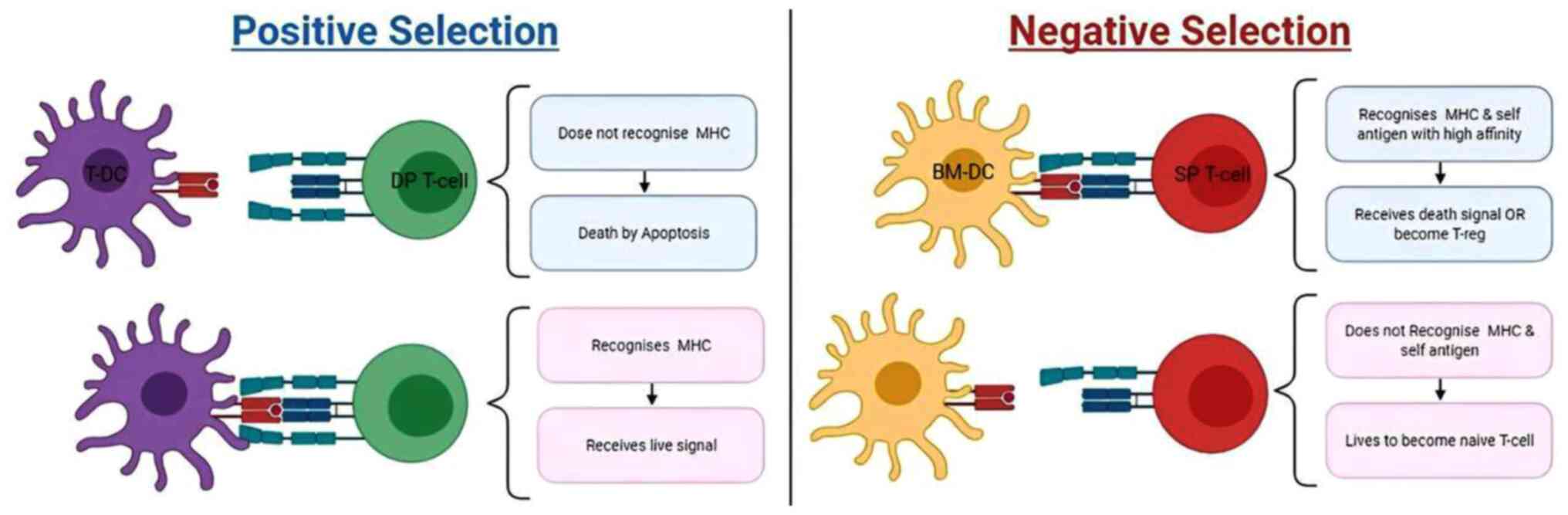
Positive selection processes
Double-positive T cells can interact with epithelial
thymic cells that present MHCs: Class one and two (MHCI and MHCII).
This process ensures MCH restriction. If the DP T cell does not
bind to MHC within 4 days, the fox receptors on their surface will
be activated to initiate apoptosis (11). The second positive selection takes
place in the medulla of the thymus gland following the negative
selection processes. Both T cell groups, that is, T helper (Th) or
T-cytotoxic (Tc), are determined in the second positive selection
process. Cells that interact with MHCI become Tc cells, while those
that recognize MHCII develop into Th cells. Signals that are
received through CD8 shut down CD4 expression; hence, the first
received signal determines the fate of T cells. T cells that bind
to MHCI and MHCII with equal affinity are eliminated. The absence
of MHC expression impedes the development of the corresponding T
cell lineage.
Recent studies highlight that bare lymphocyte
syndrome, a severe immunodeficiency disorder, arises from defective
MHC expression, leading to impaired antigen presentation and T cell
maturation (12). Consequently,
failure of T cell type production emerges. Mice that express
defective CD8 that cannot bind to MHCI do not proceed to produce
mature Tc cells. It has been reported that there is a gene that is
involved in the development of multiple systems. This gene
represents a mammalian equivalent of Notch, which directs the T
cell to become a Tc cell (13).
This gene is also of great importance for directing the lymphoid
progenitor to form T cell precursors. Abnormality in this gene may
cause a shortage of T cytotoxic cells (14).
Negative selection process
The lucky T cells that successfully pass the
positive selection exam undergo a negative selection test before
they finish their thymic training. This process ensures
self-antigen tolerance (15). In
negative selection, self-antigens are presented by APC to T cells.
T cells that strongly bind to self-antigens undergo apoptosis to
maintain self-tolerance; failure in this process contributes to
autoimmunity (16).
This process is referred to as the central tolerance
mechanism. The differential avidity hypothesis states that the MHC
complex delivers both positive and negative selection signals;
nevertheless, the signal avidity for negative selection is higher
(more signal is required to save the cell from apoptosis (11). Next, mature naïve T cells will
finally complete their stringent training and exit the thymus to
pursue their function (Fig. 1).
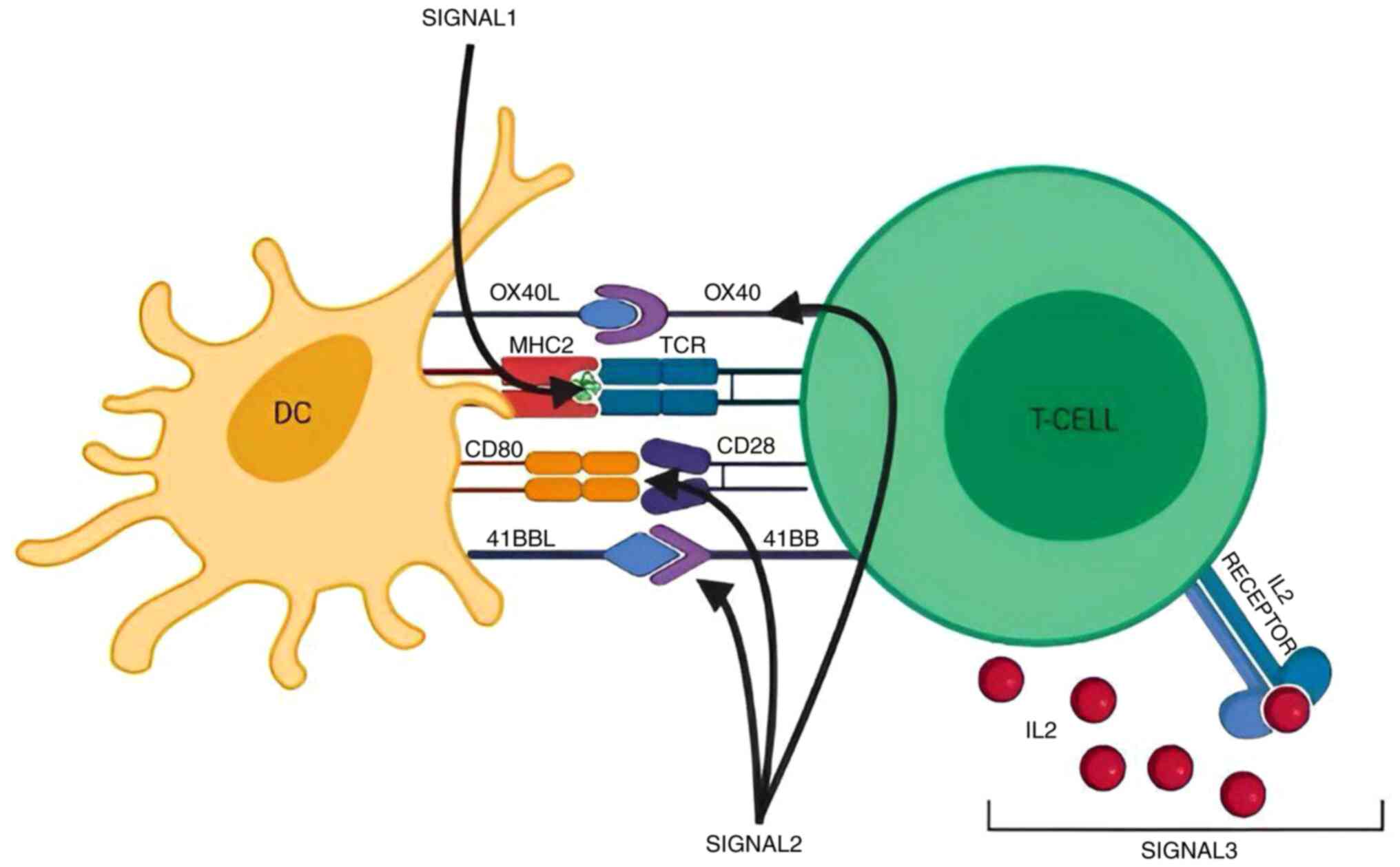
3. T cell activation
Background
It is a process in which a naïve T cell gets
activated to form a mature T cell. The T cell activation process
undergoes rigorous control. Once activated, it proliferates and
secretes cytokines that modulate immune response. Naïve Th cells
engage with antigens bound to MHCII, while cytotoxic T (Tc) cells
recognize MHCI-associated antigens, a critical step in adaptive
immunity. Three signals are required for the activation of T cells.
In signal one, the TCR binds to antigens presented by MHC on APC.
Following the first signal, T cells require co-stimulatory
signaling to avoid anergy (a state of functional unresponsiveness
to antigens, crucial for immune regulation). The final signal is
the production of cytokines that stimulate T cell proliferation
(Fig. 2) (17,18).
Signal one
This signal usually takes place in the secondary
lymphoid organ for Th cells. The TCR binds to peptides presented to
them by MHC molecules on the surface of APCs. The CD4 and CD8
molecules then interact with the beta chain of the MHC molecule,
stabilizing the binding of TCR-MHC and helping in intracellular
signaling (19).
Signal two
The co-stimulatory signal is mediated by an
interaction between CD28 on the surface of Th cells and the B7.1
(CD80) or B7.2 (CD86) proteins on the surface of APCs. This
interaction actuates T cell proliferation. In addition, the
interaction between CD28 and B7 effectuates the production of
CTLA-4, which lessens the signal by competing with CD28 for
B7(20). This represents a negative
feedback mechanism that wanes the immune response. Tc cells demand
signals from co-stimulatory molecules other than CD28, such as CD70
and CD137. Other signals that are expressed on the surface of T
cells only after the recognition of antigens include OX40,
inducible T-cell co-stimulator and 4-1BB (21). These receptors bind to their ligands
on APC, which are also not constitutively present on APCs.
This hinders the T cell activation bound to MHC in
the absence of an antigen, which is the main role of these signals.
The omission of these signals turns off T cell activation, and the
dearth of signal two leads to anergy in naïve T cells (22), which means a reduction of immune
response to a specific pathogen. T cells that develop anergy after
TCR engagement are tolerant to that particular antigen (23).
Signal three
Immediately upon successful accomplishment of signal
one and signal two, T cells encounter more signals to determine
their responder phenotypes; these signals are cytokines. Th cells
under the effect of IL-12 become Th1, under the effect of IL-4
become Th2, and under the effect of IL-23 and IL-16 become Th17
(24,25). Regarding Tc cells, signal three is
mediated by IL-12 or adjuvant. Signals one and two alone can
mediate Tc proliferation, in case of high antigen levels.
On the other hand, a low antigen level mandates the
presence of signal three for cell proliferation to take place.
Nevertheless, T cells that proliferate due to high antigen levels
without the guidance of a third signal, fail to ensure cytolytic
function (26). Hence, the fate of
proliferated Tc cells depends on third signal exposure (25). Tc cells that lack their cytolytic
function are considered tolerant (27). Out-of-order signaling may occur in
certain settings. Usually, the third signal does not take place
before the first two signals. This is largely attributed to low
cytokine receptor expression in naïve T cells juxtaposed with their
antigen-experienced fellow (28,29).
Signal three thwarting alters Th cell proliferation
but does not affect that of Tc cells. When Th cells are exposed to
high levels of cytokines, for example in therapeutic administration
immunotherapy to treat cancer, ephemeral paralysis supervenes
(30). This is largely due to the
activation of the suppressor of cytokine signal 3 (COCS3) pathway.
The COCS3 pathway hinders the piling up of Th cells quantity in
vivo and in vitro, consequently affecting the adaptive
immune response (31). Some studies
showed that naïve Th cells may exhibit a response to cytokines
despite the scarcity of Th proliferation (29,32).
This response is mediated by activating factor upregulation or
signaling molecule phosphorylation. The sequel of this response
remains ambiguous and not fully understood. In recent studies, T
cell modulation has been introduced in autoimmune disease
management after it was wildly accepted in oncological treatments.
A trial was perfomred in the UK including patients with severe
systemic lupus erythematosus (SLE) who received genetically
engineered T cells to express chimeric antigen receptor (CAR)
designed to target CD19 receptor present on autoreactive B cells.
This technique is commonly known as CAR-T cell therapy. The trial
showed promising outcomes in halting the autoantibody release from
autoreactive B cells. However, it poses some risks and drawbacks,
such as recurrent infections, a need for hospitalization and the
risk for the regeneration of autoreactive B cells (33,34).
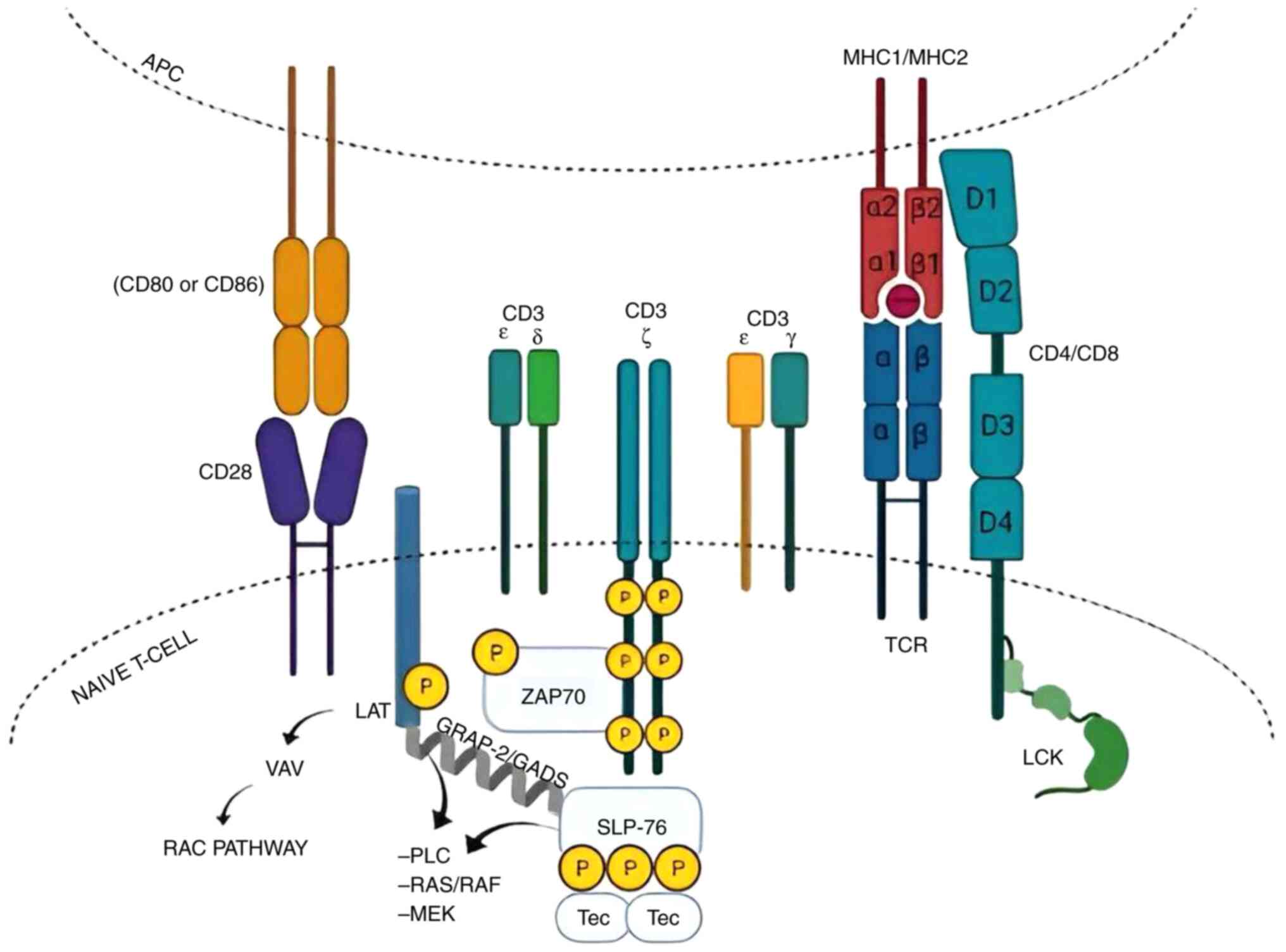
4. T cell signal transduction
T cell proximal signal transduction
pathways. T cell receptor first signal transduction
TCR possesses a dwarf intracellular domain, so the
role of neighboring CD3 subunits is indispensable for signal
transduction (35). When the T
cells encounter their specific antigen presented in APC, the TCR
interacts with the MHC molecule and antigen. This is followed by
the binding of CD8/CD4 co-receptor δ 4 subunits to MHC1/MHCII β
subunits, which prompt the TCR intracellular signaling pathway
(36). Considering that none of the
TCR integrates display autogenous kinase activity, non-receptor
tyrosine kinases PTK are demanded.
The discovery of this PTK has enhanced the
understanding of the TCR singling criterion. When co-receptors
CD4/CD8 interact with MHC molecules, they facilitate the
recruitment of key signaling kinases, Lck and Fyn, essential for
downstream TCR activation (37,38).
Lck and Fyn are members of the Sarcoma family kinases (SFK). SFK
are non-receptor protein tyrosine kinases that are switched on by
binding to various cellular receptors. They are primary kinases
that render other protein tyrosine kinases active following their
activation by receptor binding (39). Lck appears to be the crucial
benefactor of TCR signal transduction (40). Yet, SFK members exhibit functional
redundancy, rendering the assessment of Lck and Fyn's exact degree
of contributions to TCR activation an unfathomable task (41).
Next, Src phosphorylates the immunoreceptor
tyrosine-based activation motif (ITAM) of the CD3ζ chain (42). Then, the phosphorylated ITAM
recruits ζ associated protein (ZAP70) to bind to it. In turn, the
ZAP70 protein becomes phosphorylated, which leads to its activation
(40). The active ZAP70
phosphorylates the LAT (43,44).
Subsequently, LAT protein activates Vav protein, which in turn
signals the activation of the Rho/RAC GTPase pathway. RAC pathway
mediates miscellaneous T cell function; for example, fostering
actin organization (45).
In addition, Vav1 regulates the phosphorylation of
receptors and tyrosine kinases through a negative feedback
mechanism. LAT also musters and activates Src homology 2
(SH2)-domain-containing leukocyte protein of 76kDa (SLP-76)
protein. The Activated SLP-76 derives the allelic exclusion of T
cells (46). Furthermore, active
SLP-76 binds to LAT through growth factor receptor-bound protein
2(47). Finally, interleukin 2
inducible T cell kinase (ITK) binds to SLP-76. The association
between the ITK and SLP-76 mediates several intracellular pathways,
such as phospholipase C, RAS/RAF and MEK (48,49).
All of these pathways conglomerate together and ultimately lead to
the increased transcription of factors such as nuclear factor of
activated T cells (NFAT) and activator protein 1, which promote T
cell functions such as differentiation and cytokine release
(49) (Fig. 3). In addition, recent breakthroughs
have shown that LAT signaling is further regulated by phase
separation. The process starts by the activation and recruitment of
phospholipase Cγ1 (PLCγ1) following TCR engagement with APCs. PLCγ1
then facilitates phase separation of LAT by cross linking its
molecules via SH2 domains, which eventually leads to the formation
of phase-separated condensates. These condensates serve as a
protector against tyrosine phosphatase, which prevents the
dephosphorylation of LAT, which in turn helps sustain and amplify
TCR signal transduction. In addition, the CD3ε subunit of the TCR
forms condensates with Lck to enhance phosphorylation. More
importantly, phosphorylated CD3ε recruits C-terminal Src kinase, a
kinase that inhibits Lck function and dissolves concentrates, which
makes this mechanism a natural on-off switch of the process
(50,51).
Costimulatory signal transduction pathway.
Costimulatory signals ensure the effectiveness of T cell
activation. The central canon of T cell activation states that a
paucity of costimulatory signals causes anergy or apoptosis
(21). Among costimulatory
receptors, CD28 is particularly crucial in enhancing TCR signaling
and promoting robust immune responses, as recent studies
demonstrate (52). As soon as CD28
binds to its ligand on the surface of APC, the intracellular
production of phosphatidylinositol 3,4,5 triphosphate (PIP3)
increases (53).
PIP3 aids the phosphorylation of Akt by PDK1
kinases. Next, Akt promotes the expression of NF-κB. Therefore, it
boosts Bcl-xl survival genes (54).
Activated Akt lessens the activity of glycogen synthase kinase 3 to
boot. GSK ameliorates NFAT seclusion from the nucleus (55). It has been suggested that Akt also
inhibits pivotal scaffolding protein essential for calcineurin
access to the NFAT.
Hence, IL-2 production increases due to delayed
nuclear export and calcineurin hindrance. In general, the CD28
amplifies T cell responses and cytokine production, serving as a
key modulator of immune activation and longevity (56). This is particularly true when it
comes to IL-2, which induces gene expression of antiapoptotic
BCL-xL, decreases the time needed for naïve T-cell stimulation,
increases cell adhesion and braces germinal center formation. Which
explains why CD28 knockout mice showed faulty immune responses
(57,58). The inhibition of the CD28 receptor
may be used to treat autoimmune diseases and prevent allograft
rejection and graft vs. host disease (59,60).
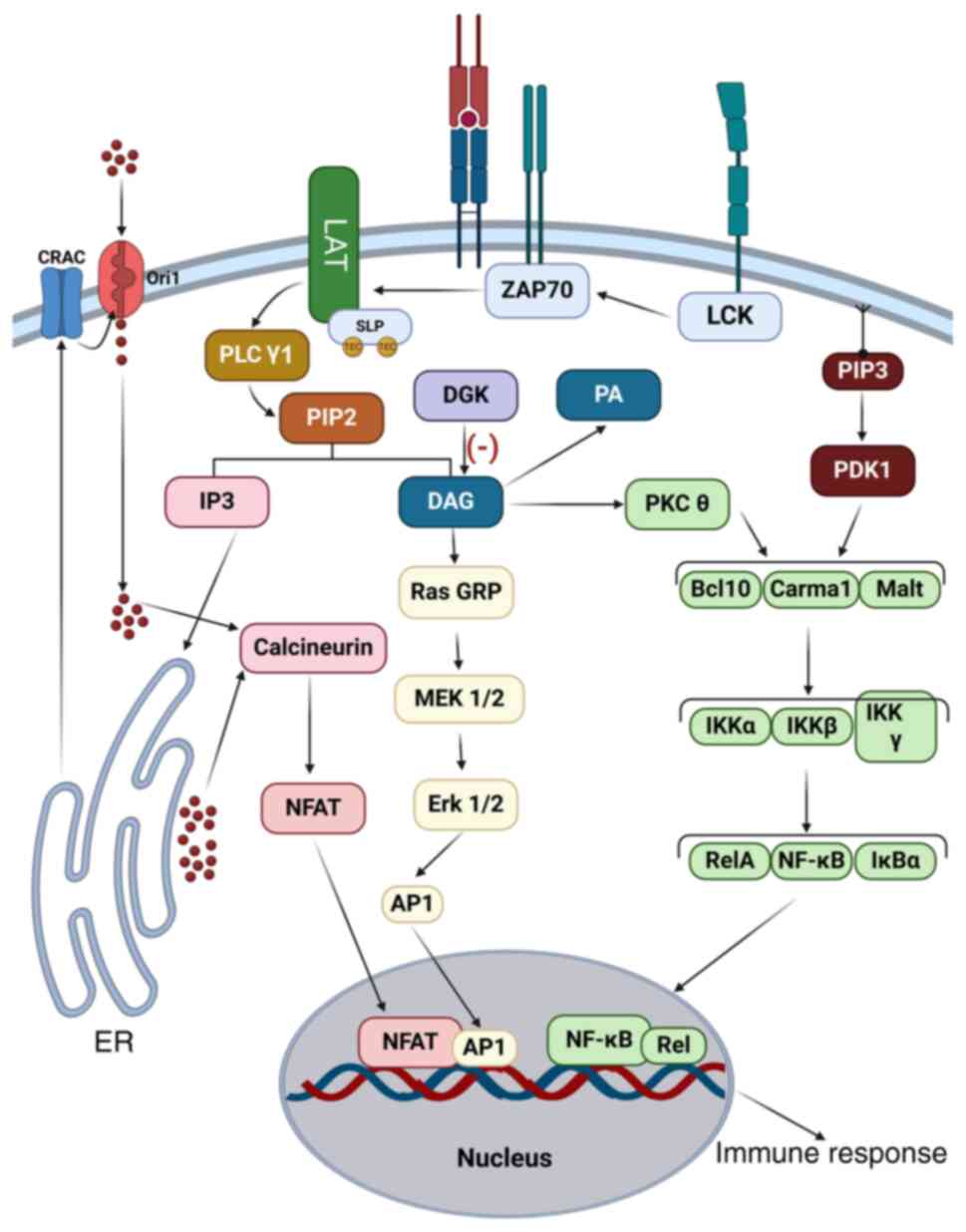
Second messengers inositol
triphosphate (IP3) and diacylglycerol (DAG)
Phospholipase Cγ1 represents a cardinal junction
between proximal and distal transduction. Tec and SLP-76
phosphorylate and consequently activate PLCγ1. Furthermore, Rlk
which is another PTK, contributes to PLCγ1 activation. The complete
absence of Rlk and Tec in mouse models annihilates PLC1γ (61). PLCγ hydrolyzes membrane-bound
phosphatidyl 4,5 bisphosphates (PIP2), generating IP3 and DAG at an
equal ratio (62). IP3 activates
the IP3-calcium-NFAT pathway. DAG collaborates with raised
cytosolic calcium caused by IP3 and directs protein kinase C to the
membrane to be activated (Fig. 4)
(63).
Distal signal transduction.
IP3-calcium-NFAT pathway
The IP3 generated by PLCγ1 advances toward its
receptors in the endoplasmic reticulum. This leads to a transient
upswing of calcium (64).
Intracellular calcium signaling is tightly regulated by ORAI1
channels, facilitating T cell activation and immune response
modulation (65). Next, free
calcium coheres to calcineurin and eventually activates it through
phosphorylation. Thereupon, calcineurin dephosphorylates
cytoplasmic NFAT by serine/threonine phosphatase enzyme (66). Calcineurin can be inhibited by
cyclosporin and FK506 immunosuppressant drugs (67). The NFAT is then imported into the
nucleus. Once inside, NFAT interacts with myriad transcription
factors of the AP-1 (Fos/Jun) family protein, and DNA composite
elements that accommodate their binding sites. Next,
NFAT/Fos-Jun/DNA, highly stable triple amalgamates, regulate the
transcription of variable inducible cytokine genes (68,69).
However, NFAT can solely bind to DNA in absentia of
AP-1, by a poorly understood mechanism. Once inside, NFAT interacts
with myriad transcription factors of the AP-1 (Fos/Jun) protein
family and DNA composite elements that accommodate their binding
sites. Subsequently, NFAT/AP-1/DNA, a highly stable triple
amalgamate, regulates the transcription of variable inducible
cytokine genes. AP-1 is derived from DAG signaling. However, NFAT
can solely bind to DNA in absentia of AP-1, by a poorly understood
mechanism (70).
Imbalances in calcium-calcineurin-NFAT signaling can
lead to T cell anergy, regulated in part by E3 ubiquitin ligase and
its downstream pathways. The dysregulation of NFAT activation and
import or impaired calcium signaling, which are due to ORAI1
mutation causing aberrant calcineurin-NFAT activation, bring about
severe combined immunodeficiency syndrome (71). This can be managed partially by
lithium chloride, as it thwarts the nuclear export of NFAT
(72).
DAG-kinase pathways
Membrane-bound DAG represents the second generated
molecule of PIP2 through PLCγ1 hydrolysis (62). DAG recruits Ras-guanyl releasing
protein 1 (Ras-GRP1) and activates Ras by converting GDP to GTP
(73) Active Ras-GRP1 consequently
activates the RAF1-MEK-ERK pathway, leading to increased cytokine
release and cell proliferation (74).
In addition, Ras-GRP1 supports the development of αβ
and γδ cell linages that secret IL-17 and increase CD8 cell
expansion (75,76). SLE patient and mouse models have
exhibited an abnormal expression of Ras and Ras-GRP1, revealing a
link between this pathway and SLE pathogenesis (77).
The other mission for DAG is accomplished by the
activation of protein kinase (PK)Cθ by attracting it to the immune
synapse and then phosphorylating it (78). Consequently, active PKC mediates the
TCR-dependent activation of NF-κB (79). NF-κB signaling plays a pivotal role
in T cell differentiation, regulating IL-2 production and balancing
Th cell subsets (80). The
impairment of these pathways results in various diseases, such as
lymphoma, autoimmune diseases and flawed T cell activation
(81,82). DAG and its downstream metabolites
are essential for T cell function, with DAG kinase ζ acting as a
key modulator in this signaling cascade. At the same time, DGK
kinase ζ converts DAG to phosphatidic acid in numerous lymphoid
tissues, particularly within T cell compartments (83).
5. Conclusion
T-cell research is rapidly advancing, unveiling
groundbreaking therapeutic applications such as CAR-T cell therapy
for autoimmune diseases and novel cancer immunotherapies. Despite
these strides, several challenges remain. A key limitation of
current knowledge is the incomplete understanding of long-term
T-cell fate, particularly in the context of engineered therapies.
Additionally, the intricate role of T-cell metabolism in immune
regulation is still not fully elucidated, and comprehensive
clinical data on emerging treatments remain limited. Another
pressing area of investigation is the phenomenon of T-cell
exhaustion, where prolonged activation leads to diminished immune
responses. Deciphering the precise mechanisms underlying exhaustion
and fine-tuning signaling pathways are crucial steps toward
optimizing therapeutic strategies.
Moving forward, future research should prioritize
enhancing CAR-T cell persistence to improve long-term efficacy,
unraveling the metabolic influences that govern T-cell function,
and developing next-generation immune checkpoint modulators to
fine-tune immune responses. These efforts will drive the evolution
of personalized immunotherapies, offering more precise and durable
treatment options for immune-mediated diseases and cancer. As our
understanding of T-cell regulation continues to expand, new
opportunities will emerge to harness the immune system more
effectively, striking a delicate balance between robust defense and
immune tolerance. Ultimately, these advancements will shape the
future of immunotherapy, transforming patient outcomes and
redefining modern medicine.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
All authors contributed significantly to the
development of the present review article. IO contributed to the
conceptualization, writing the original draft and critical
revisions. AA contributed to literature review, data collection and
initial manuscript drafting. SM contributed to the manuscript
editing and quality assurance. MB contributed to the supervision,
manuscript review and approval of the final version. All authors
read and approved the final version of the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Danzaki K, Matsui Y and Ikawa T:
‘Molecular mechanisms of T cell development and differentiation.’.
Front Immunol. 10(3184)2019.
|
|
2
|
Wilson D, Burn J, Scambler P and Goodship
J: DiGeorge syndrome: Part of CATCH 22. J Med Genet. 30:852–856.
1993.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bosma GC, Custer RP and Bosma MJ: A severe
combined immunodeficiency mutation in the mouse. Nature.
301:527–530. 1983.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Mombaerts P, Iacomini J, Johnson RS,
Herrup K, Tonegawa S and Papaioannou VE: RAG-1-deficient mice have
no mature B and T lymphocytes. Cell. 68:869–877. 1992.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ding Y, Li X and Zhang Y: Thymic stromal
interactions in T cell development and immunity: Insights from
animal models. Immunol Rev. 310:45–56. 2023.
|
|
6
|
Takahama Y: Journey through the thymus:
Stromal guides for T cell development and selection. Nat Rev
Immunol. 6:127–135. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
Hadden JW: Thymic endocrinology. Int J
Immunopharmacol. 14:345–352. 1992.PubMed/NCBI View Article : Google Scholar
|
|
8
|
McVay LD, Jaswal SS, Kennedy C, Hayday A
and Carding SR: The generation of human γδ T cell repertoires
during fetal development. J Immunol. 160:5851–5860. 1998.PubMed/NCBI
|
|
9
|
Schlissel MS: Regulating antigen-receptor
gene assembly. Nat Rev Immunol. 3:890–899. 2003.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Rothenberg EV and Taghon T: Molecular
genetics of T cell development. Annu Rev Immunol. 23:601–649.
2005.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Surh CD and Sprent J: T cell apoptosis
detected in situ during positive and negative selection in the
thymus. Nature. 372:100–103. 1994.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Patel DR, Akhter A and Channabasappa N:
‘Bare lymphocyte syndrome: Clinical presentation and advances in
molecular diagnostics.’. Curr Allergy Asthma Rep. 20(21)2020.
|
|
13
|
Robey E, Chang D, Itano A, Cado D,
Alexander H, Lans D, Weinmaster G and Salmon P: An activated form
of Notch influences the choice between CD4 and CD8 T cell lineages.
Cell. 87:483–492. 1996.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yasutomo K: The cellular and molecular
mechanism of CD4/CD8 lineage commitment. J Med Invest. 49:1–6.
2002.PubMed/NCBI
|
|
15
|
Klein L, Kyewski B, Allen PM and Hogquist
KA: Positive and negative selection of the T cell repertoire: what
thymocytes see (and don't see). Nat Rev Immunol. 14:377–391.
2014.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Leung MW, Shen S and Lafaille JJ: ‘T cell
tolerance and autoimmunity: Emerging concepts.’. Ann Rev Immunol.
40:117–142. 2022.
|
|
17
|
Smith-Garvin JE, Koretzky GA and Jordan
MS: T cell activation. Annu Rev Immunol. 27:591–619.
2009.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kumar BV, Connors TJ and Farber DL: Human
T cell development, localization, and function throughout life.
Immunity. 48202-213:2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Lanzavecchia A and Sallusto F: Dynamics of
T lymphocyte responses: Intermediates, effectors, and memory cells.
Science. 290:92–97. 2000.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Coyle AJ and Gutierrez-Ramos JC: The
expanding B7 superfamily: Increasing complexity in costimulatory
signals regulating T cell function. Nat Immunol. 2:203–209.
2001.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Croft M: Costimulation of T cells by OX40,
4-1BB, and CD27. Cytokine Growth Factor Rev. 14:265–273.
2003.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Guedan S, Posey AD Jr, Shaw C, Wing A, Da
T, Patel PR, McGettigan SE, Casado-Medrano V, Kawalekar OU,
Uribe-Herranz M, et al: Enhancing CAR T cell persistence through
ICOS and 4-1BB costimulation. JCI insight. 3(e96976)2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Martinez-Llordella M, Esensten JH and
Turka LA: ‘Mechanisms of T cell anergy and implications for
immunotherapy.’. Nat Rev Immunol. 19:531–548. 2019.
|
|
24
|
Cui W and Kaech SM: Generation of effector
CD8+ T cells and their conversion into memory T cells. Immunity.
236:151–66. 2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Geginat J, Sallusto F and Lanzavecchia A:
Cytokine-driven proliferation and differentiation of human naive,
central memory and effector memory CD4+ T cells. Pathol Biol
(Paris). 51:64–66. 2003.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Mescher MF, Curtsinger JM, Agarwal P,
Casey KA, Gerner M, Hammerbeck CD, Popescu F and Xiao Z: Signals
required for programming effector and memory development by CD8+ T
cells. Immunol Rev. 211:81–92. 2006.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Curtsinger JM, Lins DC and Mescher MF:
Signal 3 determines tolerance versus full activation of naive CD8 T
cells: Dissociating proliferation and development of effector
function. J Exp Med. 197:1141–1151. 2003.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kirberg J, Berns A and von Boehmer H:
Peripheral T cell survival requires continual ligation of the T
cell receptor to major histocompatibility complex-encoded
molecules. J Exp Med. 186:1269–1275. 1997.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Brocker T: Survival of mature CD4 T
lymphocytes is dependent on major histocompatibility complex class
II-expressing dendritic cells. J Exp Med. 186:1223–1232.
1997.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Sckisel GD, Bouchlaka MN, Monjazeb AM,
Crittenden M, Curti BD, Wilkins DE, Alderson KA, Sungur CM, Ames E,
Mirsoian A, et al: Out-of-sequence signal 3 paralyzes primary CD4+
T cell-dependent immunity. Immunity. 43:240–250. 2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Tamiya T, Kashiwagi I, Takahashi R,
Yasukawa H and Yoshimura A: Suppressors of cytokine signaling
(SOCS) proteins and JAK/STAT pathways: Regulation of T cell
inflammation by SOCS1 and SOCS3. Arterioscler Thromb Vasc Biol.
31:980–985. 2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Takeda S, Rodewald HR, Arakawa H,
Bluethmann H and Shimizu T: MHC class II molecules are not required
for survival of newly generated CD4+ T cells, but affect their
long-term life span. Immunity. 5:217–228. 1996.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Mackensen A, Müller L and Mougiakakos D:
CAR T cell therapy finds its way into autoimmunity. Nat Rev
Immunol. 22:71–72. 2022.
|
|
34
|
Schwab N, Schneider-Hohendorf T and Wiendl
H: Therapeutic uses of T cell modulation in autoimmunity and
beyond. J Clin Investig. 133(e164529)2023.
|
|
35
|
Irving BA and Weiss A: The cytoplasmic
domain of the T cell receptor zeta chain is sufficient to couple to
receptor-associated signal transduction pathways. Cell. 64:891–901.
1991.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kane LP, Lin J and Weiss A: Signal
transduction by the TCR for antigen. Curr Opin Immunol. 12:242–249.
2000.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lo WL, Shah NH, Ahsan N, Horkova V,
Stepanek O, Salomon AR, Kuriyan J and Weiss A: Lck promotes
Zap70-dependent LAT phosphorylation by bridging Zap70 to LAT. Nat
Immunol. 19:733–741. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Tan YX, Manz BN and Freedman TS:
‘Regulation of T cell receptor signaling by Lck and Fyn.’. Nat
Commun. 12(5032)2021.
|
|
39
|
Samelson LE, Patel MD, Weissman AM,
Harford JB and Klausner RD: Antigen activation of murine T cells
induces tyrosine phosphorylation of a polypeptide associated with
the T cell antigen receptor. Cell. 46:1083–1090. 1986.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Malissen B, Grégoire C, Malissen M and
Roncagalli R: Integrative biology of T cell activation. Nat
Immunol. 15:790–797. 2014.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Parsons SJ and Parsons JT: Src family
kinases, key regulators of signal transduction. Oncogene.
23:7906–7909. 2004.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Guy CS and Vignali DA: Organization of
proximal signal initiation at the TCR: CD3 complex. Immunol Rev.
232:7–21. 2009.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Finco TS, Kadlecek T, Zhang W, Samelson LE
and Weiss A: LAT is required for TCR-mediated activation of
PLCgamma1 and the Ras pathway. Immunity. 9:617–626. 1998.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zhang W, Sloan-Lancaster J, Kitchen J,
Trible RP and Samelson LE: LAT: The ZAP-70 tyrosine kinase
substrate that links T cell receptor to cellular activation. Cell.
92:83–92. 1998.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Dombroski D, Houghtling RA, Labno CM,
Precht P, Takesono A, Caplen NJ, Billadeau DD, Wange RL, Burkhardt
JK and Schwartzberg PL: Kinase-independent functions for Itk in
TCR-induced regulation of Vav and the actin cytoskeleton. J
Immunol. 174:1385–1392. 2005.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Yokosuka T, Sakata-Sogawa K, Kobayashi W,
Hiroshima M, Hashimoto-Tane A, Tokunaga M, Dustin ML and Saito T:
Newly generated T cell receptor microclusters initiate and sustain
T cell activation by recruitment of Zap70 and SLP-76. Nat Immunol.
6:1253–1262. 2005.PubMed/NCBI View
Article : Google Scholar
|
|
47
|
Yablonski D, Kuhne MR, Kadlecek T and
Weiss A: Uncoupling of nonreceptor tyrosine kinases from PLC-gamma1
in an SLP-76-deficient T cell. Science. 281:413–416.
1998.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Mak TW and Saunders ME: The immune
response. Part I: Basic Immunology 373-401, 2006.
|
|
49
|
Chandler NJ, Call MJ and Call ME: T cell
activation machinery: Form and function in natural and engineered
immune receptors. Int J Mol Sci. 21(7424)2020.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Su X, Wang X, Wang Y, Zhang X, Qi J and
Ding Y: Phase separation of CD3ε-Lck condensates amplifies TCR
signaling but is dynamically terminated by Csk recruitment. Nat
Commun. 14(4723)2023.
|
|
51
|
Zeng C, Hou X, Yan X, Li M, Xu X and Jiang
S: Phospholipase Cγ1 promotes phase separation of LAT to regulate T
cell receptor signaling. Nat Commun. 12(3211)2021.
|
|
52
|
Sharpe AH and Freeman GJ: ‘The B7-CD28
superfamily: Emerging therapeutic targets.’. Nat Rev Immunol.
20:289–306. 2020.
|
|
53
|
Garçon F, Patton DT, Emery JL, Hirsch E,
Rottapel R, Sasaki T and Okkenhaug K: CD28 provides T cell
costimulation and enhances PI3K activity at the immune synapse
independently of its capacity to interact with the p85/p110
heterodimer. Blood. 111:1464–1471. 2008.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Boise LH, Minn AJ, Noel PJ, June CH,
Accavitti MA, Lindsten T and Thompson CB: CD28 costimulation can
promote T cell survival by enhancing the expression of Bcl-XL.
Immunity. 3:87–98. 1995.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Kamphorst AO, Wieland A, Nasti T and Yang
S: ‘Immunoregulation by CD28 and its role in immune checkpoint
therapy.’. J Immunother Cancer. 8(e001021)2020.
|
|
56
|
Bjørgo E and Taskén K: Novel mechanism of
signaling by CD28. Immunol Lett. 129:1–6. 2010.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Green JM, Noel PJ, Sperling AI, Walunas
TL, Gray GS, Bluestone JA and Thompson CB: Absence of B7-dependent
responses in CD28-deficient mice. Immunity. 1:501–508.
1994.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Ferguson SE, Han S, Kelsoe G and Thompson
CB: CD28 is required for germinal center formation. J Immunol.
156:4576–4581. 1996.PubMed/NCBI
|
|
59
|
Ogawa S, Nitta K, Hara Y, Horita S, Nihei
H and Abe R: CD28 knockout mice as a useful clue to examine the
pathogenesis of chronic graft-versus-host reaction. Kidney Int.
58:2215–2220. 2000.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Beyersdorf N, Ding X, Blank G, Dennehy KM,
Kerkau T and Hünig T: Protection from graft-versus-host disease
with a novel B7 binding site-specific mouse anti-mouse CD28
monoclonal antibody. Blood. 112:4328–4336. 2008.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Lucas JA, Miller AT, Atherly LO and Berg
LJ: The role of Tec family kinases in T cell development and
function. Immunol Rev. 191:119–138. 2003.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Zhong XP, Guo R, Zhou H, Liu C and Wan CK:
Diacylglycerol kinases in immune cell function and self-tolerance.
Immunol Rev. 224:249–264. 2008.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Gupta S: Mechanisms of transmembrane
signaling in human T cell activation. Mol Cell Biochem. 91:45–50.
1989.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Oh-hora M and Rao A: Calcium signaling in
lymphocytes. Curr Opin Immunol. 20:250–258. 2008.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Prakriya M, Feske S, Gwack Y, Srikanth S,
Rao A and Hogan PG: Orai1 is an essential pore subunit of the CRAC
channel. Nature. 443:230–233. 2006.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Nagaleekar VK, Diehl SA, Juncadella I,
Charland C, Muthusamy N, Eaton S, Haynes L, Garrett-Sinha LA,
Anguita J and Rincón M: IP3 receptor-mediated Ca2+ release in naive
CD4 T cells dictates their cytokine program. J Immunol.
181:8315–8322. 2008.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Liu J, Farmer JD Jr, Lane WS, Friedman J,
Weissman I and Schreiber SL: Calcineurin is a common target of
cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell.
66:807–815. 1991.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Macián F, López-Rodríguez C and Rao A:
Partners in transcription: NFAT and AP-1. Oncogene. 20:2476–2489.
2001.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Rooney JW, Sun YL, Glimcher LH and Hoey T:
Novel NFAT sites that mediate activation of the interleukin-2
promoter in response to T cell receptor stimulation. Mol Cell Biol.
15:6299–6310. 1995.PubMed/NCBI View Article : Google Scholar
|
|
70
|
López-Rodríguez C, Aramburu J, Rakeman AS
and Rao A: NFAT5, a constitutively nuclear NFAT protein that does
not cooperate with Fos and Jun. Proc Natl Acad Sci USA.
96:7214–7219. 1999.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Hogan PG, Lewis RS and Rao A: ‘Molecular
basis of calcium signaling in T cells.’. Nature Reviews Immunology.
19:532–548. 2019.
|
|
72
|
Feske S, Draeger R, Peter HH and Rao A:
Impaired NFAT regulation and its role in a severe combined
immunodeficiency. Immunobiology. 202:134–150. 2000.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Ebinu JO, Stang SL, Teixeira C, Bottorff
DA, Hooton J, Blumberg PM, Barry M, Bleakley RC, Ostergaard HL and
Stone JC: RasGRP links T cell receptor signaling to Ras. Blood.
95:3199–3203. 2000.PubMed/NCBI
|
|
74
|
Zhong XP, Hainey EA, Olenchock BA, Zhao H,
Topham MK and Koretzky GA: Regulation of T cell receptor-induced
activation of the Ras-ERK pathway by diacylglycerol kinase zata. J
Biol Chem. 277:31089–31098. 2002.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Shin J, O'Brien TF, Grayson JM and Zhong
XP: Differential regulation of primary and memory CD8 T cell immune
responses by diacylglycerol kinases. J Immunol. 188:2111–2117.
2012.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Chen Y, Ci X, Gorentla B, Sullivan SA,
Stone JC, Zhang W, Pereira P, Lu J and Zhong XP: Differential
requirement of RasGRP1 for γδ T cell development and activation. J
Immunol. 189:61–71. 2012.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Mor A, Philips MR and Pillinger MH: The
role of Ras signaling in lupus T lymphocytes: Biology and
pathogenesis. Clin Immunol. 125:215–223. 2007.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Szamel M and Resch K: T-cell antigen
receptor-induced signal-transduction pathways: Activation and
function of protein kinases C in T lymphocytes. Eur J Biochem.
228:1–15. 1995.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Hayden MS and Ghosh S: . NF-kappaB in
immunobiology. Cell Res. 21:223–244. 2011.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Dower NA, Stang SL, Bottorff DA, Ebinu JO,
Dickie P, Ostergaard HL and Stone JC: RasGRP is essential for mouse
thymocyte differentiation and TCR signaling. Nat Immunol.
1:317–321. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
81
|
Krappmann D, Emmerich F, Kordes U,
Scharschmidt E, Dörken B and Scheidereit C: Molecular mechanisms of
constitutive NF-kappaB/Rel activation in Hodgkin/Reed-Sternberg
cells. Oncogene. 18:943–953. 1999.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Staudt LM: Oncogenic activation of
NF-kappaB. Cold Spring Harb Perspect Biol.
2(a000109)2010.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Merida I, Andrada E, Gharbi SI and
Avila-Flores A: ‘DAG signaling in T cells: From membrane biophysics
to immune function.’. Front Immunol. 11(3122)2020.
|