Introduction
Glioblastomas are among the most chemo-resistant
types of human cancers to treat clinically. The refractiveness of
glioblastomas to chemotherapy treatment regimens may be attributed
in part to the activation of a number of cytoprotective mechanisms
in response to chemotherapeutic agents (1,2). One
such cytoprotective mechanism that has emerged and received
considerable attention as a contributing factor to the therapeutic
resistance of a number of types of human cancer in general, and in
glioblastomas in particular, is the unfolded protein endoplasmic
reticulum stress response (3,4). The
unfolded protein endoplasmic reticulum stress response
fundamentally functions as an adaptive cellular program essential
for normal cellular function and survival, that is triggered by the
accumulation of unfolded proteins in the endoplasmic reticulum due
to a number of stress inducers, such as hypoxia, oxidative injury,
glucose deprivation and aberrant calcium levels. However, if the
stress applied to the endoplasmic reticulum is excessive and
exceeds its ability to maintain homeostatic control, apoptotic cell
death ensues (3,4). Therefore, assessing the efficacy of
agents that invoke endoplasmic reticulum stress for their utility
as potential chemotherapeutic drugs that promote glioblastoma cell
death should be given practical consideration. In support of this,
a number of investigational studies have demonstrated the
anti-tumorigenic effects of novel endoplasmic reticulum stress
inducers in leukemia (5), stomach
(6) and prostate cancer (7). Furthermore, recent studies have
established that glucose regulated protein 78 (GRP78), an
endoplasmic reticulum chaperone with anti-apoptotic properties and
a significant role in the unfolded protein endoplasmic reticulum
stress response signaling pathway, is overexpressed in malignant
gliomas (8–10).
To this end, in the present study, we examined two
endoplasmic reticulum stress inducers, thapsigargin and
tunicamycin, for their anti-tumor properties on glioblastomas.
Thapsigargin, an active component found in root extracts of the
umbelliferous plant, Thapsia garganica, acts as a stress
inducer by increasing the intracellular calcium concentration via
the inhibition of calcium uptake into the endoplasmic reticulum by
blocking its ATP-dependent calcium pump (11), while tunicamycin, a nucleoside
antibiotic produced by several Streptomyces species, imposes
cellular stress by inhibiting protein N-linked glycosylation, the
first step in protein glycosylation (12). This study reveals that the
endoplasmic reticulum stress inducers, thapsigargin and
tunicamycin, promote glioblastoma cell death as a consequence of
inducing the pro-apoptotic proteins, C/EBP homologous protein
(CHOP) and caspase 3.
Materials and methods
Cells, conditions and reagents
U373 and A172 glioblastoma cells were purchased from
the American Type Culture Collection (Manassas, VA, USA). All cell
lines were maintained in Dulbecco's modified Eagle's medium (DMEM)
(Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum
(Invitrogen), 2 mM L-glutamine (Invitrogen), 100 nM MEM
non-essential amino acids (Invitrogen) and penicillin-streptomycin
(Invitrogen) at 37°C and 5% CO2. Thapsigargin and
tunicamycin were purchased from Tocris (Ellisville, MO, USA).
Crystal violet cell proliferation
assay
Dose response
Cells were plated in 12-well plates, treated with 1,
5 and 10 μM thapsigargin or tunicamycin and allowed to incubate for
48 h [vehicle controls were treated with dimethyl sulfoxide
(DMSO)]. The tissue culture medium was then removed, and the cell
monolayer was fixed with 100% methanol for 5 min and stained with
0.5% crystal violet in 25% methanol for 10 min. The cells were then
washed three times for 5 min each with distilled water to remove
excess dye and allowed to dry overnight at room temperature. The
incorporated dye was then solubilized in 0.1 M sodium citrate
(Sigma-Aldrich, St. Louis, MO, USA) in 50% ethanol. Subsequently,
100 μl of treated and control samples were transferred to 96-well
plates and optical densities were read at 540 nm using an X-mark
microplate absorbance spectrophotometer (BioRad, Hercules, CA,
USA).
Time course analysis. Cells were plated in
96-well plates for 24 h, treated with 1 μM thapsigargin or
tunicamycin and allowed to incubate for 1, 3 and 6 days at 37°C. At
the end of each time-point the cells were stained with crystal
violet, solubilized with sodium citrate, and the optical densities
were read as described above.
Clonogenic survival
Cells were plated for 24 h, treated with 1 μM and
250 nM thapsigargin, tunicamycin or DMSO (vehicle) and allowed to
incubate at 37°C for 10–14 days. At the termination of the
incubation period, cells were fixed with absolute methanol, stained
with 1% crystal violet for 10 min, rinsed in tap water and allowed
to dry. Colonies, consisting of ≥50 cells, were then counted to
determine the surviving fraction.
Cell motility
Motility assays were conducted according to the
manufacturer's instructions (Cell Biolabs Inc., San Diego, CA,
USA). A cell suspension containing 0.5–1.0×106 cells/ ml
was prepared in serum-free medium with the vehicle (DMSO), 1 μM
thapsigargin or 1 μM tunicamycin, while 500 μl of medium containing
10% fetal bovine serum were added to the lower chamber of the
migration plate. A total of 300 μl of cell suspension containing
vehicle, 1 μM thapsigargin or tunicamycin was then added to the
inside of each insert and allowed to incubate for 24 h at 37°C and
5% CO2. Subsequently, non-migratory cells were removed
from plate inserts (according to the manufacturer's instructions)
and migratory cells were counterstained with cell staining solution
(Cell Biolabs Inc.).
Western blotting
Cells were plated in serum-free DMEM for 24 h,
treated with 1 μM thapsigargin, 1 μM tunicamycin or the vehicle,
and allowed to incubate for 24 and 48 h. The cells were then lysed
in lysis buffer (pH 6.8) containing 60 mM Tris and 2% SDS. Protein
concentrations were determined using the Bradford method.
Subsequently, protein samples were electrophoresed in a 4–12%
Tris-HCl polyacrylamide gel, transferred to nitrocellulose
membranes and immunoblotted with antibodies against CHOP (Cell
Signaling, Danvers, MA, USA). Protein levels were detected using a
horseradish peroxidase conjugated secondary antibody and the
chemiluminescence detection system (Pierce, Rockford, IL, USA).
Detection of caspase activity
Cells were plated in serum-free DMEM for 24 h,
treated with 1 μM thapsigargin, 1 μM tunicamycin or the vehicle and
allowed to incubate for 48 h. Cells were lysed in lysis buffer (pH
6.8) containing 60 mM Tris and 2% SDS, and protein concentrations
were determined using the Bradford method. Subsequently, caspase 3
activity assays were conducted according to manufacturer's
instructions using 30 μg of protein (Promega, Madison, WI,
USA).
Results
Endoplasmic reticulum stress inducers
impede glioblastoma cell production
Eliciting a hyper-stress response in the endoplasmic
reticulum as a means of promoting anti-tumor cell behavior due to
the accumulation of unfolded proteins or an unstable physiological
cellular environment, such as increased intracellular calcium
concentration, has been demonstrated in a variety of types of human
cancer (13–17). In this study, we performed a
comparative assessment of two endoplasmic reticulum stress
inducers, thapsigargin and tunicamycin, with markedly different
modes of stressing the endoplasmic reticulum for their anti-tumor
properties on glioblastomas. Thapsigargin and tunicamycin were
first examined for their effects on glioblastoma cells in
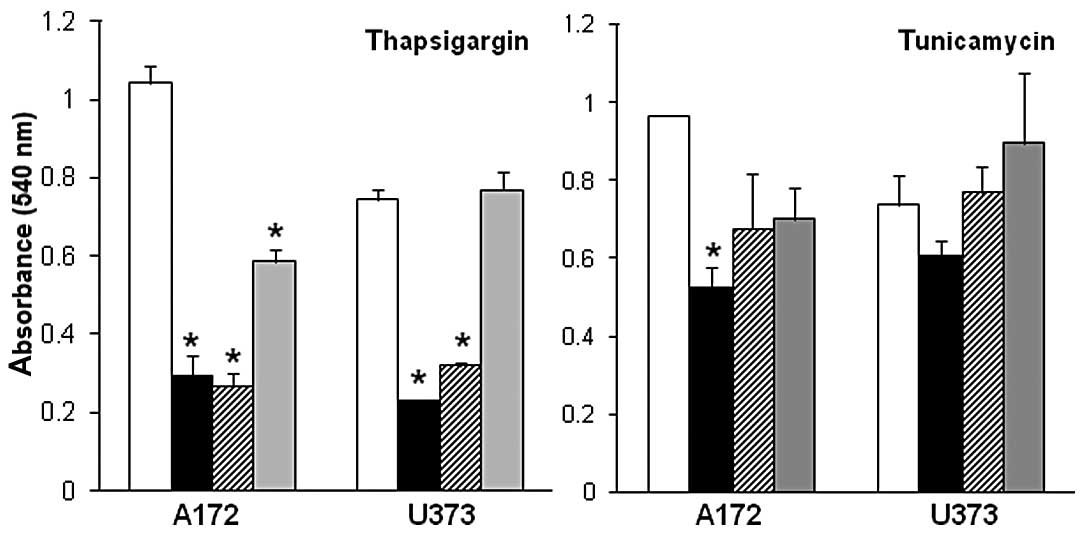
dose-response experiments. These experiments revealed a
dose-dependent decrease in glioblastoma cell proliferation in A172
and U373 cells exposed to increasing concentrations (1–10 μM) of
thapsigargin or tunicamycin, as compared to the vehicle-treated
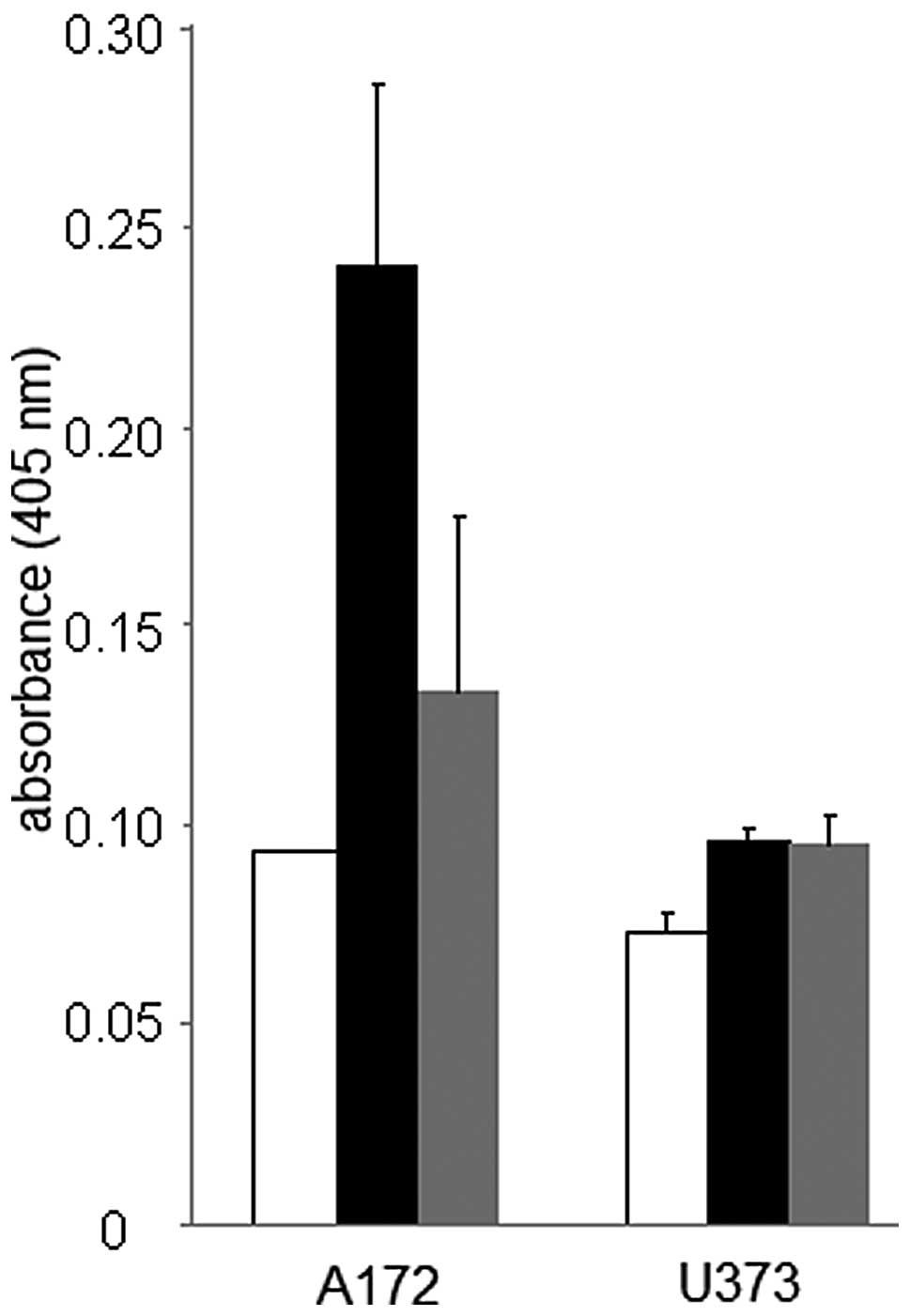
control cells (Fig. 1). Dose
response data also revealed that the inhibitory effects on A172 and
U373 glioblastoma cell proliferation were more pronounced in the
cells treated with 5–10 μM thapsigargin as compared to the cells
treated with 5–10 μM tunicamycin. Additionally, A172 cells appeared
to be more sensitive to endoplasmic reticulum stress as indicated
by a 44±6 and 27±11% reduction in cell proliferation (compared to
controls) when treated with 1 μM thapsigargin or tunicamycin,
respectively, in comparison to the U373 cells treated with either
endoplasmic reticulum stress inducer at the same concentration.
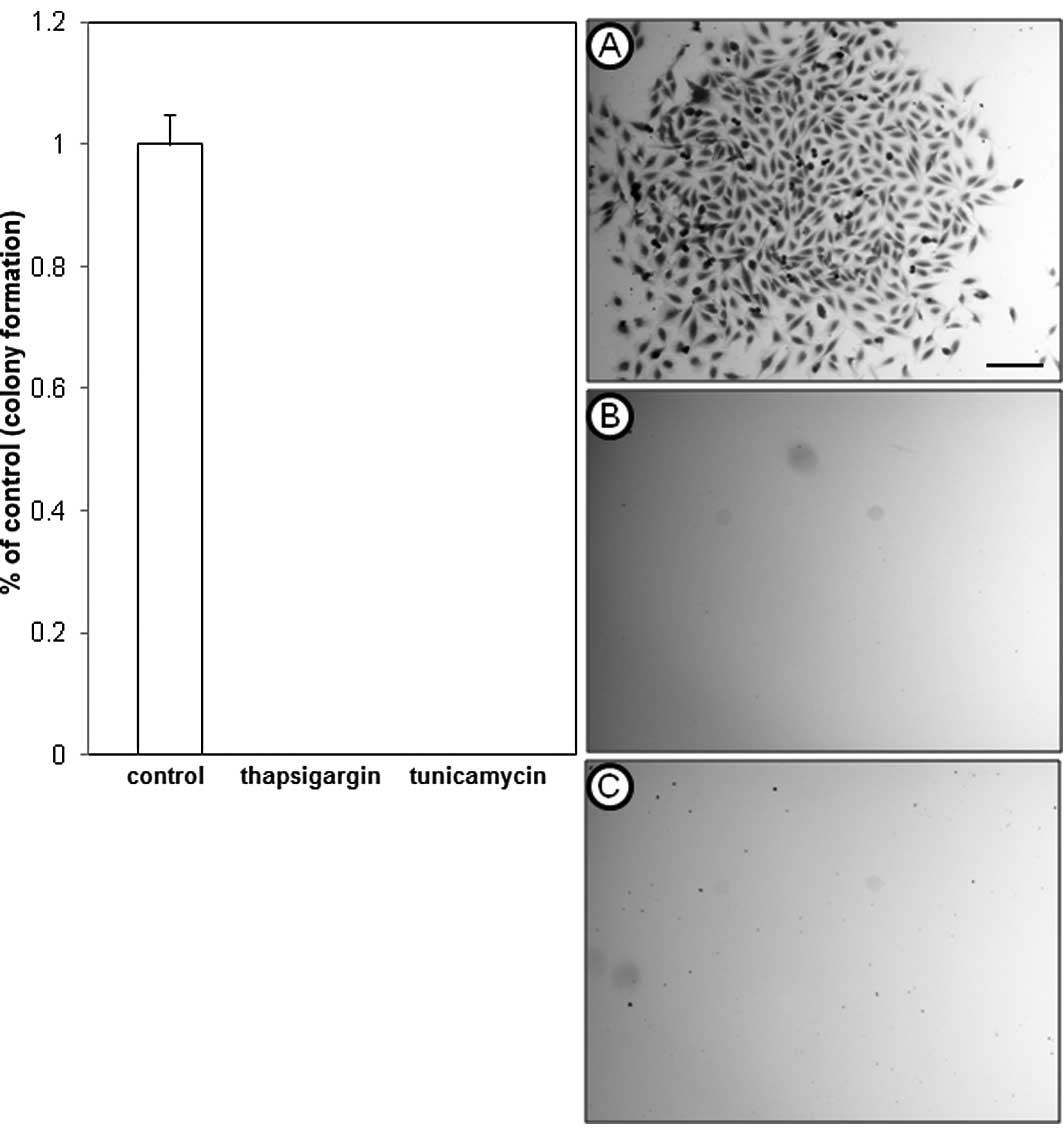
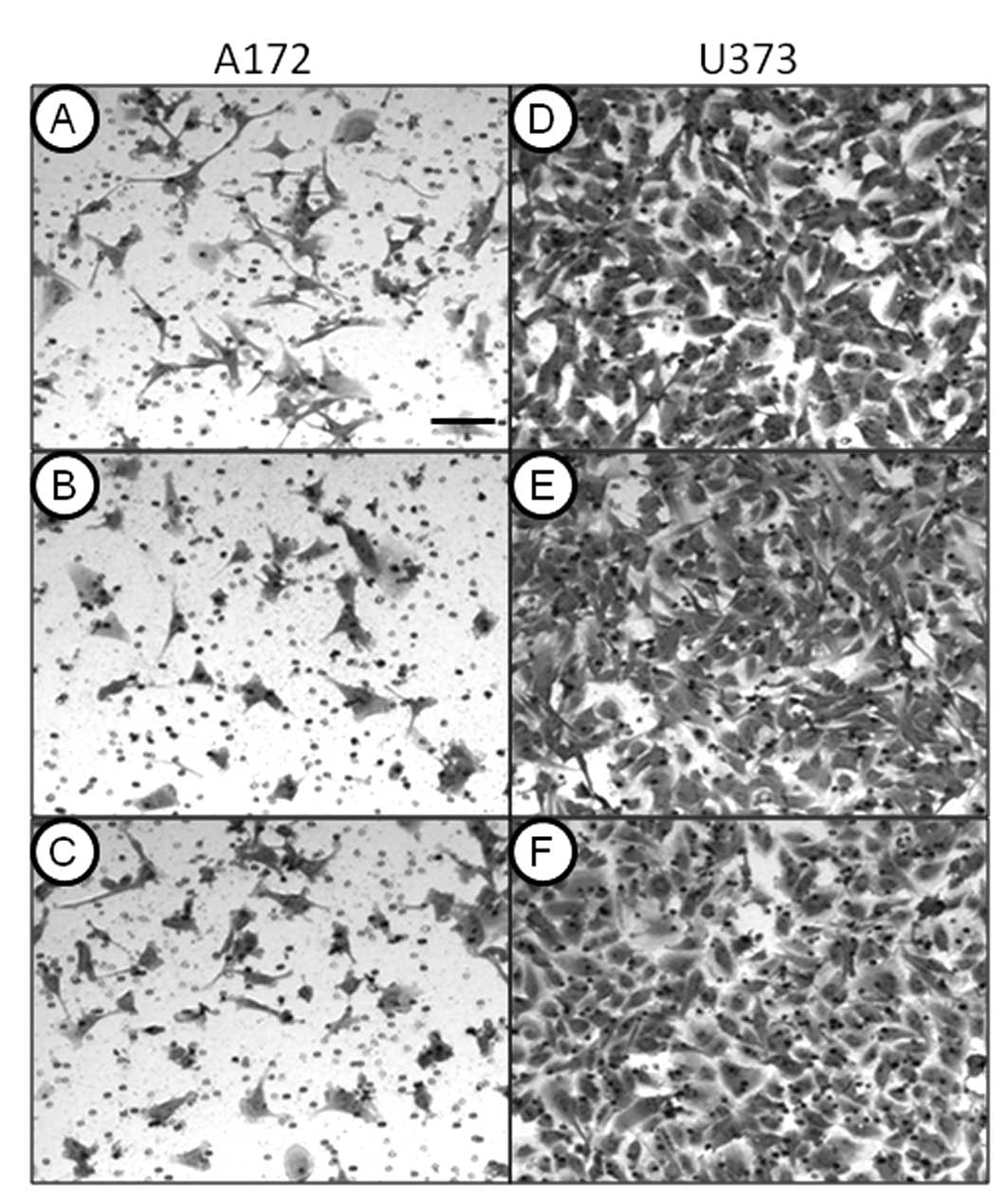
To further examine the inhibitory effects of
thapsigargin and tunicamycin on glioblastoma cell production,
clonogenic survival assays that measure cellular reproductive
capacity at low cell plating densities were performed by treating
A172 and U373 cells with 1 μM thapsigargin or tunicamycin. This
concentration was selected for its non-lethal effects on U373
cells, and showed an inhibitory concentration greater than 50
(>IC50) in A172 cells, as shown in the dose-response
experiments. Clonogenic survival experiments revealed that in
comparison to the vehicle-treated control cells, thapsigargin or
tunicamycin completely abrogated colony formation of A172 (Fig. 2) and U373 (Fig. 3) cells.
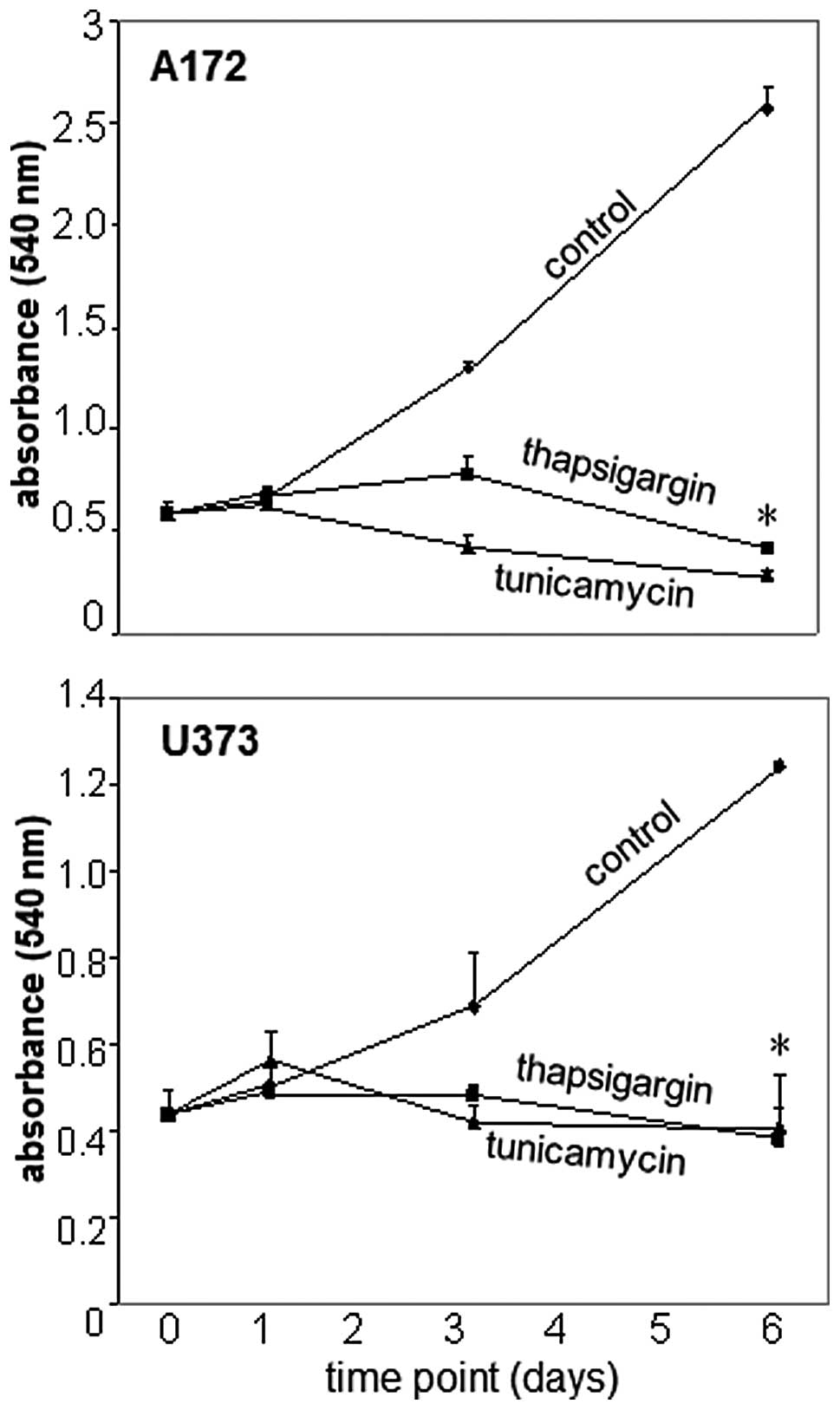
In addition to clonogenic survival experiments,
time-course experiments were performed as a means to determine the
cellular behavior underlying the inhibition of glioblastoma cell
proliferation in response to thapsigargin or tunicamycin exposure
(Fig. 4). Analysis of glioblastoma
cell proliferation was conducted over a 6-day period post-exposure
to the vehicle, thapsigargin or tunicamycin. The most demonstrative
effects of thapsigargin or tunicamycin on A172 and U373 cell
production were observed on day 6, with a statistically significant
(p<0.05) decrease in glioblastoma cell proliferation as compared
to the vehicle-treated control cells examined at the same
time-point (Fig. 4). Time course
data also displayed a 28±4 and 52±4% reduction in A172 cell
proliferation 6 days post-treatment with thapsigargin or
tunicamycin, respectively, in comparison to A172 cell proliferation
observed on day 0 (Fig. 4). A
comparative assessment between these same time-points in U373 cells
also revealed a decrease in cell proliferation 6 days
post-treatment with either thapsigargin or tunicamycin when
compared to U373 cell proliferation on day 0 (Fig. 4). Taken together, the time course
data presented here suggest that the overall inhibitory effect of
thapsigargin and tunicamycin was due to glioblastoma cell death.
These results are similar to those from other studies on several
types of human cancer that also demonstrated that thapsigargin
(18–21) and tunicamycin (22–24)
promoted tumor cell death.
ER stress inducers increase CHOP
expression and caspase 3 activity
It is well established that the inability of the
endoplasmic reticulum to activate compensatory mechanisms essential
for cellular survival in response to stress will subsequently lead
to apoptotic cell death (3,4).
CHOP, a basic-leucine zipper (bZIP) transcription factor, and
caspase 3, a cysteine protease, are pro-apoptotic proteins known to
play prominent roles in endoplasmic reticulum stress-induced cell
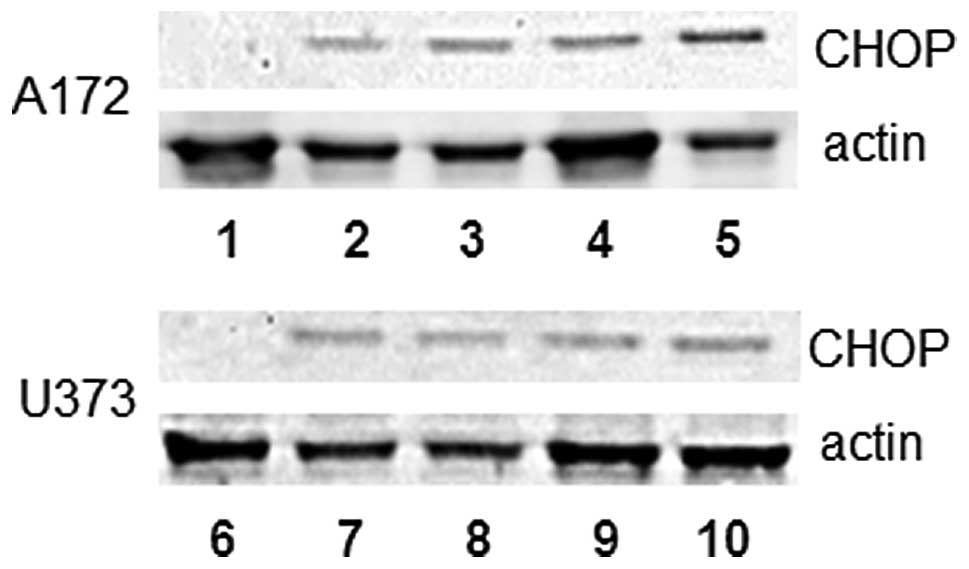
death (25,26). We therefore evaluated the protein
expression levels of CHOP and caspase 3 in thapsigargin- and
tunicamycin-treated glioblastoma cells. A temporal analysis using
immunoblotting procedures revealed an up-regulation of CHOP protein
levels at 24 and 48 h in A172 and U373 cells treated with 1 μM
thapsigargin or tunicamycin (Fig.
5). Caspase 3 activity assessment displayed a 2.6- and
1.43-fold increase in caspase 3 activity at 48 h in A172 cells
treated with thapsigargin or tunicamycin, respectively, while a
1.32- and 1.30-fold increase in caspase 3 activity was observed at
the same time-point in U373 cells treated with thapsigargin or
tunicamycin (Fig. 6). Although our
observations are consistent with previous ones on colon cancer,
leukemia and neuroblastomas, which also demonstrated that
thapsigargin or tunicamycin invoked CHOP expression or caspase 3
activity (24,27–30),
few studies have shown the induction of both of these pro-apoptotic
proteins in the same tumor type in response to single exposures of
endoplasmic reticulum stress inducers with different modes of
action. This suggests that glioblastomas are susceptible to
endoplasmic reticulum stress-induced cell death by diverse
physiological stressors.
Effects of ER stress on glioblastoma
cell motility
Glioblastomas are notoriously invasive tumors with
high rates of recurrence, which are major factors contributing to
their therapeutic refractiveness during clinical treatment
(surgery, chemo- and radiotherapy) regimens. Few studies to date
have investigated the role of the unfolded protein endoplasmic
reticulum stress response in tumor cell motility and invasion.
However, a study by Chiu et al (31) on head and neck cancers revealed
that silencing the function of the endoplasmic reticulum chaperone
protein, GRP78, using siRNA reduced the metastatic potential of
these cancers; thus, providing evidence for a role of the unfolded
protein endoplasmic reticulum stress response in the metastatic
invasion of tumor cells. Therefore, in this study, we examined the
effects of thapsigargin and tunicamycin on glioblastoma cell
motility, a prerequisite cellular program for invasive tumor cells.
In contrast to the findings by Chiu et al (31), we did not observe antagonistic
effects of thapsigargin or tunicamycin on glioblastoma cell
motility (Fig. 7), which is likely
due to variations in the experimental approach and the differential
mechanisms targeted.
Discussion
Targeting the unfolded protein endoplasmic reticulum
stress response is a relatively avant-garde yet practical
therapeutic approach for the treatment of human cancers. Human
tumors and their microenvironment are in a continuous flux of
imbalance due to the presence of abnormally folded proteins and
physiological instability, relating to fluctuations in pH and ion
concentration, all of which invoke stress on tumor cells. As a
survival mechanism, tumor cells respond via the activation of
stress responders, such as GRP78/binding immunoglobulin protein
(BiP), inositol-requiring enzyme 1α (IRE1α), protein kinase
RNA-like endoplasmic reticulum kinase (PERK) and activating
transcription factor 6 (ATF6) that collectively underlie the
unfolded protein endoplasmic reticulum stress response and confer
tumor cell cytoprotection (3,4). The
involvement of cytoprotective stress responders in glioblastomas
has been demonstrated in a number of recent investigations that
together establish that high levels of GRP78 expression correlate
with increased survival and drug resistance (8–10) in
these tumors. However, studies to date have failed to demonstrate
that antagonizing stress responder function, particularly GRP78,
promotes glioblastoma cell death.
The dichotomy of the unfolded protein endoplasmic
reticulum stress response is that stress exceeding the endoplasmic
reticulum's capacity to promote cell survival will consequently
result in cell death. To this end, in this study, we demonstrate
that two endoplasmic reticulum stress inducers, thapsigargin and
tunicamycin, promote glioblastoma cell death. Our findings are
consistent with those from previous studies on several human
cancers that also showed that thapsigargin (prostate, breast,
leukemia, and melanoma) (18–20,30,32)
and tunicamycin (neuroblastoma and melanoma) induced tumor cell
death (24,33).
It is well established that the mechanism affiliated
with endoplasmic reticulum stress-induced cell death is the
activation of the transcription factor, CHOP, a downstream
pro-apoptotic component of the IRE1α, PERK and ATF6 stress
responder pathways (25).
Concomitantly with glioblastoma cell death, we observed a
significant increase in CHOP expression in response to endoplasmic
recticulum stress inducers. These findings parallel those observed
by Rosati et al (22) and
Oda et al (24) who also
detected increased CHOP expression levels in leukemia and
neuroblastoma cells, respectively, in response to thapsigargin and
tunicamycin. Although effector molecules of CHOP still remain
somewhat elusive, its overexpression has been shown to lead to a
decrease in the pro-survival protein, Bcl-2 (34), providing evidence that the
pro-apoptotic functions of CHOP are associated with
mitochondria-dependent mechanisms of cell death. Our data support
this premise and were substantiated by observations of increased
caspase 3 activity in glioblastoma cells treated with thapsigargin
or tunicamycin.
In spite of the pro-apoptotic effects of endoplasmic
reticulum stress inducers on glioblastoma cell production observed
in this study, thapsigargin and tunicamycin failed to impair
glioblastoma cell motility. This is in contrast to experimental
data by Chiu et al who demonstrated that silencing GRP78
function reduced cell motility and prevented tumor cell invasion of
head and neck cancers (31). The
differential cell motility responses observed in this study and by
Chiu et al are likely attributed to divergent cellular and
physiological mechanisms targeted in the unfolded protein
endoplasmic reticulum stress response between our two studies, and
further emphasizes the pleiotropic effects of the endoplasmic
reticulum stress response on cell behavior.
Taken together, we provided evidence that
hyper-stressing the endoplasmic reticulum, by using endoplasmic
reticulum stress inducers with markedly different modes of action,
is a viable approach for the promotion of killing glioblastomas.
Furthermore, our study suggests that endoplasmic reticulum stress
inducers exert their anti-tumorigenic effects on proliferating
cells, a selective advantage for treating clinical glioblastomas
which typically reside in regions of the human brain that contain
dormant cells.
Acknowledgements
This work was supported by grant
number P20MD002731 from the National Institute on Minority Health
and Health Disparities (MOF). The content is solely the
responsibility of the authors and does not necessarily represent
the official views of the National Institute on Minority Health and
Health Disparities or the National Institutes of Health.
References
|
1.
|
C KrakstadM ChekenyaSurvival signaling and
apoptosis resistance in glioblastomas: opportunities for targeted
therapeuticsMol Cancer9135201010.1186/1476-4598-9-13520515495
|
|
2.
|
D KögelS FuldaM MittelbronnTherapeutic
exploitation of apoptosis and autophagy for glioblastomaAnticancer
Agents Med Chem10438449201020879985
|
|
3.
|
G WangZ YangK ZhangEndoplasmic reticulum
stress response in cancer: molecular mechanism and therapeutic
potentialAm J Transl Res26574201020182583
|
|
4.
|
GC ShoreFR PapaSA OakesSignaling cell
death from the endoplasmic reticulum stress responseCurr Opin Cell
Biol2143149201110.1016/j.ceb.2010.11.00321146390
|
|
5.
|
S LustB VanhoeckeM Van GeleJ BoelensH Van
MelckebekeM KailehW Vanden BergheG HaegemanJ PhilippéM BrackeF
OffnerXanthohumol activates the proapoptotic arm of the unfolded
protein response in chronic lymphocytic leukemiaAnticancer
Res1037973805200919846911
|
|
6.
|
X HuangZ ZhangL JiaY ZhaoX ZhangK
WuEndoplasmic reticulum stress contributes to vitamin E
succinate-induced apoptosis in human gastric cancer SGC-7901
cellsCancer
Lett296123131201010.1016/j.canlet.2010.04.00220435408
|
|
7.
|
P RavananR SanoT PritiS OgasawaraSI
MatsuzawaM CuddySK SinghGS RaoP KondaiahJC ReedSynthetic
triterpenoid cyano enone of methyl boswellate (CEMB) activates
intrinsic, extrinsic and endoplasmic reticulum stress cell death
pathways in tumor cell linesMol Cancer
Ther1016351643201110.1158/1535-7163.MCT-10-0887
|
|
8.
|
P PyrkoAH SchönthalF HofmanTC ChenAS
LeeThe unfolded protein response regulator GRP78/BiP as a novel
target for increasing chemosensitivity in malignant gliomasCancer
Res6798099816200710.1158/0008-5472.CAN-07-062517942911
|
|
9.
|
JJ VirreyD DongC StilesJB PattersonL PenM
NiAH SchonthalTC ChenFM HofmanAS LeeStress chaperone GRP78/BiP
confers chemoresistance to tumor-associated endothelial cellsMol
Cancer Res612681275200810.1158/1541-7786.MCR-08-006018708359
|
|
10.
|
HK LeeC XiangS CazacuS FinnissG
KazimirskyN LemkeNL LehmanSA RempelT MikkelsenC BrodieGRP78 is
overexpressed in glioblastomas and regulates glioma cell growth and
apoptosisNeuro
Oncol10236243200810.1215/15228517-2008-00618403493
|
|
11.
|
P SabałaM CzarnyJP WoronczakJ
BarańskaThapsigargin: potent inhibitor of Ca2+ transport
ATP-ases of endoplasmic and sarcoplasmic reticulumActa Biochim
Pol403093191993
|
|
12.
|
K IsonoNucleoside antibiotics: structure,
biological activity, and biosynthesisJ Antibiot
(Tokyo)4117111739198810.7164/antibiotics.41.17113061990
|
|
13.
|
CN HancockLH StockwinB HanRD DivelbissJH
JunSV MalhotraMG HollingsheadDL NewtonA copper chelate of
thiosemicarbazone NSC 689534 induces oxidative/ER stress and
inhibits tumor growth in vitro and in vivoFree Radic Biol
Med50110121201110.1016/j.freeradbiomed.2010.10.69620971185
|
|
14.
|
SK MinSK LeeJS ParkJ LeeJY PaengSI LeeHJ
LeeY KimHO PaeSK LeeEC KimEndoplasmic reticulum stress is involved
in hydrogen peroxide induced apoptosis in immortalized and
malignant human oral keratinocytesJ Oral Pathol
Med37490498200810.1111/j.1600-0714.2008.00679.x18631371
|
|
15.
|
Y WatanabeH TsuchiyaT SakabeS MatsuokaY
AkechiY FujimotoK YamaneR IkedaR NishioK TerabayashiCD437 induces
apoptosis in ovarian adenocarcinoma cells via ER stress
signalingBiochem Biophys Res
Commun366840847200810.1016/j.bbrc.2007.12.02818082618
|
|
16.
|
JH JooG LiaoJB CollinsSF GrissomAM
JettenFarnesol-induced apoptosis in human lung carcinoma cells is
coupled to the endoplasmic reticulum stress responseCancer
Res6779297936200710.1158/0008-5472.CAN-07-093117699800
|
|
17.
|
A YacoubHA HamedJ AllegoodC MitchellS
SpiegelMS LesniakB OgretmenR DashD SarkarWC BroaddusPERK-dependent
regulation of ceramide synthase 6 and thioredoxin play a key role
in mda-7/IL-24-induced killing of primary human glioblastoma
multiforme cellsCancer
Res7011201129201010.1158/0008-5472.CAN-09-404320103619
|
|
18.
|
C JackischHA HahmB TombalD McCloskeyK
ButashNE DavidsonSR DenmeadeDelayed micromolar elevation in
intracellular calcium precedes induction of apoptosis in
thapsigargin-treated breast cancer cellsClin Cancer
Res6284428502000
|
|
19.
|
JK HuangCT ChouHT ChangSS ShuCC KuoJY
TsaiWC LiaoJL WangKL LinYC LuEffect of thapsigargin on
Ca2+ fluxes and viability in human prostate cancer
cellsJ Recept Signal Transduct Res312472552011
|
|
20.
|
DJ Vander GriendL AntonySL DalrympleY XuSB
ChristensenSR DenmeadeJT IsaacsAmino acid containing thapsigargin
analogues deplete androgen receptor protein via synthesis
inhibition and induce the death of prostate cancer cellsMol Cancer
Ther8134013492009
|
|
21.
|
Y KanekoA TsukamotoThapsigargin-induced
persistent intracellular calcium pool depletion and apoptosis in
human hepatoma cellsCancer
Lett79147155199410.1016/0304-3835(94)90253-48019972
|
|
22.
|
E RosatiR SabatiniG RampinoF De FalcoM Di
IanniF FalzettiK FettucciariA BartoliI ScrepantiP MarconiNovel
targets for endoplasmic reticulum stress-induced apoptosis in
B-CLLBlood11627132723201010.1182/blood-2010-03-27562820628148
|
|
23.
|
L GirnitaM WangY XieG NilssonA DricuJ
WejdeO LarssonInhibition of N-linked glycosylation down-regulates
insulin-like growth factor-1 receptor at the cell surface and kills
Ewing's sarcoma cells: therapeutic implicationsAnticancer Drug
Des156772200010888037
|
|
24.
|
T OdaY KosugeM ArakawaK IshigeY
ItoDistinct mechanism of cell death is responsible for
tunicamycin-induced ER stress in SK-N-SH and SH-SY5Y cellsNeurosci
Res602939200810.1016/j.neures.2007.09.00518029041
|
|
25.
|
S OyadomariM MoriRoles of CHOP/GADD153 in
endoplasmic reticulum stressCell Death
Differ11381389200410.1038/sj.cdd.440137314685163
|
|
26.
|
I TabasD RonIntegrating the mechanisms of
apoptosis induced by endoplasmic reticulum stressNat Cell
Biol1318490201110.1038/ncb0311-18421364565
|
|
27.
|
H YamaguchiK BhallaHG WangBax plays a
pivotal role in thapsigargin-induced apoptosis of human colon
cancer HCT116 cells by controlling Smac/Diablo and Omi/HtrA2
release from mitochondriaCancer Res6314831489200312670894
|
|
28.
|
Y KitamuraA MiyamuraK TakataM IndenD
TsuchiyaK NakamuraT TaniguchiPossible involvement of both
endoplasmic reticulum-and mitochondria-dependent pathways in
thapsigargin-induced apoptosis in human neuroblastoma SH-SY5Y
cellsJ Pharmacol Sci92228236200310.1254/jphs.92.228
|
|
29.
|
MK DahmerCaspases-2, -3, and -7 are
involved in thapsigargin-induced apoptosis of SH-SY5Y neuroblastoma
cellsJ Neurosci Res80576583200510.1002/jnr.2047115825194
|
|
30.
|
XQ FengY YouJ XiaoP
ZouThapsigargin-induced apoptosis of K562 cells and its
mechanismZhongguo Shi Yan Xue Ye Xue Za Zhi142530200616584585
|
|
31.
|
CC ChiuCY LinLY LeeYJ ChenTF KuoJT ChangCT
LiaoHM WangTC YenCR ShenSK LiaoAJ ChengGlucose-regulated protein 78
regulates multiple malignant phenotypes in head and neck cancer and
may serve as a molecular target of therapeutic interventionMol
Cancer Ther727882797200810.1158/1535-7163.MCT-08-0172
|
|
32.
|
LH ChenCC JiangKA KiejdaYF WangRF ThorneXD
ZhangP HerseyThapsigargin sensitizes human melanoma cells to
TRAIL-induced apoptosis by up-regulation of TRAIL-R2 through the
unfolded protein
responseCarcinogenesis2823282336200710.1093/carcin/bgm173
|
|
33.
|
A DricuM CarlbergM WangO LarssonInhibition
of N-linked glycosylation using tunicamycin causes cell death in
malignant cells: role of down-regulation of the insulin-like growth
factor 1 receptor in induction of apoptosisCancer
Res575435481997
|
|
34.
|
KD McCulloughJL MartindaleLO KlotzTY AwNJ
HolbrookGadd153 sensitizes cells to endoplasmic reticulum stress by
down-regulating Bcl2 and perturbing the cellular redox stateMol
Cell Biol2112491259200110.1128/MCB.21.4.1249-1259.200111158311
|