Introduction
Autophagy is a prevalent phenomenon in eukaryotic
cells and occurs following myocardial ischemia and subsequent
reperfusion (1). Low levels of
autophagy play a protective role in cardiomyoctyes, whereas high
levels or chronic autophagy are able to facilitate cell injury and
result in the death of cells that cannot be repaired (2). Determining how to effectively inhibit
autophagy and attenuate myocardial damage has become a
cardiovascular disease research hotspot (3,4).
Apelin is highly expressed in the cardiovascular system, where it
dilates blood vessels to reduce blood pressure, improves heart
function, inhibits cardiomyocyte apoptosis and protects against
myocardial ischemia reperfusion injury (5,6). We
have previously shown that Apelin-13 also inhibits cardiomyocyte
autophagy (7), but the underlying
mechanism remains unclear. In the present study, the possible
mechanism responsible for autophagy following glucose deprivation
(GD) in cardiomyoctyes was investigated.
Autophagy is important for clearing injured
organelles following hypoxia-ischemia, nutritional deficiencies,
oxidative stress and infection. While autophagy plays a protective
role, excessive autophagy may result in type II programmed cell
death (2). Hence, chronic
autophagy may lead to irreversible damage. The pathways involved in
autophagy progression are complicated, with three pathways
positively identified thus far: i) type I
phosphatidylinositol-3-kinase (PI3K)/Akt signaling, ii) mammalian
target of rapamycin (mTOR) signaling and iii) type III PI3K
signaling. The first pathway inhibits autophagy. mTOR signaling
plays a significant regulatory role in cell growth and receives
multiple upstream signals, including those of type I PI3K, IGF-1/2
and mitogen-activated protein kinase (MAPK). Moreover, it inhibits
autophagy by perceiving changes in nutrient and energy levels. The
structure of type III PI3K is similar to that of type I PI3K,
however, it presents with a reversed role. Autophagosome formation
may be interfered with or blocked using PI3K inhibitors, including
3-MA, wortmannin and LY294002 (8).
Tatemoto et al(9) first extracted and purified Apelin
from calf stomach secretions using reverse pharmacology. Apelin was
determined to be an endogenous ligand of the orphan G
protein-coupled receptor angiotensin type 1 receptor-associated
protein (APJ). Apelin is expressed ubiquitously, with particularly
high levels in the cardiovascular system and with the ability to
dilate blood vessels to reduce blood pressure, improve heart
function, inhibit cardiomyocyte apoptosis and protect against
myocardial ischemia reperfusion injury (3,5,6). We
have previously shown that Apelin-13 also inhibits cardiomyocyte
autophagy (7), however the
underlying mechanism remains unclear. Another study suggested that
PI3K/Akt signaling may be involved (10). In the present study, the effect of
Apelin-13 on autophagy was studied further and the role of PI3K/Akt
signaling was assessed. Our previously established in vitro
neonatal rat cardiomyocyte model of GD was employed (10) and transmission electron microscopy
(TEM), PI3K activity assays and western blotting were performed.
Collectively, our findings support the hypothesis that Apelin-13
inhibits cardiomyocyte autophagy by modulating PI3K/Akt
signaling.
Materials and methods
Reagents
Specimens were obtained from Sprague Dawley (SD) 1–3
day postnatal rat pups (Experimental Animal Center of Liaoning
Medical College, Jinzhou, China). Dulbecco’s modified Eagle’s
medium (DMEM):F12 (1:1) culture medium, calf serum,
5-bromodeoxyuridine (BrdU) and phosphate-buffered saline (PBS)
balanced salt solution were all purchased from Hyclone (Logan, UT,
USA). Trypsin (Beyotime Co., Jiangsu, China), Apelin-13 and
triciribine (Sigma, St. Louis, MO, USA) were also used.
Anti-phosphotyrosine antibody was purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA.) Rabbit
anti-human-β-actin, LC3, Akt, p-Akt (Ser473), mTOR and p-mTOR
(Ser2448) antibodies were purchased from Cell Signaling Technology
(Danvers, MA, USA). The study was approved by the ethical committee
of The Third Affilated Hospital of Liaoning Medical University
(Jinzhou, China).
Culture and identification of
myocardiocytes
The protocol to culture and identify the
myocardiocytes was employed as described previous (11). The experiments were approved by the
Institutional Animal Care and Use Committee. Briefly, SD rats at
postnatal days 1–3 were sacrificed and their hearts removed under
sterile conditions, rinsed in PBS balanced salt solution three
times and then cut into 1-mm3 sections. The samples were
placed into a magnetic stirrer at 37°C with 0.06% trypsin until
they were completely digested. The digestion was stopped using calf
serum. The resulting cell suspension was centrifuged at 1,200 rpm
for 10 min and resuspended in DMEM:F12 (1:1) medium containing 15%
fetal calf serum. The resulting single-cell suspension was filtered
through a 200 mesh filter and the cell density was adjusted to
5×105/ml prior to inoculation in a 25-cm3
flask. The flasks were then placed in an incubator at 37°C with 5%
CO2. The fibroblasts were removed by replacing the
culture medium after 90 min when the myocardiocytes had adhered to
the flask. The cell suspension was added with 5-BrdU to a final
concentration of 0.1 ml/l and adjusted to 5×105/ml. The
culture medium was replenished every 24 h. Subsequent to being
cultured for 72 h, the cells that grew in a good condition with
regular contraction were used for the experiments. Monoclonal
antibodies against α-sarcomeric actin were used to characterize the
myocardiocytes.
Experimental groups and model
establishment
The cardiomyocytes cultured for 72 h were divided
into 5 groups: i) Control group, the culturing of the cells
continued without any treatment for 12 h subsequent to changing the
medium; ii) GD group, the culturing of the cells continued for 12 h
subsequent to the replacement of the medium with a glucose-free
DMEM:F12 (1:1) medium; iii) GD+Apelin-13 group, the cells were
pretreated with 1 μmol/l Apelin-13 for 30 min prior to the
change to a glucose-free medium; iv) GD+Triciribine+Apelin-13
group, the cells were pretreated with 1 μmol/l triciribine
for 30 min and 1 μmol/l Apelin-13 for 30 min in succession
prior to the change to a glucose-free medium; and v) Triciribine
group, the cells were pretreated with 1 μmol/l triciribine
for 30 min followed by 12 h in a normal culture medium.
Autophagosome observation by TEM
Next, the culture medium was apirated and the cells
were gently washed three times with PBS buffer prior to harvesting
the cells with cell scratches. The samples were placed in a tube
and centrifuged at 1,800 rpm for 30 min and the supernatant was
carefully absorbed. The cells were then fixed with 3%
glutaraldehyde and 1% osmium tetroxide for 3 h. Following rinsing
with PBS for 30 min, the samples were dehydrated with ethanol and
isopropanol, embedded in epoxy resin and prepared under a
dissecting microscope. An ultrathin sectioning machine (Leica EM
UC6, Leica Microsystems, Mannheim, Germany) was used to prepare the
1-μm sections, then the samples were double stained with
uranyl acetate and lead citrate and the cardiomyocytes were
observed using TEM (JEM-1200EX, JEOL Ltd., Tokyo, Japan). Images
were captured and 10 randomly selected fields of vision from each
group were used to quantify the area of the autophagosomes to the
total cytoplasmic area.
PI3K activity assay using
immunoprecipitation lipid kinase analysis
The PI3K activity was measured as previously
described (12). Briefly, the
total proteins were extracted and incubated with
anti-phosphotyrosine antibodies for 1 h. The proteins were
subsequently incubated with a G-protein-coated agarose bead
suspension for 4–12 h. The G-protein-coated agarose beads were
washed 3 times with a buffer containing 100 mM Tris (pH 7.5) and
500 mM LiCl, followed by a single wash with buffer containing 10 mM
Tris-HCl, 100 mM NaCl, 1 mM EDTA and then a further single wash in
the PI3K analysis buffer. The washed antigen-antibody complex was
pre-incubated with 50 μl PI3K buffer and 20 μl lipid
substrate mixture. The enzyme reaction was carried out using 10
μl γ32P-ATP mixture. After 10 min, the reaction
was quenched by adding an equal volume (80 μl) of stop
solution. The mixture was extracted twice using phosphorylated
chloroform-methanol (1:1). The resulting mixture was analyzed using
thin-layer chromatography and autoradiography. Radioactivity was
detected using a FJ-2115 automatic liquid scintillation counter.
The mean radioactivity in the control group was set as 100% and the
radioactivity levels were compared among the groups.
Western blotting for the detection of
LC3, Akt, p-Akt, mTOR, p-mTOR and PI3K
The cardiomyocytes were washed twice in cold PBS
then gently mixed in a lysis buffer [25 mM Tris-HCl (pH 7.4), 150
mM NaCl, 2 mM EDTA, 1% Triton-X-100, 1% sodium deoxycholate, 0.1%
sodium dodecyl sulfate and protease inhibitor cocktail] and
incubated for 20 min on ice. The samples were centrifuged at 12,000
rpm for 20 min at 4°C and the supernatant was collected and
subjected to a bicinchoninic acid assay for protein quantification.
Samples consisting of 50 μg total protein were separated
using sodium dodecyl sulfate polyacrylamide gel electrophoresis and
the proteins were transferred to membranes using a semi-dry method.
The membranes were then blocked for 1 h with 5% bovine serum
albumin in PBS with Tween-20 (BSA-PBST) at room temperature. The
blots were then incubated with primary antibodies against LC3,
PI3K, Akt, p-Akt, mTOR, p-mTOR or β-actin at 4°C overnight. The
next day, the blots were washed and incubated in the appropriate
secondary antibodies at room temperature for 1 h. Subsequent to
being washed, the blots were developed and gray scale scanning
(iBox Scientia 500/600, UVP, Upland, CA, USA) was performed. The
expression levels of LC3, PI3K, Akt, p-Akt, mTOR, and p-mTOR
proteins were normalized to β-actin.
Statistical analysis
All analyses were performed with SPSS 13.0 software
(SPSS Inc., Chicago, IL, USA). The measurement data are presented
as mean ± standard deviation (SD) and the multi-group comparisons
were made with a one-way factor analysis of variance and the Least
Significant Difference post hoc test. Values of P<0.05 were
considered to indicate a statistically significant difference. The
TEM image selection was performed randomly.
Results
Observation of autophagosomes under
TEM
Autophagosomes are mono- or bilayer membrane
structures that engulf regions of the cytoplasm that contain
degraded or abandoned organelles. In the present study, the
cardiomyocytes in each group contained varying numbers of
autophagic vacuole-like structures under TEM. The number of
autophagosomes in the GD group was significantly increased compared
with in the Co group. The GD+Apelin-13 group had markedly fewer
autophagosomes than the GD group. Conversely, the addition of
triciribine, as in the GD+Apelin-13+Triciribine group, increased
the number of autophagosomes compared with the GD+Apelin-13 group.
There was no significant difference observed between the
Triciribine and Co groups (P>0.05; Fig. 1).
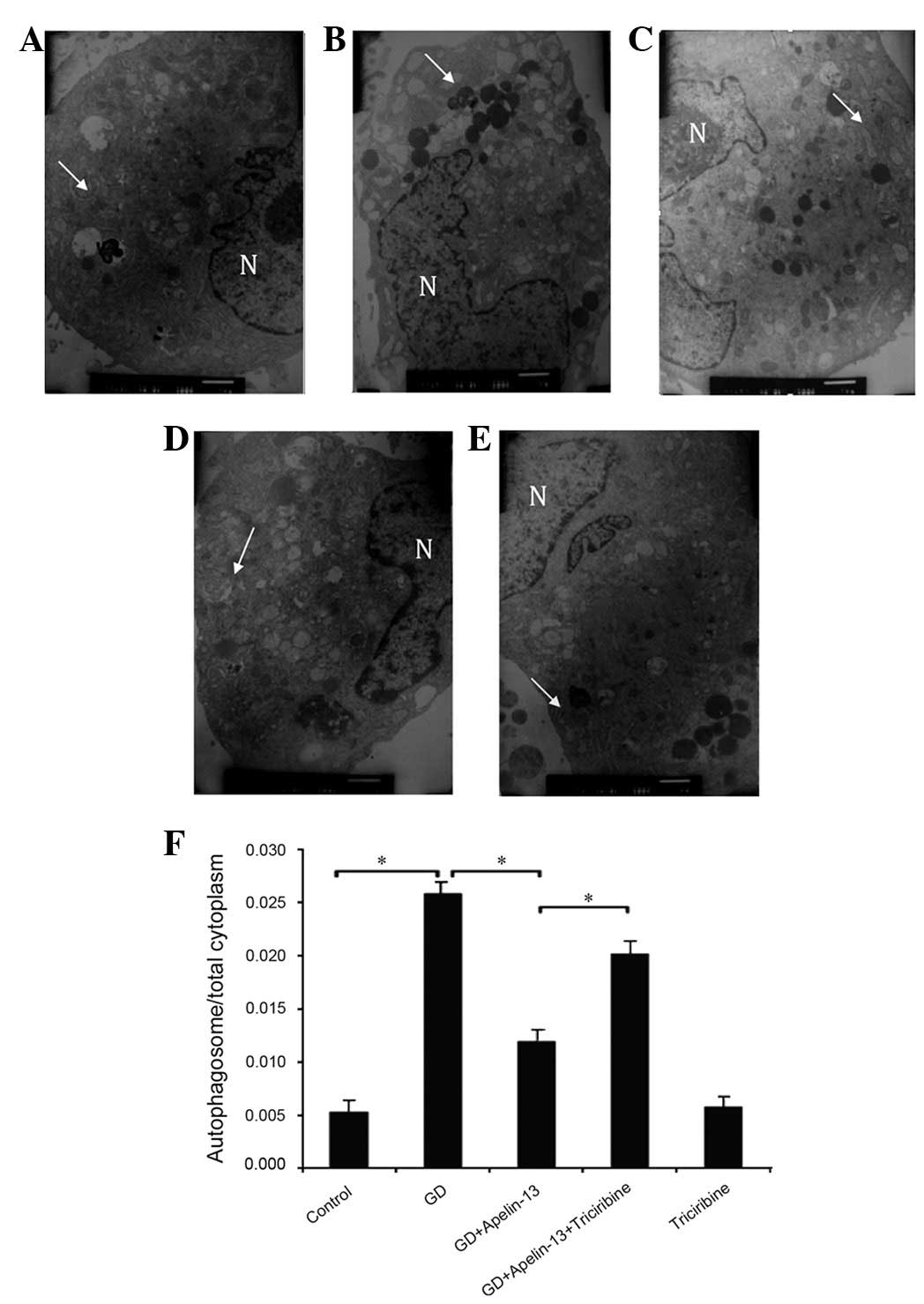 | Figure 1.Myocardial cell autophagosomes under
TEM (×8,000). (A) Co group, (B) GD group, (C) GD+Apelin-13 group,
(D) GD+Apelin-13+Triciribine group and (E) Triciribine group;
arrows indicate the bilayered membrane structure of the
autophagosomes wrapped around the cytoplasmic components during
cellular degradation. GD significantly increased the number of
autophagosomes, while pretreatment with Apelin-13 significantly
decreased the number of autophagosomes compared with the GD group.
Using Apelin-13 and triciribine resulted in a significantly greater
number of autophagosomes than when using Apelin-13 alone. There was
no significant difference in autophagosome number between the
Apelin-13 and Co groups. (F) Quantitation of the autophagosome to
cytoplasmic ratio by group, which indirectly shows the degree of
autophagy (*P<0.05). TEM, transmission electron
microscopy; Co, control; GD, glucose deprivation; N, nuclei. |
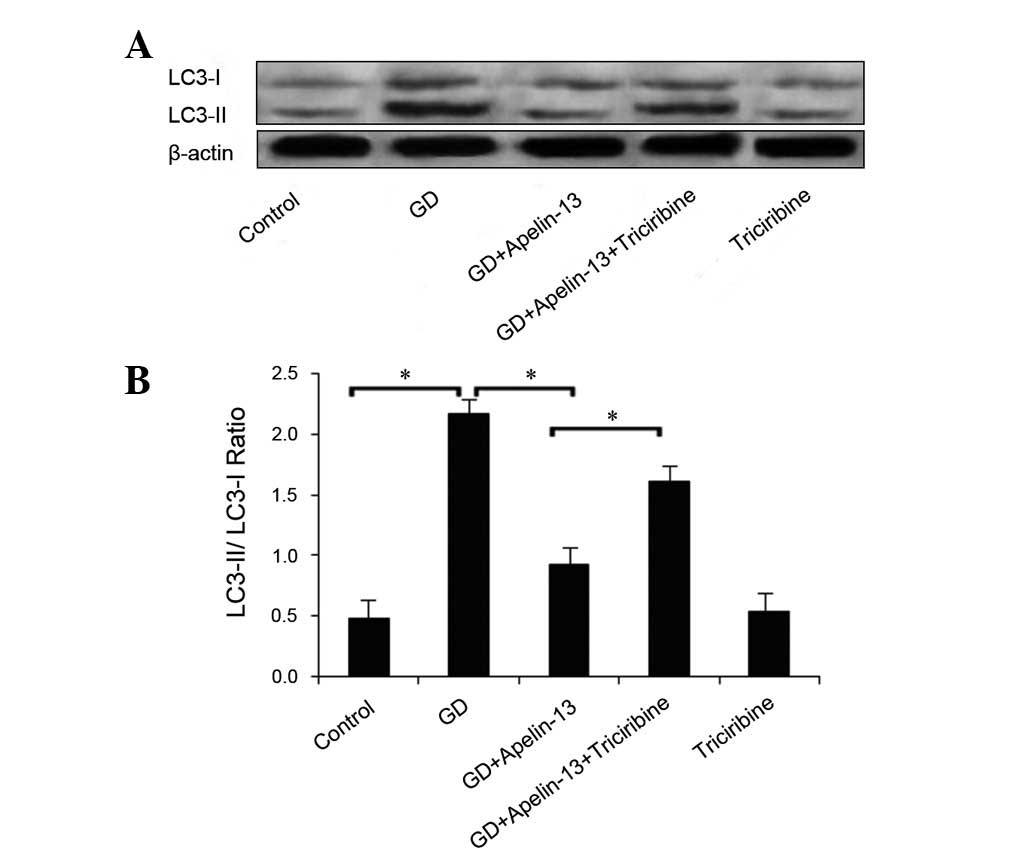
Autophagy-related protein LC3
expression
LC3 is an autophagic marker that exists in two
forms: type I and type II. Prior to the occurrence of autophagy,
LC3 is processed to form a soluble type I LC3 protein and autophagy
induces further processing (ubiquitination) of LC3-I, inducing it
to bind phosphatidyl ethanolamine on the surface of the autophagy
layer to form type II LC3. The LC3-II/LC3-I ratios positively
correlate with the autophagosome number (13), thereby providing an indirect
measure of autophagy. The present study used the LC3-II/LC3-I ratio
to assess the effect of Apelin-13 on LC3 protein expression; a
larger ratio indicated enhanced autophagy in the mammalian cells.
Fig. 2 shows the results of this
analysis for each group. Compared with the Co group, the ratio of
LC3-II/LC3-I in the GD group increased significantly (P<0.05).
Conversely, the ratio was significantly decreased in the
GD+Apelin-13 group compared with the GD and
GD+Apelin-13+Triciribine groups (both P<0.05). A significant
difference was not observed between the Triciribine and Co groups
(P>0.05; Fig. 2).
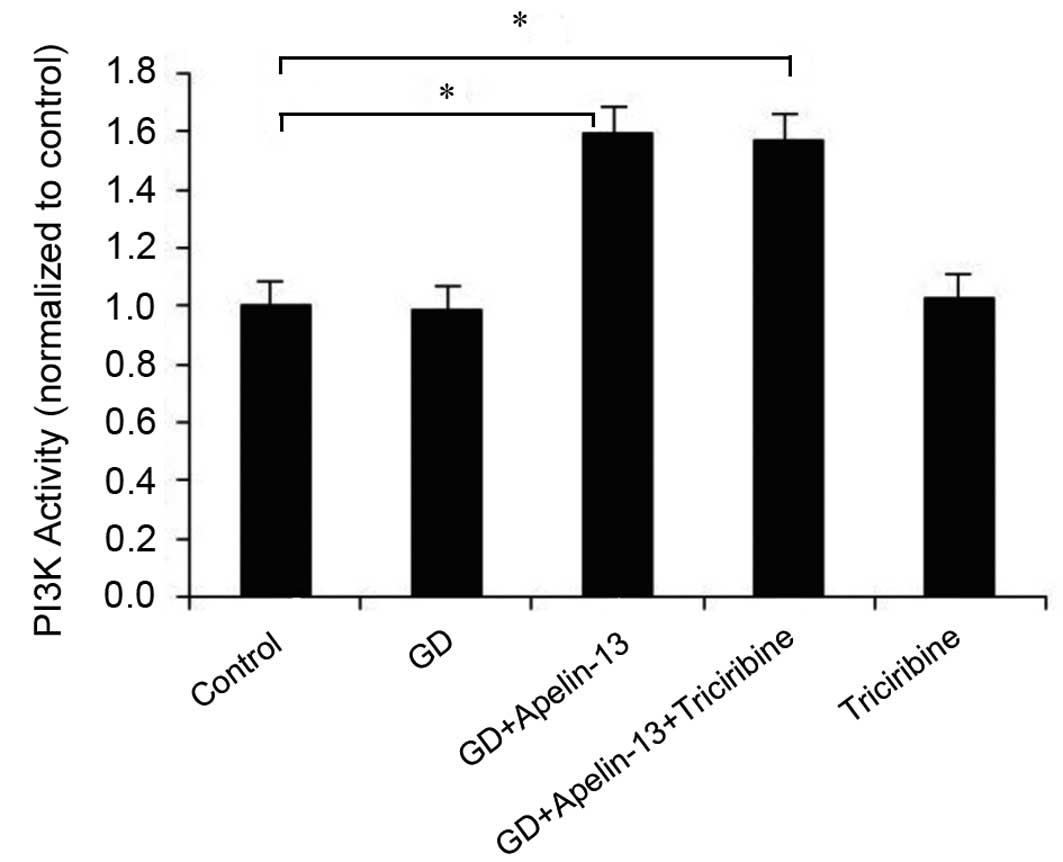
PI3K activity
PI3K activities in the GD+Apelin-13 and GD+Apelin-13
+Triciribine groups were significantly greater (1.59- and 1.56-fold
greater, respectively) than those observed in the Co group. These
differences were statistically significant (P<0.05). However,
the PI3K activities in the GD and Triciribine groups were not
significantly different from the Co group (P>0.05; Fig. 3).
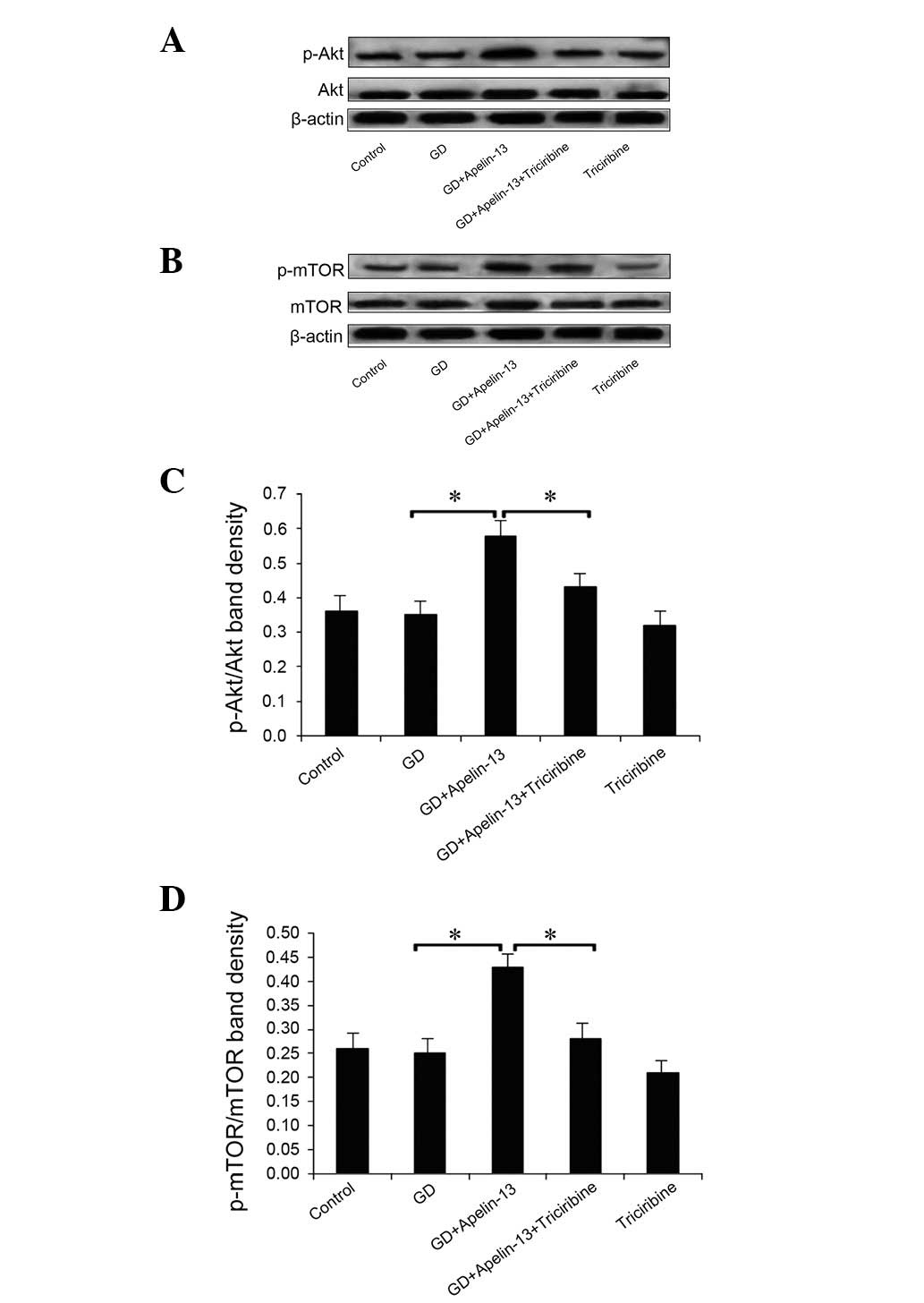
Expression levels of PI3K/Akt/mTOR
pathway-related proteins
Compared with the GD group, PI3K and phosphorylated
mTOR (p-mTOR) expression levels in the GD+Apelin-13 and
GD+Apelin-13+Triciribine groups were significantly increased
(P<0.05). There was no significant difference in PI3K expression
between the GD+Apelin-13 and GD+Apelin-13+Triciribine groups
(P>0.05), but p-mTOR expression was decreased significantly in
the GD+Apelin-13+Triciribine group compared with the GD+Apelin-13
group (P<0.05; Fig. 4).
Compared with the Co group, the p-Akt and p-mTOR
expression levels in the GD group were slightly, but not
significantly, decreased (P>0.05). The Apelin-13 pretreatment
significantly increased the p-Akt and p-mTOR levels (P<0.05),
which were inhibited by the triciribine pretreatment (P<0.05).
However, the total expression levels of the Akt and mTOR proteins
were not significantly changed by the treatments (P>0.05;
Fig. 5).
Discussion
Autophagy occurs in eukaryotic cells at a low level
under normal conditions. Cell autophagy may be initiated to remove
damaged organelles when stimulated by ischemia and hypoxia,
nutrient deficiency, oxidative stress, infection and other factors.
Normally, autophagy is protective, but excessive activation of
autophagy may lead to type II programmed cell death (2). Therefore, ensuring the timely and
appropriate cessation of autophagy prevents injured cells from
irreversible injury. In the present study, cardiomyocyte autophagy
was induced by culturing the cell in a sugar- and serum-free
culture medium (7).
Apelin was originally purified from bovine gastric
secretions by Tatemoto et al(9) using a reversed pharmacological
method. Apelin was ultimately determined to be the endogenous
ligand of the orphan G protein-coupled receptor, APJ (14). Apelin is highly expressed in the
cardiovascular system where it exerts protective effects, including
the dilation of blood vessels to reduce blood pressure (15), improvements to heart function, the
inhibition of myocardial ischemia injury (1) and the promotion of angiogenesis
(16).
We previously demonstrated that Apelin-13 is able to
inhibit GD-induced cardiomyocyte autophagy in a
concentration-dependent fashion and that 1 μmol/l Apelin-13
is the optimal concentration for this inhibition (7). Previously, it has been demonstrated
that Apelin-13 is able to inhibit acute ischemia-induced
myocardiocyte apoptosis by activating the PI3K/Akt singaling
pathway (10). Other studies have
shown that the small molecule triciribine is able to inhibit Akt
activity (17,18). Therefore, in the present study
cells were pretreated with 1 μmol/l Apelin-13 and
administered the Akt inhibitor triciribine 30 min prior to
observing cardiomyocyte autophagy and protein expression levels to
investigate whether Apelin-13 is able to inhibit excessive
autophagy of the GD cardiomyocytes by altering PI3K-Akt
signaling.
TEM imaging is the gold standard for assessing
autophagy (19,20). In the cells with active autophagy,
the present study observed damaged organelles with swelling or
degenerated mitochondria, which were indicated by a vacuolar
bilayer membrane-like structure around the mitochondria, as well as
residual bodies in autophagic lysosomes that were not degraded.
Calculating the ratio of autophagosomes to the total cytoplasmic
area is considered as the appropriate method for quantifying
autophagy. However, LC3 is the homolog of autophagy-related gene 8
(ATG8) and is therefore a useful indirect marker of autophagy.
It was reported that in cardiomyocytes cultured in
GD medium, the ATP levels significantly decreased and the AMP/ATP
ratios increased, which led to activation of AMP-activated protein
kinase (AMPK). Moreover, activated AMPK inhibited mTOR in
cardiomyocytes, which led to mTOR activation and to negatively
regulated cell autophagy (1,13,21,22).
In the present study, GD significantly increased the number of
autophagosomes and the LC3-II/LC3-I ratio in neonatal rat
myocardiocytes. Exogenous administration of Apelin-13 attenuated
the GD-induced increase in the autophagosomes and the LC3-II/LC3-I
ratio, indicating that Apelin-13 inhibited autophagy. Triciribine
partially reversed the Apelin-13-mediated cardiomyocyte
improvements, but triciribine alone did not exert any significant
effect on cardiomyocytes compared with the Co group (P>0.05).
Overall, Apelin-13 was observed to effectively inhibit
cardiomyocyte autophagy and this effect was likely related to
increased PI3K-Akt signaling.
The mechanism of autophagy is relatively complex and
involves numerous diverse signaling pathways, including mTOR
(4,23). mTOR is an evolutionarily highly
conserved serine/threonine kinase which is involved in the
regulation of cell responses in changed nutrition conditions and a
number of energy-related regulatory pathways. mTOR is the gating
mechanism of autophagy and negatively regulates the process
(23). Therefore, mTOR activation
may be indirectly inhibited using triciribine to inhibit Akt and
promote autophagy. Activation of extracellular receptors leads to
the phosphorylation of tyrosine kinase receptors and the
phosphorylated sites recognize and combine with the p85 subunit of
PI3K to activate type I PI3K signaling. This in turn produces
phosphatidylinositol 4,5-bisphosphate and 3,4,5-trisphosphate to
activate Akt, which activates mTOR and inhibits autophagy (24). In the present study, the PI3K
activity and the levels of p-Akt and p-mTOR were significantly
increased by Apelin-13, however, the effect was partially inhibited
by triciribine. Combined with the TEM results, these results lead
to the hypothesis that Apelin-13 inhibited cardiomyocyte autophagy
by activating the PI3K-Akt signaling pathway and by simultaneously
activating the downstream signaling molecule mTOR.
Autophagy plays a significant role in immunity,
infection, inflammation, tumors and cardiovascular and
neurodegenerative diseases (25).
There are numerous studies with regard to the involvement of
autophagy in tumors, neurodegeneration and immune conditions, but
it is relatively less studied in cardiovascular diseases. In the
present study, the mechanisms underlying the regulatory effect of
Apelin-13 on GD-induced autophagy in cardiomyocytes were assessed.
The results confirmed that Apelin-13 is able to partially attenuate
GD-induced autophagy by increasing the expression levels and
activation of the components of the PI3K/Akt/mTOR signaling
pathway. These findings suggest that Apelin-13-related drugs may
provide new methods for protecting cardiomyoctyes from injury, but
full understanding of the specific pathways that inhibit autophagy
requires further study.
Acknowledgements
This study was supported by The
Scientific Research Item of Education Department of Liaoning
Province of Universities (No. 2009A453).
References
|
1.
|
Matsui Y, Takagi H, Qu X, et al: Distinct
roles of autophagy in the heart during ischemia and reperfusion:
roles of AMP-activated protein kinase and Beclin 1 in mediating
autophagy. Circ Res. 100:914–922. 2007. View Article : Google Scholar
|
|
2.
|
Kroemer G and Levine B: Autophagic cell
death: the story of a misnomer. Nat Rev Mol Cell Biol. 9:1004–1010.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Sorli SC, van den Berghe L, Masri B,
Knibiehler B and Audigier Y: Therapeutic potential of interfering
with apelin signalling. Drug Discov Today. 11:1100–1106. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Dutta D, Calvani R, Bernabei R,
Leeuwenburgh C and Marzetti E: Contribution of impaired
mitochondrial autophagy to cardiac aging: mechanisms and
therapeutic opportunities. Circ Res. 110:1125–1138. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Quazi R, Palaniswamy C and Frishman WH:
The emerging role of apelin in cardiovascular disease and health.
Cardiol Rev. 17:283–286. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Tycinska AM, Lisowska A, Musial WJ and
Sobkowicz B: Apelin in acute myocardial infarction and heart
failure induced by ischemia. Clin Chim Acta. 413:406–410. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Ren X and Zhang Z: Effect of Apelin-13 on
glucose deprivation-induced rat myocardial cell autophagy.
Guangdong Yi Xue. 32:3176–3177. 2011.(In Chinese).
|
|
8.
|
Zhu S, Sun F, Li W, et al: Apelin
stimulates glucose uptake through the PI3K/Akt pathway and improves
insulin resistance in 3T3-L1 adipocytes. Mol Cell Biochem.
353:305–313. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Tatemoto K, Hosoya M, Habata Y, et al:
Isolation and characterization of a novel endogenous peptide ligand
for the human APJ receptor. Biochem Biophys Res Commun.
251:471–476. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Zhang Z, Yu B and Tao GZ: Apelin protects
against cardiomyocyte apoptosis induced by glucose deprivation.
Chin Med J (Engl). 122:2360–2365. 2009.PubMed/NCBI
|
|
11.
|
Xin Y, Xu X and Huang Y: Isolation,
primary culture and identification of cardiac fibroblasts and
cardiac myocytes of neonatal mouse. Xinjiang Yi Xue Yuan Xue Bao.
28:541–547. 2011.(In Chinese).
|
|
12.
|
Folli F, Saad MJ, Backer JM and Kahn CR:
Insulin stimulation of phosphatidylinositol 3-kinase activity and
association with insulin receptor substrate 1 in liver and muscle
of the intact rat. J Biol Chem. 267:22171–22177. 1992.PubMed/NCBI
|
|
13.
|
Wohlgemuth SE, Seo AY, Marzetti E, Lees HA
and Leeuwenburgh C: Skeletal muscle autophagy and apoptosis during
aging: effects of calorie restriction and life-long exercise. Exp
Gerontol. 45:138–148. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Tatemoto K: Search for an endogenous
ligand of the orphan G protein-coupled receptor - discovery of
apelin, a novel biologically active peptide. Nihon Rinsho.
58:737–746. 2000.(In Japanese).
|
|
15.
|
Galanth C, Hus-Citharel A, Li B and
Llorens-Cortès C: Apelin in the control of body fluid homeostasis
and cardiovascular functions. Curr Pharm Des. 18:789–798. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Tempel D, de Boer M, van Deel ED, et al:
Apelin enhances cardiac neovascularization after myocardial
infarction by recruiting aplnr+ circulating cells. Circ Res.
111:585–598. 2012.PubMed/NCBI
|
|
17.
|
Balasis ME, Forinash KD, Chen YA, et al:
Combination of farnesyltransferase and Akt inhibitors is
synergistic in breast cancer cells and causes significant breast
tumor regression in ErbB2 transgenic mice. Clin Cancer Res.
17:2852–2862. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Dieterle A, Orth R, Daubrawa M, et al: The
Akt inhibitor triciribine sensitizes prostate carcinoma cells to
TRAIL-induced apoptosis. Int J Cancer. 125:932–941. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Chen Y, Azad MB and Gibson SB: Methods for
detecting autophagy and determining autophagy-induced cell death.
Can J Physiol Pharmacol. 88:285–295. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Martinet W, De Meyer GR, Andries L, Herman
AG and Kockx MM: In situ detection of starvation-induced autophagy.
J Histochem Cytochem. 54:85–96. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Arad M, Seidman CE and Seidman JG:
AMP-activated protein kinase in the heart: role during health and
disease. Circ Res. 100:474–488. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Hardie DG: AMP-activated/SNF1 protein
kinases: conserved guardians of cellular energy. Nat Rev Mol Cell
Biol. 8:774–785. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Yu L, McPhee CK, Zheng L, et al:
Termination of autophagy and reformation of lysosomes regulated by
mTOR. Nature. 465:942–946. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Gottlieb RA, Finley KD and Mentzer RM Jr:
Cardioprotection requires taking out the trash. Basic Res Cardiol.
104:169–180. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Cuervo AM: Autophagy: in sickness and in
health. Trends Cell Biol. 14:70–77. 2004. View Article : Google Scholar : PubMed/NCBI
|