Introduction
Angiotensin-II receptor blockers (ARBs) are
representative anti-hypertensive drugs. They are effective in left
ventricular dysfunction and have reno-protective effects in the
reduction of proteinuria and atrial reverse-remodeling effects as
electrophysiological stabilizers (1). Studies concerning the
reverse-remodeling effect are relatively rare and a number do not
agree with each other in certain regards. Atrial fibrillation has a
clear association with atrial remodeling. Angiotensin-converting
enzyme lowers bradykinin levels, which increases fibrosis and
collagen deposition in atrial tissue (2). Goette et al (3) demonstrated that the decrease in
bradykinin levels is due to angiotensin-converting enzyme-dependent
extracellular signal-regulated kinases (Erk1/Erk2). The activated
angiotensin-II receptor activates mitogen-activated protein kinase.
As a result of histological changes, atrial enlargement occurs and
the atrium may be a substrate of atrial fibrillation (4). Electrical remodeling is another
mechanism of atrial remodeling. Angiotensin activation induces
myocyte calcium overload, which causes prolongation of the
refractory period, depolarization-delay and an increase in
automaticity. This also creates substrates for atrial fibrillation.
In a similar manner, the renin-angiotensin-aldosterone system has a
marked correlation with atrial remodeling which is closely
associated with the development and maintenance of atrial
fibrillation.
It is well known that hemodynamic overload in the
atrium is one of the most important factors for atrial fibrosis
(5) and structural changes, such
as atrial enlargement and fibrosis, are associated with atrial
dysfunction (6,7). Li et al (7) reported that electrical inhomogeneity
due to atrial fibrosis had a significant role in atrial
fibrillation induction and maintenance in a canine heart failure
model. Gap junctions build signal propagation channels to
neighboring myocytes. The geometrical distortion creates an
inhomogeneous electrophysiological network and the subsequent
consolidation of atrial fibrillation. These histological changes
accompany atrial fibrosis and the expression of the proteins
connexin 43 and connexin 40 (8).
However, these results showed wide variations, with opposing
results, even within the same models. Consequently, these studies
were unable to define a definite causal relationship between
arrhythmia and connexin (9).
Losartan has been reported to prevent left
ventricular systolic dysfunction in a rat myocardial infarction
model (10). The
renin-angiotensin-aldosterone system is associated with the
pathological mechanism of atrial fibrillation. Kumagai et al
(1) reported that this ARB is able
to prevent atrial electrical remodeling in canine models, in which
rapid atrial pacing was used to induce atrial fibrillation.
The present study was performed to evaluate the
reverse remodeling effect of an ARB by studying echocardiographic
results, expression of cardiac connexin and atrial fibrillation
inducibility in a heart failure model.
Materials and methods
Experimental animals and reagents
Male Sprague-Dawley rats (Jung-Ang Lab Animal Inc.,
Seoul, Korea) weighing ∼260 g were used in the experiments. Each
rat was isolated and housed individually in a ventilated
microisolator-cage rack system, in which day and night were set at
12 hourly intervals. The rats were fed with standard rodent
provender and distilled water. The experiments were performed in
accordance with the guidelines of the Chonnam National University
Hospital Animal Subject Institutional Review Board (Gwangju,
Korea). The amount of provender for each subject was estimated for
5 days of adaptation. The ground medicine was mixed into the ground
provender, which was congealed for one day at 35°C.
A total of 38 rats were divided into sham (n=8),
heart failure (n=15) and heart failure-angiotensin-II receptor
blocker (ARB) (n=15) groups. Due to 14 rats not surviving surgery,
each group ultimately contained 8 rats. Losartan potassium (30
mg/kg) was administered to the heart failure-ARB group for 4 weeks
(10).
Heart failure model
The ischemic heart failure model was induced using
the method reported by Johns and Olson (11). Anesthesia was induced using an
intramuscular injection of ketamine (50 mg/kg) and xylazine (5
mg/kg). Artificial ventilation was performed with a small animal
mechanical ventilator (Harvard Apparatus, Holliston, MA, USA)
following tracheal intubation. The rat heart was exposed after left
thoracotomy in a supine position. Myocardial infarction was induced
via ligation of the left anterior descending coronary artery in the
heart failure and heart failure-ARB groups. The chest was closed
immediately. In the sham group, only the thoracotomy and closure
were performed. The rats were allowed to recover for 4 weeks.
Echocardiography
Echocardiography was performed under anesthesia
using an intramuscular injection of ketamine (50 mg/kg) and
xylazine (5 mg/kg) prior to surgery and 4 weeks after surgery. A 15
MHz linear probe (Sequoia C512; Acuson-Siemens, Mountain View, CA,
USA) was used to perform transthoracic echocardiography. The left
ventricular end systolic diameter (LVESd), left ventricular end
diastolic diameter (LVEDd), left atrial diameter (LAD) in the
parasternal long axis view and left ventricular ejection fraction
(LVEF) were measured (Fig. 1A and
B). All echocardiograpies were performed in blinded state.
Induction of atrial fibrillation
After follow-up echocardiography, atrial
fibrillation induction was performed under anesthesia. Following a
10-min equilibration period after anesthesia, a 4F electrode
catheter (St. Jude Medical, Inc., St. Paul, MN, USA) was inserted
into the esophagus and positioned to ensure constant atrial
capture. A BLOOM 215B DTU stimulator, Prucka recording system,
Grass S-8800 2-channel stimulator (Grasstech, Quincy, MA, USA),
MP150 and ECG100C ×2 (Biopac, Goleta, CA, USA) were used for
stimulation and recording. Atrial pacing was performed at twice the
diastolic threshold (10 V, 500 Ω, 20 mA), using 2 poles on the
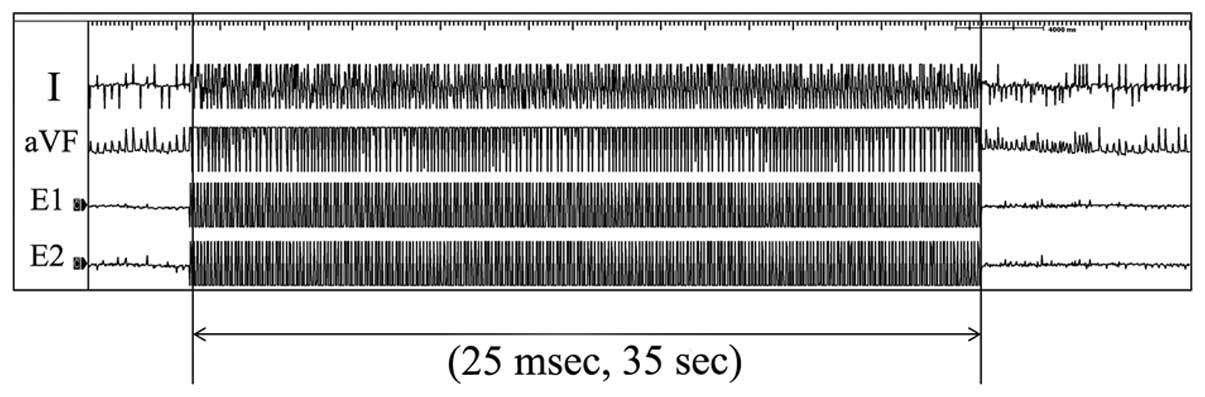
pacing catheter (pulse width, 5 msec). To induce atrial
fibrillation, burst stimulation at a frequency of 25 msec intervals
was performed for 35 sec in each of the rats (Fig. 2). The rats were allowed 5 min of
recovery in the sinus rhythm between stimulations for respiratory
and circulatory recovery. The induction test was performed 5 times
in each rat. Atrial fibrillation inducibility and duration were
measured.
Histological analysis
Following the atrial fibrillation induction test,
all rats were sacrificed. For 5 of the 8 rats in each group, the
hearts were fixed using 10% formaldehyde perfusion into the
abdominal aorta. Following formaldehyde perfusion, the hearts were
excised immediately. The excised hearts were fixed in 10%
formaldehyde. Hematoxylin and eosin (HE) and Masson’s trichrome
staining, as well as immunohistochemical staining for connexin 43
(using rabbit polyclonal anti-C×43 antibody, 1/100; Zymed
Laboratories, CliniSciences, Montrouge, France) were performed.
Fibrosis and connexin expression were quantified using automatic
computer graphics software (Image J®).
Western blot analysis
For the western blot analysis of connexin 43, the
two atria from 3 of the 8 rats in each group were excised following
sacrifice via CO2 inspiration. Western blot analysis was
performed on lyates from frozen tissue samples. NP buffer (1%) was
used as the lysis buffer. Protein extracts (50 μg) were
used. Connexin 43 was detected using rabbit polyclonal anti-C×43
antibody (1/100, Zymed Laboratories).
Statistical analysis
Statistical analysis was performed using the SPSS
15.0 software for Windows®. Mann-Whitney tests were used
for comparisons between the groups. Intra-group analysis was
performed using paired t-tests. P<0.05 was considered to
indicate a statistically significant difference.
Results
Heart failure model
The rats were divided into sham (n=8), heart failure
(n=15) and heart failure-ARB groups (n=15). After left anterior
descending artery ligation, 14 rats died. A total of 8 rats were
successfully maintained in each group for 4 weeks following
surgery. There was no significant difference in weight gain between
the heart failure and heart failure-ARB groups for all 4 weeks
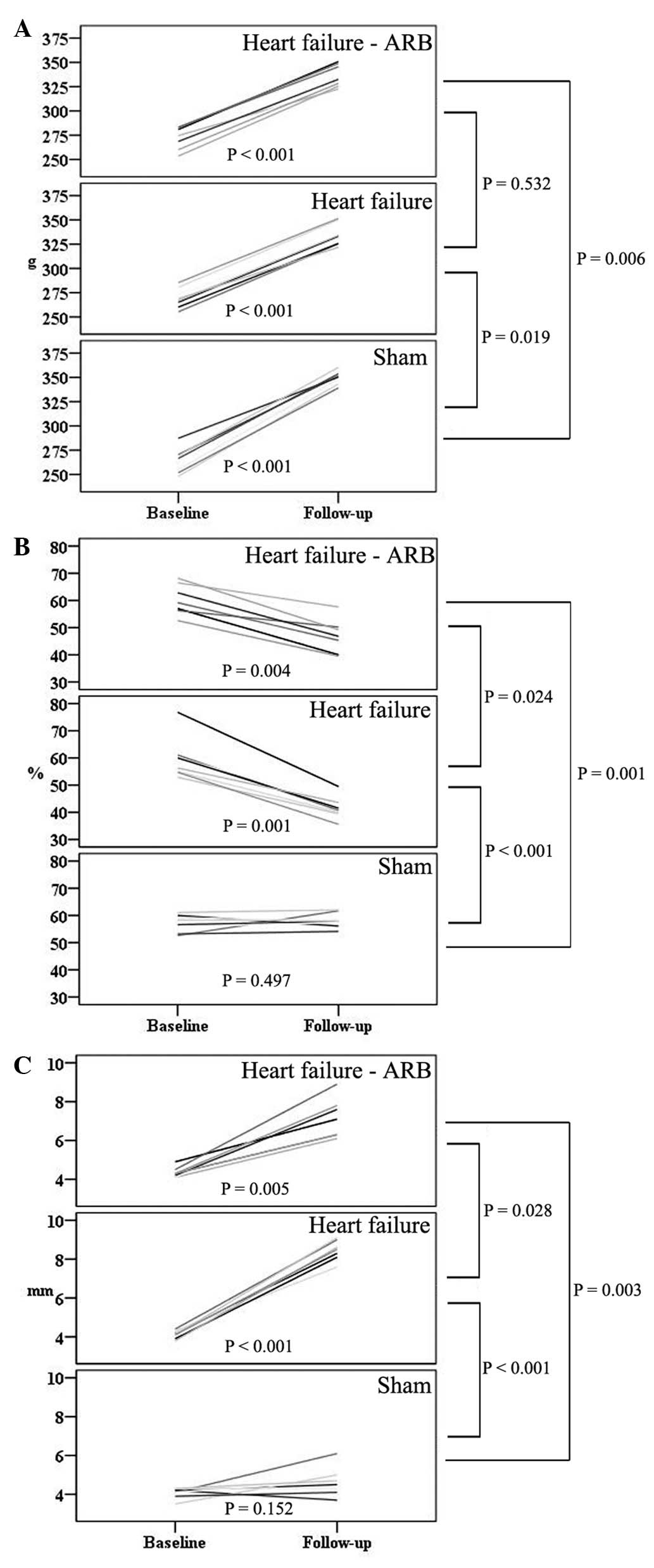
(Fig. 3A).
Echocardiography
The baseline LVEFs in the sham, heart failure and
heart failure-ARB groups were 56.6±2.78, 62.6±8.28 and 59.3±4.13%,
respectively. The follow-up LVEFs were 58.2±2.08, 43.0±3.82 and
47.4±6.76%, respectively. The reduction in the LVEF of the heart
failure-ARB group was significantly smaller than that in the heart
failure group (P= 0.024; Fig. 3B).
The baseline LADs in the sham, heart failure and heart failure-ARB
groups were 4.0±0.29, 4.2±0.18 and 4.3±0.33 mm, respectively. The
follow-up LADs were 4.8±0.87, 8.5±0.39 and 7.3±1.19 mm,
respectively. The increase in the LAD of the heart failure-ARB
group was significantly smaller than that in the heart failure
group (P=0.028; Fig. 3C). The size
of the left atrial appendage in the heart failure-ARB group was
smaller than that in the heart failure group.
Atrial fibrillation inducibility
The atrioventricular block cycle lengths in the
sham, heart failure and heart failure-ARB groups were 107±8.3,
124±11.4 and 116±11.4 msec, respectively (Fig. 4A). The inducibility of atrial
fibrillation in the sham, heart failure and heart failure-ARB
groups was 80±14, 91±11 and 92±10%, respectively. The durations of
induced atrial fibrillation in the sham, heart failure and heart
failure-ARB groups were 5213±2840.2, 12960±5088.8 and 16790±7112.8
msec, respectively (Fig. 4B).
Histology, immunohistochemical staining
and western blotting
The gross sizes of the excised hearts in the heart
failure and heart failure-ARB groups were larger than those in the
sham group. The area of infarction was pale and thin (Fig. 5).
In the HE staining (Fig. 5) of the heart failure group,
significant dilatation of the atria and ventricles was evident, as
was marked thinning of the wall of the infarcted area. The HE
staining showed the destructive features of the myocardial
contraction unit, abnormal sarcomeres and clear interstitial
fibrosis. By contrast, the atrial myocytes were preserved
relatively intact in the myocardial contraction unit with normal
sarcomeres in the heart failure-ARB group (Fig. 6B and D).
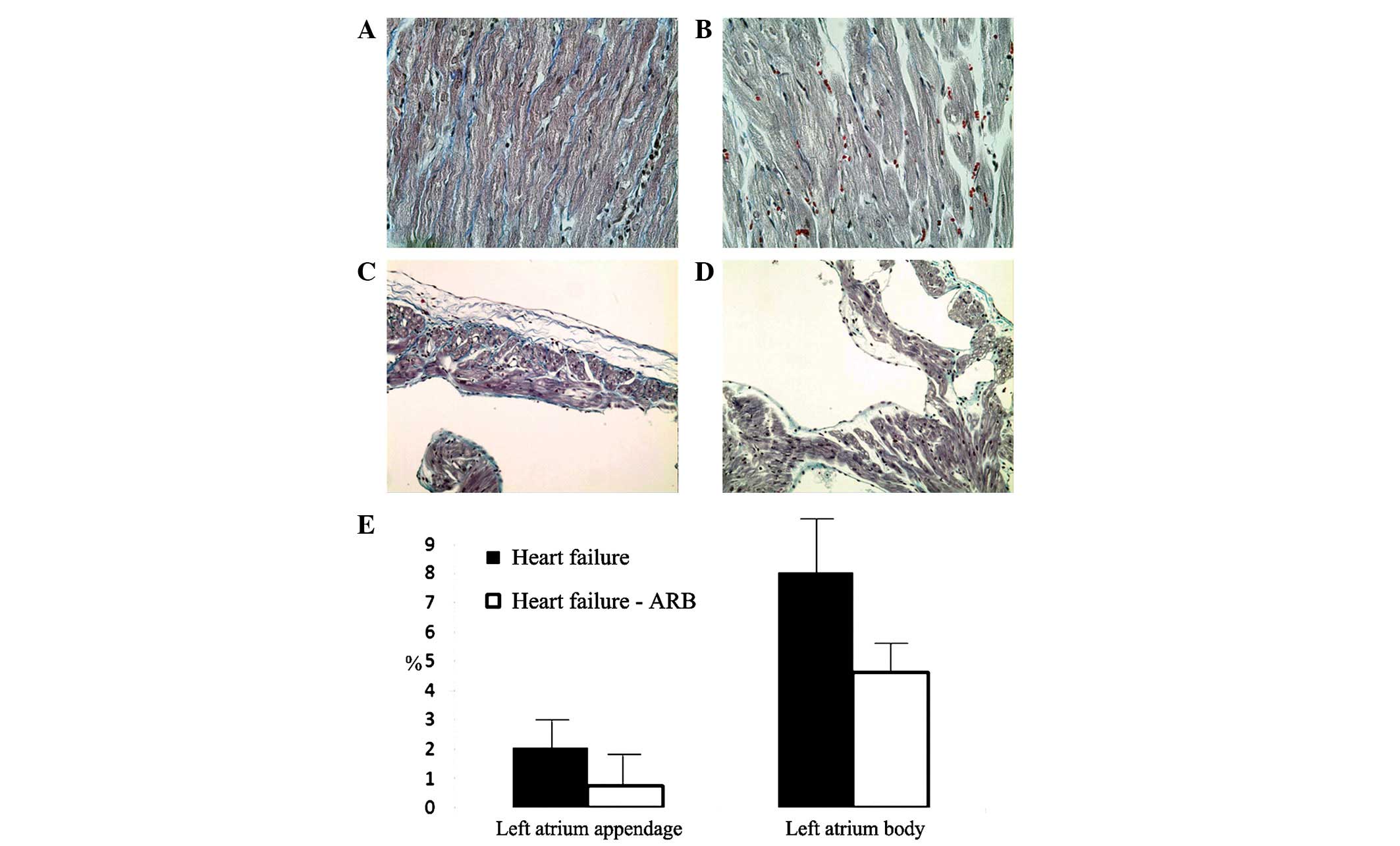
The Masson’s trichrome staining revealed severe
fibrosis of the infarcted ventricular area and more fibrosis of the
atria in the heart failure group (Fig.
6A and B). The left atrial appendage areas of fibrosis in the
heart failure and heart failure-ARB groups were 2.074 and 0.761%,
respectively, according to Masson’s trichrome staining. The left
atrial body areas of fibrosis in the heart failure and heart
failure-ARB groups were 8.009 and 4.611%, respectively (Fig. 6E).
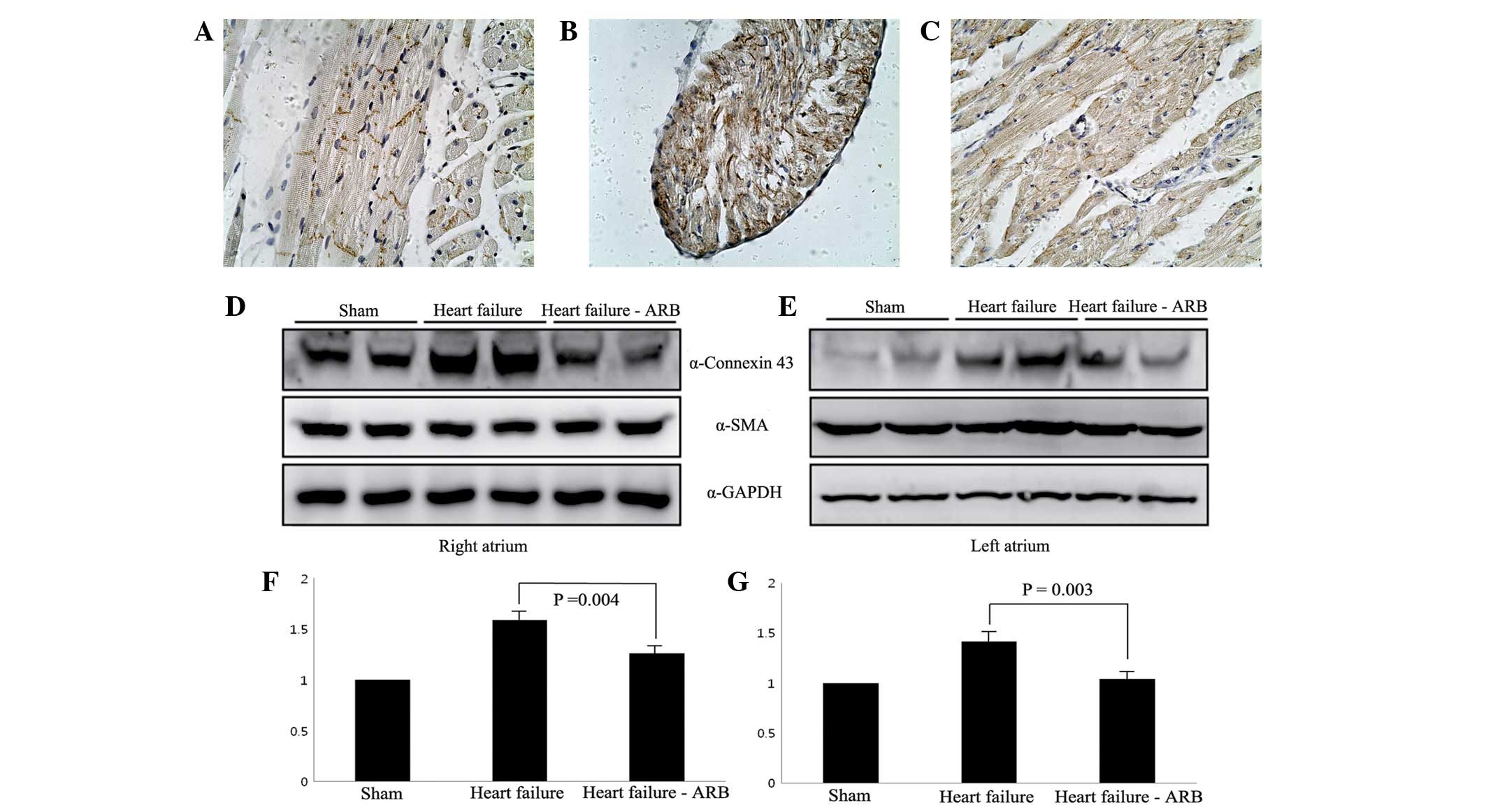
The immunohistochemical staining revealed that
connexin 43 protein was present in the intercalated discs in the
sham group (Fig. 7A). Abundant
plasmalemmal distribution of connexin 43 was observed in the heart
failure group, while connexin 43 staining was not observed in the
intercalated discs (Fig. 7B). In
the heart failure-ARB group, a small amount of connexin 43 protein
staining was observed in the intercalated discs (Fig. 7C).
Western blotting for connexin 43 showed that the
connexin 43 bands of the heart failure group were more marked than
those of the sham group and the heart failure-ARB group had weaker
connexin 43 bands compared with the heart failure group (Fig. 7D–G).
Discussion
Atrial fibrillation is highly associated with atrial
remodeling. It is a relatively common arrhythmia, which may be
paroxysmal and persistent. It develops with or without underlying
heart diseases. Atrial fibrillation has clinical significance due
to probable hemodynamic instabilities and thromboembolic events.
The multiple-wavelet hypothesis has been the accepted theory for
the mechanism of atrial fibrillation, whereby multiple wavelets
conflict and collide, then create small wave-lets repeatedly
(12).
Atrial fibrillation is induced by initial abnormal
electrical activity and is maintained in the atrium with irregular
reentry. These irregular electrical reentries are associated with
structural and electrical remodeling. The ion channel-like protein
connexin appears to be important in remodeling (13). This remodeling is frequent in
myocardial infarction. The reason why atrial fibrillation develops
frequently in old ventricular myocardial infarction is not fully
understood (14). The current
understanding is that the old ventricular myocardial infarction
causes atrial structural and electrical remodeling, which act as
substrates for atrial fibrillation. Old ventricular myocardial
infarctions may induce electrical inhomogeneity and sympathetic
over-distribution in the atrium without atrial infarction. These
conditions appear to be sufficient to induce atrial fibrillation
(14). With regard to this view,
the rat ischemic heart failure model was induced by myocardial
infarction via left coronary artery ligation in the present study.
This was to create similar substrates to the theoretical conditions
for atrial fibrillation maintenance. In humans, the dilatation of
the infarcted ventricle develops alongside hypertrophy of the
non-infarcted myocardium within months or years (15). In the present study, the thinning
of the infarcted area and hypertrophy of the non-infarcted area
developed in 28 days. The rapidity of these changes appears to be
due to the high left ventricular systolic pressure, similar to that
of humans, and the difference in myocardial thickness between
humans and rats.
Atrial systolic function may not be recovered even
when cardioversion is performed chemically or electrically. We
suggest that the cause of this phenomenon is that structural and
electrical remodeling of the atrium may not be reversed quickly.
The fact that persistent atrial fibrillation induces chronic
hibernation-like changes in the atrial myocardium may explain the
slow reversal (6). Inhomogeneity
of the gap junction is one of the structural remodelings in chronic
atrial fibrillation. We propose that gap junction inhomogeneity is
the result of atrial dilatation and fibrillation itself so that
atrial fibrillation begets further atrial fibrillation.
Consequently, atrial dilatation and fibrillation may be induced in
ventricular failure. The remodeling of gap junctions may affect the
conduction properties and subsequently contribute to the
maintenance of atrial fibrillation. It is not clear how significant
a role the remodeling has in the induction of atrial fibrillation
(13). We hypothesize that gap
junction remodeling is able to reduce conduction velocity in the
atrium.
Thomas et al (16) reported that a 38% reduction in
ventricular conduction velocity was observed when connexin 43
levels were reduced by 50%, indicating that connexin 43 has a
significant role in electrical conduction between ventricular
myocytes. By contrast, the electrical conduction velocity in the
atrium was not affected by the reduction in connexin 43 levels but
was affected by reductions in connexin 40 levels. Consequently, the
authors suggested that connexin 43 is ventricle-specific and
connexin 40 is atrium-specific. However, since their report, it has
been recognized that connexin 40 and connexin 43 are associated
with atrial fibrillation.
In the present study, connexin 43 exhibited an
abundant plasmalemmal distribution in the heart failure group and a
smaller connexin 43-stained area in the heart failure-ARB group.
Additionally, western blotting showed that the connexin 43 bands of
the heart failure group were more marked than those of the heart
failure-ARB group. We hypothesize that the change in the quantity
and distribution of connexin 43 is the result of changes in
dephosphorylated connexin 43 levels in myocardial infarction or
ischemic heart failure. Connexin 43 has multiple electrophoretic
isoforms, including a non-phosphorylated and phosphorylated form.
Phosphorylation regulates the assembly, degradation and gating of
connexin 43 (17). During heart
failure, large amounts of connexin 43 are hypophosphorylated and
contribute to reducing the gap junction functions (18), causing electrical uncoupling, which
promotes arrhythmogenicity. It appears that ARBs inhibit the
dephosphorylation of connexin 43, thus reducing the levels of
non-functional gap junctions in the rat heart failure model, and
that this phosphorylation is important in gap junction coupling.
The phosphorylation of connexin 43 is regulated by several protein
kinases, including mitogen-activated protein kinase. Notably, the
mechanism by which ARBs inhibit atrial fibrosis is due to the
effect of this kinase. ARBs bind to the angiotensin-II receptor and
inhibit mitogen-activated protein kinase; this inhibition, in turn,
inhibits collagen accumulation, fibroblast proliferation and
apoptosis (19). Also, the
blocking of the angiotensin-II receptor inhibits electrical
remodeling, by inhibiting calcium overload in the atrium.
Certain studies have reported the changes in
connexin quantity or arrangement of various species (20) and the results have been extremely
variable. The studies have reported conflicting results, even
within the same models. We considered that this variability of
results was due to the use of different species. Consequently, we
reviewed several studies which used rat models. Hoyano et al
(21) reported the downregulation
of connexin 43 in an autoimmune myocarditis model while Reil et
al (22) reported no change in
connexin 43 levels in an aldosterone infusion model. Additionally,
Rucker-Martin et al (23)
reported the upregulation of connexin 43 in a heart failure
model.
The results of the present study are compatible with
the results of Rucker-Martin et al. Regardless of the
connexin results, it is important for physicians to know whether
ARBs have potential as an upstream therapy in atrial fibrillation.
However, in the present induction study of atrial fibrillation, it
was not possible to not identify a significant difference between
heart failure and heart failure-ARB groups. Certain studies have
reported that candesartan is able to prevent atrial fibrillation,
and thus inhibit atrial structural remodeling (1). In a canine model study, the
inhibition of the angiotensin-II receptor prevented the shortening
of the atrial effective refractory period during rapid atrial
pacing. This evidence demonstrates the association between
angiotension II and atrial electrical characteristics (24). The 2010 European Society of
Cardiology guidelines recommend that angiotensin converting enzyme
inhibitors and angiotensin-II receptor blockers should be
considered for the prevention of new-onset atrial fibrillation in
patients with heart failure and reduced ejection fractions
(25). The guidelines also suggest
that there is now little reason to consider the use of upstream
therapy for the prevention of atrial fibrillation recurrence in
patients with little or no underlying heart disease (26).
The present study had several limitations. Losartan
potassium is lipid-soluble, so if it is dissolved in water, an
uneven distribution may be expected. However, ground medicine and
provender were mixed for each rat individually. Also, whether the
rats had eaten all their provender was checked daily.
Echocardiography is a sonographer-dependent
technique. As such, there is a probability of inter-observer
variation or open labeling. In the present study, all
echocardiography was performed in a blinded state to attenuate such
concerns.
The fact that an angiotensin converting enzyme
inhibitor is able to inhibit left ventricular dilatation in
patients with ventricular dysfunction following myocardial
infarction has been well established by animal experiments and
clinical trials (27,28). In the present experiment, the
atrial response was evaluated in a left ventricular dysfunction
model. Consequently, the atrial response may have been a secondary
result from the improvement of left ventricle function. The
prolonged inhibition of angiotensin converting enzyme and
angiotensin-II receptor is able to reduce ventricular hypertrophy,
recover coronary artery resistance and reduce ventricular fibrosis
(29). Thus, in the present study,
the angiotensin-II receptor blocking effect may have been the
result of inhibition of not only atrial remodeling itself, but also
left ventricular remodeling. In a heart failure model, it may be
difficult to differentiate between these two effects. However, they
may be differentiated in transgenic animal models, such as connexin
knockouts or overexpression of Rho-A, Junctin, CRE modulator,
angiotensin converting enzyme, constitutive TGF-b1 and Kir2.1.
However, even in such models, there have been a number of
contradictory results.
In conclusion, ARBs have a small preventive effect
in atrial fibrillation if there is no underlying heart disease such
as heart failure. However, ARBs also have an atrial reverse
remodeling effect. It was revealed that, in a rat ischemic heart
failure model, the ARB losartan had structural and histological
atrial reverse-remodeling effects. It should be kept in mind that
atrial fibrillation is a result of complex molecular, mechanical
and electrical remodeling. Thus, we suggest that gap junctions may
not be surrogate targets for atrial remodeling. The role of ARBs as
electrical stabilizers requires further study.
Acknowledgements
This study was supported by a grant
from the Chonnam National University Hospital Biomedical Research
Institute (CRI 11 083-32).
References
|
1
|
Kumagai K, Nakashima H, Urata H, Gondo N,
Arakawa K and Saku K: Effects of angiotensin II type 1 receptor
antagonist on electrical and structural remodeling in atrial
fibrillation. J Am Coll Cardiol. 41:2197–2204. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wollert KC and Drexler H: The
kallikrein-kinin system in post-myocardial infarction cardiac
remodeling. Am J Cardiol. 80:158A–161A. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Goette A, Staack T, Röcken C, et al:
Increased expression of extracellular signal-regulated kinase and
angiotensin-converting enzyme in human atria during atrial
fibrillation. J Am Coll Cardiol. 35:1669–1677. 2000. View Article : Google Scholar
|
|
4
|
Novo G, Guttilla D, Fazio G, Cooper D and
Novo S: The role of the renin-angiotensin system in atrial
fibrillation and the therapeutic effects of ACE-Is and ARBS. Br J
Clin Pharmacol. 66:345–351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Boixel C, Fontaine V, Rücker-Martin C, et
al: Fibrosis of the left atria during progression of heart failure
is associated with increased matrix metalloproteinases in the rat.
J Am Coll Cardiol. 42:336–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ausma J, Wijffels M, Thoné F, Wouters L,
Allessie M and Borgers M: Structural changes of atrial myocardium
due to sustained atrial fibrillation in the goat. Circulation.
96:3157–3163. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li D, Fareh S, Leung TK and Nattel S:
Promotion of atrial fibrillation by heart failure in dogs: atrial
remodeling of a different sort. Circulation. 100:87–95. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dhein S: Role of connexins in atrial
fibrillation. Adv Cardiol. 42:161–174. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gollob MH: Cardiac connexins as candidate
genes for idiopathic atrial fibrillation. Curr Opin Cardiol.
21:155–158. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Daniëls MC, Keller RS and de Tombe PP:
Losartan prevents contractile dysfunction in rat myocardium after
left ventricular myocardial infarction. Am J Physiol Heart Circ
Physiol. 281:H2150–H2158. 2001.PubMed/NCBI
|
|
11
|
Johns TN and Olson BJ: Experimental
myocardial infarction. I. A method of coronary occlusion in small
animals. Ann Surg. 140:675–682. 1954. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jalife J: Experimental and clinical AF
mechanisms: bridging the divide. J Interv Card Electrophysiol.
9:85–92. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Takeuchi S, Akita T, Takagishi Y, et al:
Disorganization of gap junction distribution in dilated atria of
patients with chronic atrial fibrillation. Circ J. 70:575–582.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miyauchi Y, Zhou SM, Okuyama Y, et al:
Altered atrial electrical restitution and heterogeneous sympathetic
hyperinnervation in hearts with chronic left ventricular myocardial
infarction: implications for atrial fibrillation. Circulation.
108:360–366. 2003. View Article : Google Scholar
|
|
15
|
Mitchell GF, Lamas GA, Vaughan DE and
Pfeffer MA: Left ventricular remodeling in the year after first
anterior myocardial infarction: a quantitative analysis of
contractile segment lengths and ventricular shape. J Am Coll
Cardiol. 19:1136–1144. 1992.
|
|
16
|
Thomas SA, Schuessler RB, Berul CI, et al:
Disparate effects of deficient expression of connexin43 on atrial
and ventricular conduction: evidence for chamber-specific molecular
determinants of conduction. Circulation. 97:686–691. 1998.
View Article : Google Scholar
|
|
17
|
Solan JL and Lampe PD: Connexin
phosphorylation as a regulatory event linked to gap junction
channel assembly. Biochim Biophys Acta. 1711:154–163. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ai X and Pogwizd SM: Connexin 43
downregulation and dephosphorylation in nonischemic heart failure
is associated with enhanced colocalized protein phosphatase type
2A. Circ Res. 96:54–63. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Savelieva I and Camm AJ: Atrial
fibrillation and heart failure: natural history and pharmacological
treatment. Europace. 5(Suppl 1): S5–S19. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kato T, Iwasaki YK and Nattel S: Connexins
and atrial fibrillation: filling in the gaps. Circulation.
125:203–206. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hoyano M, Ito M, Kimura S, et al:
Inducibility of atrial fibrillation depends not on inflammation but
on atrial structural remodeling in rat experimental autoimmune
myocarditis. Cardiovasc Pathol. 19:e149–157. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Reil JC, Hohl M, Selejan S, et al:
Aldosterone promotes atrial fibrillation. Eur Heart J.
33:2098–2108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rucker-Martin C, Milliez P, Tan S, et al:
Chronic hemodynamic overload of the atria is an important factor
for gap junction remodeling in human and rat hearts. Cardiovasc
Res. 72:69–79. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakashima H, Kumagai K, Urata H, Gondo N,
Ideishi M and Arakawa K: Angiotensin II antagonist prevents
electrical remodeling in atrial fibrillation. Circulation.
101:2612–2617. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
European Heart Rhythm Association;
European Association for Cardio-Thoracic Surgery; Camm AJ, Kirchhof
P, Lip GYH, et al: Guidelines for the management of atrial
fibrillation The Task Force for the Management of Atrial
Fibrillation of the European Society of Cardiology (ESC). Europace.
12:1360–1420. 2010. View Article : Google Scholar
|
|
26
|
Camm AJ, Lip GY, De Caterina R, et al:
2012 focused update of the ESC Guidelines for the management of
atrial fibrillation: an update of the 2010 ESC Guidelines for the
management of atrial fibrillation - developed with the special
contribution of the European Heart Rhythm Association. Europace.
14:1385–1413. 2012. View Article : Google Scholar
|
|
27
|
Nicolosi GL, Latini R, Marino P, et al:
The prognostic value of predischarge quantitative two-dimensional
echocardiographic measurements and the effects of early lisinopril
treatment on left ventricular structure and function after acute
myocardial infarction in the GISSI-3 trial. Gruppo Italiano per lo
Studio della Sopravvivenza nell’Infarto Miocardico. Eur Heart J.
17:1646–1656. 1996.
|
|
28
|
St John Sutton M, Pfeffer MA, Plappert T,
et al: Quantitative two-dimensional echocardiographic measurements
are major predictors of adverse cardiovascular events after acute
myocardial infarction. The protective effects of captopril.
Circulation. 89:68–75. 1994.
|
|
29
|
Schieffer B, Wirger A, Meybrunn M, et al:
Comparative effects of chronic angiotensin-converting enzyme
inhibition and angiotensin II type 1 receptor blockade on cardiac
remodeling after myocardial infarction in the rat. Circulation.
89:2273–2282. 1994. View Article : Google Scholar
|