Introduction
Renal cell carcinoma (RCC) is the most common type
of kidney cancer in adults. Following the occurrence of metastasis,
survival rates are very poor and the 5-year survival rate is ~20%
(1). RCC is resistant to
chemotherapy (2). At present,
treatment regimens using interferon (IFN)-α have been applied in
clinical practice to treat RCC, achieving therapeutic response
rates between 4 and 33% (3). A
previous study revealed that IFN-α mediates anticancer effects
indirectly by modulating immunomodulatory mechanisms or directly
through antiproliferative effects and inducing the differentiation
of cancer cells (4).
IFN-α exerts these effects by binding to cell
surface receptors and activating the Janus kinase (Jak) protein
family. Activated Jak1 and tyrosine kinase 2 phosphorylate signal
transducers and activators of transcription (STATs). Subsequently,
phospho-STATs translocate to the nucleus and interact with specific
regulatory elements to induce target gene transcription (5). RCC treatment has developed
significantly, as vascular endothelial growth factor (VEGF)
receptor tyrosine kinase inhibitors and drugs that inhibit
mammalian target of rapamycin (mTOR) signaling have become the
mainstay for the management of advanced RCC. These treatments have
improved progression-free survival and/or overall survival outcomes
(6). The mTOR pathway has been
reported to be central to cancer progression and rapamycin (RPM)
has been shown to suppress carcinogenesis by decreasing mTOR
activity (7). RPM may function by
stimulating the degradation of cyclin D1, which inhibits the G1 to
S-phase transition in the cell cycle (8). RPM also downregulates phospho-p70 S6
kinase (K), which is considered to be an indicator of the activated
mTOR pathway (9). The primary
substrate of p70 S6K, S6 ribosomal protein, has also been shown to
have an important role in determining cell size. Phosphorylation of
the eukaryotic translation initiation factor, 4E binding protein 1
(4E-BP1), by mTOR results in the activation of cap-dependent
translation of nuclear mRNAs by releasing the inhibition of the
eukaryotic translation initiation factor 4E (10). RPM has been shown to suppress the
growth of small cell lung cancer and pancreatic cancer cells
(11,12). In addition, mTOR inhibitors have
shown promising efficacy in early-stage trials in patients with
advanced RCC (13). A previous
study indicated that RPM may be of value to patients with RCC and
that the antitumor efficacy of RPM is achieved by cell-cycle arrest
and targeted reduction of VEGF-A and transforming growth factor-β1
(14). An additional study
revealed the synergistic effects of RPM and chemotherapeutic agents
against tumor cells; RPM was reported to increase the cytotoxicity
of cisplatin by sensitizing human promyelocytic leukemia and
ovarian cancer cells to the drug, thereby inducing apoptosis
(15). However, receptor tyrosine
kinase inhibitors only demonstrate additive effects in combination
with RPM in the treatment of prostate cancer (16). A previous study indicated that
IFN-α suppresses the phosphoinositide 3 kinase and mTOR signaling
pathways (17). Furthermore,
combining RPM with other upstream mTOR inhibitors has been shown to
induce greater growth suppression in RCC compared with that
achieved by administering the drugs alone (18). However, whether IFN-α and RPM have
a synergistic effect against RCC remains unknown.
High frequency mutations or the loss of the two
copies of the Von Hippel-Lindau (VHL) tumor suppressor gene have
been observed in RCC (19). VHL
protein is the substrate recognition component of the E3 ligase
that ubiquitinates hypoxia-inducible transcription factors (HIFs),
including HIF-1α and -2α. VHL plays a pivotal role in the
downregulation of VEGF expression (20). Previous studies have indicated that
mTOR stimulates HIF expression and RPM exhibits antiangiogenic
activity that is associated with a reduction in the production of
HIF/VEGF (21,22). However, the effect of VHL activity
on the antiproliferative ability of IFN-α and RPM in RCC remains
unknown.
Materials and methods
Cell lines and agents
Three RCC cell lines, ACHN, NC65 and A498 (ATCC,
Rockefeller, MD, USA), were cultured in complete medium consisting
of RPMI-1640 (Gibco, Gaithersburg, MD, USA) supplemented with 25 mM
hydroxyethyl piperazineethanesulfonic acid, 2 mM L-glutamine, 1%
nonessential amino acids, 100 U/ml penicillin, 100 μg/ml
streptomycin and 10% heat-inactivated fetal bovine serum. Cell
lines were maintained as monolayers on 10-cm plastic dishes and
incubated in a humidified atmosphere containing 5% CO2
at 37°C. Intron A (recombinant IFN-α2b) was purchased from Merck
& Co, Inc. (Whitehouse Station, NJ, USA) and RPM was purchased
from Sigma-Aldrich (St. Louis, MO, USA).
WST-1 assays
Effects of IFN-α and/or RPM on the RCC cells were
determined using a WST-1 assay. Exponentially growing cells were
harvested and seeded at 2,000 cells/well in a 96-well microtiter
plate. After 4 h of incubation, Intron A (10, 50, 100, 200, 400 or
800 IU/ml), RPM (1, 5, 10, 15 or 20 μM), a combination of Intron A
(50 or 100 IU/ml) and RPM (1, 5, 10, 15 or 20 μM), or
penicillin/streptomycin medium (untreated control) were added. The
cells were then continuously incubated for 72 h. WST-1 (Roche
Diagnostics, Penzberg, Germany) at a volume of 10 μl was added to
each well and the cells were incubated for an additional 2 h.
Absorbance was measured with a microculture plate reader
(Immunoreader; Japan Intermed Co., Ltd., Tokyo, Japan) at 450 nm.
The percentage of cell cytotoxicity was calculated using the
following formula: % Cytotoxicity = [1 − (absorbance of
experimental − absorbance of blank)/(absorbance of untreated
control − absorbance of blank)] × 100.
siRNA transfection
A498 cells, which lack the wild-type VHL gene, were
stably transfected using Lipofectamine 2000 (Invitrogen Life
Technologies, Carlsbad, CA, USA) with an expression vector
containing the full-length cDNA for VHL or with a blank vector
without the VHL insert. Single colonies were selected with G418 and
confirmed by cell staining, western blot analysis and cDNA
sequencing. ACHN and A498 cells were seeded in complete medium
without antibiotics and were allowed to grow until 30–50%
confluence was reached. The cells were then transfected with siRNA
oligonucleotides or scrambled siRNA control using Lipofectamine
2000. Following incubation for 72 h, gene expression was confirmed
by western blot analysis. SignalSilence mTOR siRNA I was purchased
from Cell Signaling Technology, Inc. (Beverly, MA, USA). All RNAi
target sequences and oligonucleotide sets used in the study are
shown in Table I.
 | Table IPrimer and RNAi sequences. |
Table I
Primer and RNAi sequences.
| A. Primer
sequences |
|---|
|
|---|
| Gene | Forward primer,
5′-3′ | Reverse primer,
5′-3′ | Length of PCR
products, bp |
|---|
| VHL |
AGAAGGTGGTGGCATTTTTG |
AGCAGATGCCAATGCCTTCT | 124 |
| HIF-1α |
GAAAGCGCAAGTCCTCAAAG |
CATACGGTCTTTTGTCACTG | 126 |
| HIF-2α |
TTGATGTGGAAACGGATGAA |
CTCATGGGGTTTTGGGTGAA | 110 |
| GAPDH |
GAAGGTGAAGGTCGGAGTC |
GAAGATGGTGATGGGATTTC | 226 |
|
| B. RNAi
sequences |
|
| Gene | Sense
oligonucleotide, 5′-3′ | Antisense
oligonucleotide, 5′-3′ | Target gene sequence,
5′-3′ |
|
| VHL |
CGAGCGCGCGCGAAGACUACG |
UAGUCUUCGCGCGCGCUCGGU |
ACCGAGCGCGCGCGA
AGACTACG (98–120 bp) |
| Negative
control |
GUACCGCACGUCAUUCGUAUC |
UACGAAUGACGUGCGGUACGU | |
Reverse transcription polymerase chain
reaction (RT-PCR)
Total RNA was isolated using an RNeasy mini kit
(Qiagen, Frankfurt, Germany). A first-strand cDNA synthesis kit (GE
Healthcare, Little Chalfont, UK) was used for reverse
transcription. The PCR conditions were selected according to the
manufacturer’s instructions and the expected sizes of the PCR
products were confirmed by agarose gel electrophoresis. The PCR
products were quantified with a GeneAmp 5700 Sequence Detection
system (Applied Biosystems, Inc., Foster City, CA, USA). All primer
sets used in this study are shown in Table I.
Western blot analysis
The procedures were performed as previously
described (23). Protein was
extracted and the concentration was measured using a Bradford
dye-binding protein assay (Bio-Rad Laboratories, Inc., Richmond,
CA, USA). Subsequently, SDS polyacrylamide gel electrophoresis was
performed. Anti-β-actin monoclonal antibodies (Abcam, Cambridge,
UK) were used as an internal control. Other antibodies used in the
study were all purchased from Cell Signaling Technology, Inc..
These included mTOR (7C10)/phospho-mTOR (Ser2481), p70 S6K
(49D7)/phospho-p70 S6K (Thr421/Ser424), S6 ribosomal protein
(5G10)/phospho-S6 ribosomal protein (Ser240/244) (D68F8) XP and
4E-BP1 (53H11)/phospho-4E-BP1 (Thr70) rabbit monoclonal antibodies.
Immune complexes were detected using an enhanced chemiluminescence
system (GE Healthcare) combined with image analysis. The image
analysis software used was ImageJ (NIH, Bethesda, MD, USA).
Statistical analysis
All determinations were performed in triplicate and
the results are expressed as the mean ± SD. Statistical
significance was determined using the Student’s t-test and
P<0.05 was considered to indicate a statistically significant
difference. Synergy was evaluated by isobolographic analysis, as
described by Berenbaum (24). The
fractional inhibitory concentration of each agent was equal to the
IC50 dosage of the agent in combination divided by the
IC50 dosage of the agent when used alone. An additive,
synergistic or antagonistic combination was indicated by whether
the point lies on, below or above, respectively, the straight line
joining the dosages of the two drugs that when administered alone
produce the same effect as that of the combination, as based on the
isobolographic analysis.
Results
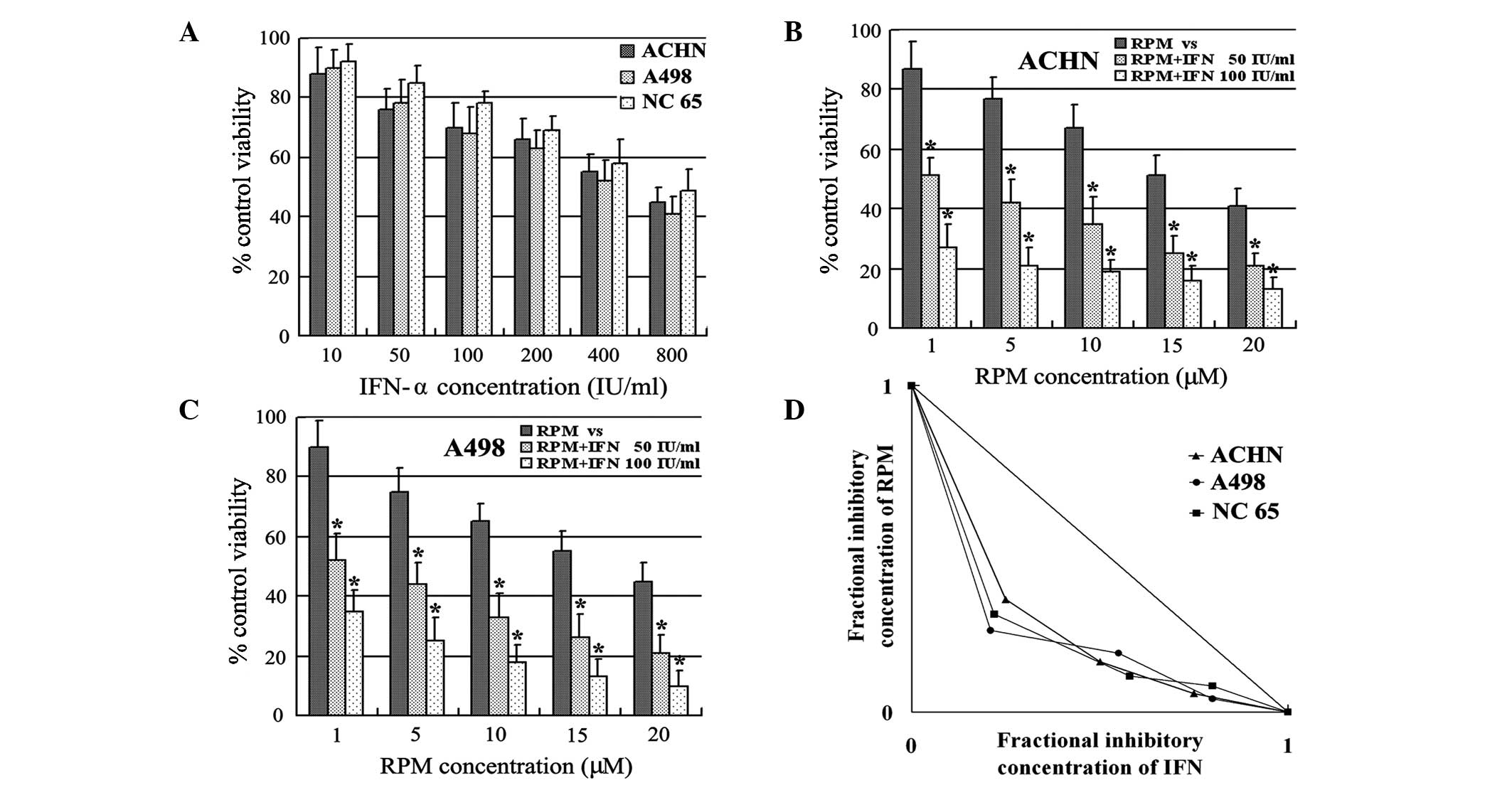
Synergistic growth suppression by IFN-α
and RPM
IFN-α administration caused dose-dependent cell
growth inhibition in the ACHN, A498 and NC65 RCC cell lines
(Fig. 1A). In addition, a
combination of IFN-α and RPM caused dose-dependent cell growth
inhibition in the RCC cell lines (Figs. 1B and C). IFN-α, at low
concentrations of 50 and 100 IU/ml, significantly increased the
susceptibility of the ACHN and A498 RCC cell lines to RPM (Fig. 1B and C). Combined treatment with
IFN-α and RPM resulted in synergistic growth suppression in all the
RCC cell lines examined in this study, as shown by isobolographic
analysis (Fig. 1D).
Suppression of mTOR pathway components by
IFN-α and/or RPM
To determine if the mTOR pathway is involved in the
synergistic effect of IFN-α and RPM against RCC cells,
phosphorylation of the mTOR pathway was evaluated following
stimulation with IFN-α and/or RPM. In the ACHN and A498 cell lines,
although 100 IU/ml IFN-α and/or 5 μM RPM did not affect the total
protein expression of mTOR, p70 S6K, S6 or 4E-BP1, it was observed
that IFN-α and RPM, alone or in combination, decreased the
phosphorylation of mTOR, p70 S6K, S6 and 4E-BP1, as determined by
western blot analysis (Fig. 2). In
addition, IFN-α significantly enhanced the RPM-induced suppression
of the mTOR pathway in these two cell lines. These results indicate
that the mTOR pathway plays a key role in the synergistic effect of
IFN-α and RPM against RCC cells.
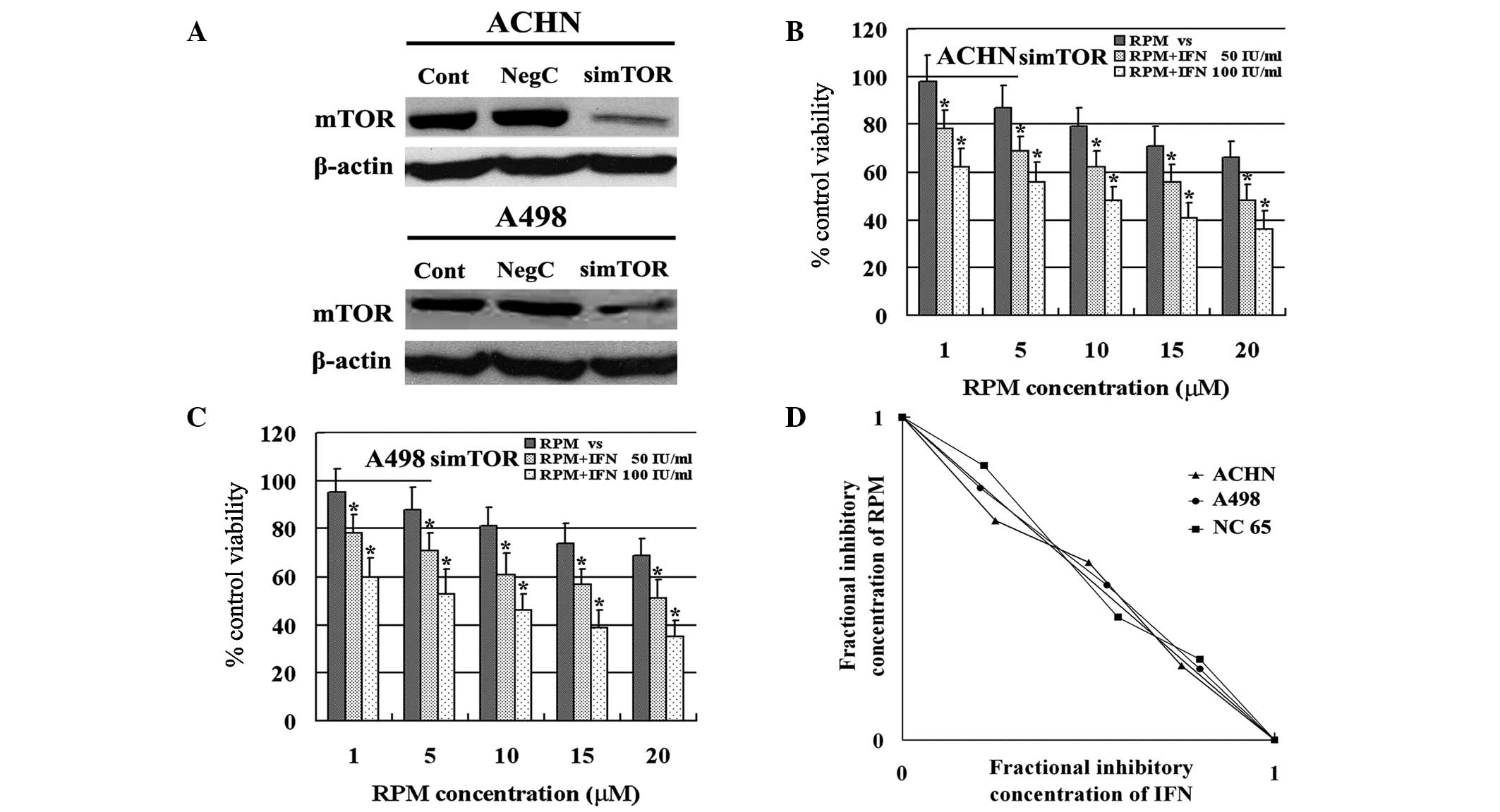
Effect of mTOR activity on the synergy of
IFN-α and RPM
The effect of mTOR activity on the synergy of IFN-α
and RPM against RCC was investigated. The expression of mTOR was
downregulated by RNAi and the results indicated that mTOR
expression was suppressed effectively in ACHN and A498 cells
(Fig. 3A). Regardless of mTOR
expression, IFN-α enhanced the susceptibility of RCC to RPM in ACHN
and A498 cells (Fig. 3B and C).
However, the synergy of the two agents was eliminated in these cell
lines, as an additive effect was indicated by isobolographic
analysis (Fig. 3D). These results
indicate that mTOR activity is necessary for the synergistic effect
of IFN-α and RPM against RCC cells.
Effect of VHL activity on the synergy of
IFN-α and RPM
The effect of VHL activity on the synergy of IFN-α
and RPM against RCC was also investigated. VHL expression was
downregulated in ACHN cells via RNAi and upregulated in A498 cells
by transfection with a VHL vector. VHL/HIF expression was confirmed
by western blot analysis and RT-PCR (Fig. 4A and B). Since VHL mediates HIF
levels via a post-translational mechanism, VHL did not alter the
mRNA expression levels of HIF-1α or -2α. Therefore, the results
indicate that regardless of VHL activity, IFN-α enhances the
susceptibility of RCC to RPM in all RCC cells tested in the study
(Fig. 4C and D). Thus, the synergy
of IFN-α and RPM does not depend on VHL activity in RCC cells.
Discussion
Although a number of clinical trials with various
combination chemotherapies have been performed in an attempt to
overcome the current limitations of advanced RCC treatment, few
have achieved favorable results or prognosis for patients with the
disease (25). Therefore, the
development of more effective combination chemotherapies for
advanced RCC is required.
Promising new combination chemotherapies are usually
identified simultaneously with advances in the understanding of
oncogenesis. RPM has previously been reported to have
immunosuppressant and anticancer effects on a large variety of
malignancies, including hepatocellular carcinoma and RCC (26,27).
In addition, RPM is well tolerated with minimal side-effects and
has shown anticancer activity in patients with androgen-independent
prostate cancer (28). Previous
studies concerning combination chemotherapy of RPM with
chemotherapeutic agents have been performed and combinations of RPM
with bevacizumab, sorafenib or 5-fluorouracil have been reported to
be promising therapeutic approaches for the treatment of
hepatocellular carcinoma (29–33).
IFN-α therapy is the most common approach for advanced RCC.
However, the synergistic effects of RPM and IFN-α against RCC
remain unclear. In the present study, the effect of a combination
of IFN-α and RPM on the inhibition of RCC cell growth was analyzed.
The results demonstrated that IFN-α and RPM caused dose-dependent
inhibition of proliferation and combined treatment with the two
agents resulted in synergistic growth suppression in all three RCC
cell lines examined. At present, IFN-α is widely administered for
the treatment of RCC. The observations of the present study
indicate that RPM may be an optimal agent to combine with IFN-α for
clinical application against RCC. Since chemotherapy is associated
with severe side-effects that usually limit the clinical
application, reducing the dosage of IFN-α or RPM is expected to
alleviate the associated side-effects but not decrease the
synergistic effects of these agents. Therefore, further clinical
trials are required to analyze the tolerance towards IFN-α and RPM
and to reveal the possible synergy of the two agents in patients
with RCC.
The underlying mechanism behind the synergy between
IFN-α and RPM in RCC cell lines was further investigated. The
molecular mechanism promoting the anticancer effects of RPM is
complex, as RPM suppresses the activity of mTOR and the
phosphorylation of its downstream effectors, p70S6K and 4E-BP1
(34). The mTOR pathway is
considered to be a central regulator in various malignant tumors.
There are two distinct functional mTOR complexes. Firstly, mTORC1
consists of mTOR and regulatory-associated protein of mTOR (Raptor)
and increases the phosphorylation of p70 S6K/4E-BP1. Secondly,
there is mTORC2, which consists of mTOR and rapamycin-insensitive
companion of mTOR and increases Akt (also known as protein kinase
B) phosphorylation (35). Akt
enhances cell growth by alleviating the tuberous sclerosis complex
1/2 suppression of mTOR, allowing the latter to function as part of
the mTOR/Raptor complex on p70 S6K and 4E-BP1 (36,37).
p70 S6K phosphorylates the S6 protein of the 40 S ribosomal subunit
(38), while translation repressor
protein 4E-BP1 inhibits translation by binding to the translation
initiation factor eIF4E (39,40).
Hyperphosphorylation of 4E-BP1 disrupts this interaction and
results in the activation of translation (41).
In the present study, the role of the mTOR pathway
in the synergistic effect of IFN-α and RPM against RCC was
investigated. The results indicated that IFN-α and RPM did not
affect protein expression in the mTOR pathway. However, each agent
individually decreased the phosphorylation of mTOR, p70 S6K, S6 and
4E-BP1 in RCC cells. In addition, IFN-α significantly enhanced the
RPM-induced suppression of the mTOR pathway, indicating that the
synergy between IFN-α and RPM against RCC depends on the
suppression of the mTOR pathway. The effect of mTOR activity on the
synergy of IFN-α and RPM was also analyzed. In RCC cells expressing
low levels of mTOR, the synergistic growth suppression of the two
agents was eliminated and an additive effect was observed. These
observations indicate that mTOR activity is important for the
synergy of IFN-α and RPM against RCC cells.
Inactivation of the VHL tumor suppressor protein is
a common event in clear cell RCC, which is the most common form of
kidney cancer. A previous study reported that, in response to
IFN-α, the exponential growth of wild-type VHL RCC cells was
inhibited more than that of VHL-null RCC cells. This observation
indicated that VHL inactivation may be involved in IFN-α resistance
and that combined immunotherapy with antiangiogenic drugs may be
beneficial for patients with a mutated VHL gene (42,43).
However, the effect of VHL activity on the synergy of IFN-α and RPM
against RCC is unknown. In the present study, A498 was used as the
VHL-null RCC cell line, while the other cell lines were wild type
for VHL. The results indicated that regardless of VHL activity,
synergy of IFN-α and RPM was observed in all RCC cells and, thus,
may be independent of VHL activity.
In conclusion, the present study demonstrated that
the mTOR pathway plays an important role in the synergistic effect
of IFN-α and RPM against RCC cells. The results indicate that
blocking the activity of mTOR may provide a novel treatment
strategy for patients with RCC. In addition, the suppression of RCC
cell growth by IFN-α and RPM may be more effective in RCC cells
with high mTOR activity.
References
|
1
|
Pantuck AJ, Zisman A and Belldegrun AS:
The changing natural history of renal cell carcinoma. J Urol.
166:1611–1623. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hartmann JT and Bokemeyer C: Chemotherapy
for renal cell carcinoma. Anticancer Res. 19:1541–1543.
1999.PubMed/NCBI
|
|
3
|
Hernberg M, Pyrhönen S and Muhonen T:
Regimens with or without interferon-alpha as treatment for
metastatic melanoma and renal cell carcinoma: an overview of
randomized trials. J Immunother. 22:145–154. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yanai Y, Horie S, Yamamoto K, et al:
Characterization of the antitumor activities of IFN-alpha8 on renal
cell carcinoma cells in vitro. J Interferon Cytokine Res.
21:1129–1136. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Darnell JE Jr, Kerr IM and Stark GR:
Jak-STAT pathways and transcriptional activation in response to
IFNs and other extracellular signaling proteins. Science.
264:1415–1421. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rathmell WK and Godley PA: Recent updates
in renal rell carcinoma. Curr Opin Oncol. 22:250–256. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kremer CL, Klein RR, Mendelson J, et al:
Expression of mTOR signaling pathway markers in prostate cancer
progression. Prostate. 66:1203–1212. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hashemolhosseini S, Nagamine Y, Morley SJ,
Desrivières S, Mercep L and Ferrari S: Rapamycin inhibition of the
G1 to S transition is mediated by effects on cyclin D1 mRNA and
protein stability. J Biol Chem. 273:14424–14429. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nozawa H, Watanabe T and Nagawa H:
Phosphorylation of ribosomal p70 S6 kinase and rapamycin
sensitivity in human colorectal cancer. Cancer Lett. 251:105–113.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ruvinsky I, Sharon N, Lerer T, et al:
Ribosomal protein S6 phosphorylation is a determinant of cell size
and glucose homeostasis. Genes Dev. 19:2199–2211. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seufferlein T and Rozengurt E: Rapamycin
inhibits constitutive p70s6k phosphorylation, cell proliferation,
and colony formation in small cell lung cancer cells. Cancer Res.
56:3895–3897. 1996.PubMed/NCBI
|
|
12
|
Grewe M, Gansauge F, Schmid RM, Adler G
and Seufferlein T: Regulation of cell growth and cyclin D1
expression by the constitutively active FRAP-p70s6K pathway in
human pancreatic cancer cells. Cancer Res. 59:3581–3587.
1999.PubMed/NCBI
|
|
13
|
Cho D, Signoretti S, Regan M, Mier JW and
Atkins MB: The role of mammalian target of rapamycin inhibitors in
the treatment of advanced renal cancer. Clin Cancer Res.
13:758s–763s. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Luan FL, Ding R, Sharma VK, Chon WJ,
Lagman M and Suthanthiran M: Rapamycin is an effective inhibitor of
human renal cancer metastasis. Kidney Int. 63:917–926. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shi Y, Frankel A, Radvanyi LG, Penn LZ,
Miller RG and Mills GB: Rapamycin enhances apoptosis and increases
sensitivity to cisplatin in vitro. Cancer Res. 55:1982–1988.
1995.PubMed/NCBI
|
|
16
|
Masiello D, Mohi MG, McKnight NC, et al:
Combining an mTOR antagonist and receptor tyrosine kinase
inhibitors for the treatment of prostate cancer. Cancer Biol Ther.
6:195–201. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu WZ, Sun HC, Shen YF, et al: Interferon
alpha 2a down-regulates VEGF expression through PI3 kinase and MAP
kinase signaling pathways. J Cancer Res Clin Oncol. 131:169–178.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Costa LJ, Gemmill RM and Drabkin HA:
Upstream signaling inhibition enhances rapamycin effect on growth
of kidney cancer cells. Urology. 69:596–602. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Linehan WM, Lerman MI and Zbar B:
Identification of the von Hippel-Lindau (VHL) gene. Its role in
renal cancer. JAMA. 273:564–570. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kaelin WG Jr: Molecular basis of the VHL
hereditary cancer syndrome. Nat Rev Cancer. 2:673–682. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guba M, von Breitenbuch P, Steinbauer M,
et al: Rapamycin inhibits primary and metastatic tumor growth by
antiangiogenesis: involvement of vascular endothelial growth
factor. Nat Med. 8:128–135. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Treins C, Giorgetti-Peraldi S, Murdaca J,
Monthouël-Kartmann MN and Van Obberghen E: Regulation of
hypoxia-inducible factor (HIF)-1 activity and expression of HIF
hydroxylases in response to insulin-like growth factor I. Mol
Endocrinol. 19:1304–1317. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu YT, Shang D, Akatsuka S, et al:
Chronic oxidative stress causes amplification and overexpression of
ptprz1 protein tyrosine phosphatase to activate beta-catenin
pathway. Am J Pathol. 171:1978–1988. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Berenbaum MC: A method for testing for
synergy with any number of agents. J Infect Dis. 137:122–130. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hattori K and Akaza H: New combination
chemotherapy in urological cancers. Gan To Kagaku Ryoho.
27:382–387. 2000.(In Japanese).
|
|
26
|
Zhang JF, Liu JJ, Lu MQ, et al: Rapamycin
inhibits cell growth by induction of apoptosis on hepatocellular
carcinoma cells in vitro. Transpl Immunol. 17:162–168. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bukowski RM: Metastatic renal cell
carcinoma: role of mammalian target of rapamycin inhibitors. Clin
Genitourin Cancer. 5:359–361. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Amato RJ, Jac J, Mohammad T and Saxena S:
Pilot study of rapamycin in patients with hormone-refractory
prostate cancer. Clin Genitourin Cancer. 6:97–102. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Z, Zhou J, Fan J, et al: Effect of
rapamycin alone and in combination with sorafenib in an orthotopic
model of human hepatocellular carcinoma. Clin Cancer Res.
14:5124–5130. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huynh H, Chow PK, Palanisamy N, et al:
Bevacizumab and rapamycin induce growth suppression in mouse models
of hepatocellular carcinoma. J Hepatol. 49:52–60. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shi Y and August DA: A new trick for an
old drug: mTOR inhibitor rapamycin augments the effect of
fluorouracil on hepatocellular carcinoma by inducing cell
senescence. Cancer Biol Ther. 7:397–398. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pectasides D, Pectasides E, Papaxoinis G,
et al: Combination chemotherapy with docetaxel, vinorelbine and
estramustine phosphate in metastatic androgen-resistant prostate
cancer: a single institution experience. Anticancer Res.
29:769–775. 2009.
|
|
33
|
Vaishampayan UN, Marur S, Heilbrun LK, et
al: Phase II trial of capecitabine and weekly docetaxel for
metastatic castrate resistant prostate cancer. J Urol. 182:317–323.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hou G, Xue L, Lu Z, Fan T, Tian F and Xue
Y: An activated mTOR/p70S6K signaling pathway in esophageal
squamous cell carcinoma cell lines and inhibition of the pathway by
rapamycin and siRNA against mTOR. Cancer Lett. 253:236–248. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fan QW and Weiss WA: Inhibition of
PI3K-Akt-mTOR signaling in glioblastoma by mTORC1/2 inhibitors.
Methods Mol Biol. 821:349–359. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fingar DC, Salama S, Tsou C, Harlow E and
Blenis J: Mammalian cell size is controlled by mTOR and its
downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 16:1472–1487.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rosner M and Hengstschläger M:
Nucleocytoplasmic localization of p70 S6K1, but not of its isoforms
p85 and p31, is regulated by TSC2/mTOR. Oncogene. 30:4509–4522.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Terzis G, Spengos K, Mascher H, Georgiadis
G, Manta P and Blomstrand E: The degree of p70 S6k and S6
phosphorylation in human skeletal muscle in response to resistance
exercise depends on the training volume. Eur J Appl Physiol.
110:835–843. 2010.PubMed/NCBI
|
|
39
|
Pullen N and Thomas G: The modular
phosphorylation and activation of p70s6k. FEBS Lett. 410:78–82.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hinnebusch AG: Translational homeostasis
via eIF4E and 4E-BP1. Mol Cell. 46:717–719. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tait S, Dutta K, Cowburn D, Warwicker J,
Doig AJ and McCarthy JE: Local control of a disorder-order
transition in 4E-BP1 underpins regulation of translation via eIF4E.
Proc Natl Acad Sci USA. 107:17627–17632. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pause A, Belsham GJ, Gingras AC, et al:
Insulin-dependent stimulation of protein synthesis by
phosphorylation of a regulator of 5′-cap function. Nature.
371:762–767. 1994.
|
|
43
|
Perier A, Fregni G, Wittnebel S, et al:
Mutations of the von Hippel-Lindau gene confer increased
susceptibility to natural killer cells of clear-cell renal cell
carcinoma. Oncogene. 30:2622–2632. 2011. View Article : Google Scholar : PubMed/NCBI
|