Introduction
The proliferation of smooth muscle cells (SMCs)
plays a pivotal role in cardiovascular diseases (1–3).
However, the underlying mechanism remains unclear. The
renin-angiotensin-aldosterone system (RAAS) is involved in
endothelial dysfunction, vascular remodeling, oxidative stress,
proinflammatory cytokine production and adhesion molecule synthesis
in the vascular wall (4). As an
important part of the RAAS, angiotensin II (AngII) is able to
stimulate the growth and proliferation of vascular SMCs by binding
to the AngII receptor Type 1. AngII activates downstream signaling
molecules, such as protein kinase C and mitogen-activated protein
kinase (MAPK), and a number of extracellular signaling molecules,
including extracellular signal-regulated kinase (ERK) 1/2, p38MAPK,
c-Jun N-terminal kinase and ERK5, to carry out its functions
(5–8).
Chemokines are low molecular-mass cytokines that,
based on the spacing of N-terminus cysteine residues, are
classified into the C, CC, CXC and CX3C families; fractalkine (Fkn;
CX3CL1) is the only member of the CX3C family identified to date
(9). Studies have shown that Fkn
is involved in various inflammatory diseases, including
cardiovascular diseases (10–12).
Sullivan et al (13) found
that the expression of Fkn was markedly increased in the mesenteric
arteries of spontaneously hypertensive rats (SHRs) and was notably
higher in female SHRs. Studies have also demonstrated that Fkn can
induce SMC proliferation, which is mediated by nuclear factor
κB-dependent inflammatory signals (14,15).
Recently, Rius et al (16)
found that by stimulating endothelial cells with 1 μmol/l AngII,
Fkn expression was upregulated in the cells. These findings
highlight the significance of Fkn in the pathogenesis of
hypertension and suggest it may play a role in vascular
remodeling.
Neferine is extracted from the herb Nelumbo
nucifera (Gaertn.), an ingredient in Traditional Chinese
Medicine. Studies have shown that N. nucifera has multiple
biological activities, including attenuating bleomycin-induced
pulmonary fibrosis (17),
enhancing insulin sensitivity in insulin-resistant rats (18) and exerting an antioxidant effect
against isoproterenol-induced oxidative stress (19). Previous studies have found that
N. nucifera leaf extract and neferine can inhibit the
proliferation of vascular SMCs; however the underlying mechanism
has yet to be elucidated (20,21).
Based on these previous findings on the role of Fkn
in the pathogenesis of various cardiovascular diseases, on the
proliferative effects of AngII on vascular SMCs and on the effects
of N. nucifera and neferine, we hypothesized that Fkn could
be involved in the AngII-induced proliferation of vascular SMCs,
and that the anti-proliferative effects of neferine on the cells
could be fundamentally associated with Fkn and AngII. The aim of
the present study was to explore the mechanisms underlying the
effects of AngII and neferine on rat aortic smooth muscle cells
(RASMCs).
Materials and methods
Materials
The RASMC line was obtained from the American Type
Culture Collection (Manassas, VA, USA). The AngII, propidium iodide
(PI) solution, bicinchoninic acid (BCA) protein assay and RNase A
were purchased from Sigma (St. Louis, MO, USA). Goat polyclonal
anti-Fkn antibody and mouse monoclonal anti-β-actin antibody were
obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Neferine, with a purity of 98.6%, was extracted from the seed
embryo of N. nucifera using a preparative high-speed
counter-current chromatography method by the Department of
Pharmacy, Xiangya Hospital (Central South University, Changsha,
China) (22).
Cell culture and experimental design
The cells were grown in Dulbecco’s Modified Eagle’s
Medium (DMEM; Hyclone, Logan, UT, USA) supplemented with 10% fetal
bovine serum (FBS; Hyclone) at 37°C in a humidified atmosphere of
5% CO2. Cells between four and seven passages were used
for all the experiments in this study. RASMCs were seeded on
96-well plates (0.2–1.0×104 cells/well) or six-well
culture plates (1×105 cells/well) and cultured for 24 h,
prior to being starved for 24 h in DMEM containing 0.1% FBS.
Different concentrations of AngII (1×10−6,
1×10−7 and 1×10−8 M) were then added and the
cells were cultured for a certain time-period according to the
experimental design (6, 12 or 24 h) to evaluate the effects of
AngII on RASMC proliferation and Fkn expression. To examine the
effect and determine a suitable concentration of neferine on the
proliferation of RASMCs, the cells were divided into four groups:
Control and neferine treatment with three different concentrations
of neferine (1, 5 and 10 μmol/l obtained by dilution with DMEM).
The cells were then cultured for another 24 h prior to an MTT assay
being performed. In order to conduct all the other experiments in
the neferine study, the cells that had reached synchronization were
divided into three groups: Control, AngII (1×10−6 M) and
neferine plus AngII (neferine at 5 μmol/l, preculture for 1 h and
the subsequent addition of AngII at 1×10−6 M). The cells
were cultured for a further 24 h prior to undergoing analyses,
including MTT assay, flow cytometry, western blotting and the
quantitative polymerase chain reaction (qPCR). For RNA interference
and cell transfection, the cells were divided into four groups:
Control, AngII (1×10−6 M), control small interfering
(si)RNA (following transfection with control siRNA, AngII at
1×10−6 M was added) and Fkn siRNA plus AngII (following
transfection with Fkn siRNA, AngII at 1×10−6 M was
added). The cells were subsequently cultured for 24 h for the MTT
assay and flow cytometry. All the experiments were performed in
triplicate. The study was approved by the Ethics Committee of
Xiangya Hospital.
MTT assay
Subsequent to finishing the cell culture according
to the experimental design, 20 μl MTT (5 mg/ml) solution was added
and incubated for 4 h. The supernatants of the cell culture were
then removed and 150 μl dimethylsulfoxide was added to each well. A
multi-well plate reader (Model Stat-Fax-2100, Awareness Technology
Inc., Palm City, FL, USA) was subsequently used to determine the
optical density (OD). The absorbance of samples was measured at 570
nm (OD570), and the absorbance at 690 nm was used as a
reference. The background absorbance of the medium was also
subtracted. Each assay was repeated in triplicate, and the mean for
each experiment was calculated.
Flow cytometric determination of cell
cycles
At the end of cell culturing, the cells were
trypsinized and washed twice with cold phosphate-buffered saline
(PBS). The cells were then fixed with ice-cold 70% ethanol at 4°C
overnight. The cells were subsequently centrifuged to remove
fixative solution and washed with PBS twice, and then stained with
50 μg/ml PI solution and RNAse for 30 min in the dark. A total of
≥10,000 cells were analyzed. The percentages of cells in different
phases of the cell cycle were measured with a
fluorescence-activated cell sorting flow cytometer (Cytomics™ FC
500; Beckman Coulter, Miami, FL, USA) and analyzed with CXP
software (Beckman Coulter). The percentage of cells in the S and
G2/M phases was used as the indicator for cell
proliferation.
RNA interference and cell
transfection
To confirm the role of Fkn in the AngII-induced
proliferation of RASMCs, Fkn siRNA was developed to silence Fkn
gene expression, as Fkn expression has been found in RASMCs under
certain stimuli (23,24). The Fkn siRNA (sense strand, 5′-GCU
GUGGUAGUAAUUCAUAdTdT-3′ and antisense strand,
3′-dTdTCGACACCAUCAUUAAGUAU-5′) was synthesized by RayBiotech
(Guangzhou, China) and was transfected into RASMCs using
Lipofectamine® 2000 (Invitrogen Life Technologies,
Carlsbad, CA, USA) according to the manufacturer’s instructions.
Transfection efficiency was determined by qPCR and western blot
analysis.
Western blot analysis
The cells were harvested at the end of culture and
the proteins were obtained by use of radioimmunoprecipitation assay
buffer containing 1 mM phenylmethylsulfonyl fluoride. The protein
concentrations were measured using a BCA protein assay. Subsequent
to being boiled in 100°C water for 5 min, equal amounts of total
protein were separated by 8% SDS-PAGE. The protein was then
transferred to polyvinylidene fluoride membranes (Millipore,
Billerica, MA, USA). The membranes were blocked with 5% evaporated
skimmed milk containing 0.1% Tween 20 for 2 h and then incubated
with primary antibodies at 4°C overnight. The membranes were
subsequently washed with Tris-buffered saline with Tween 20 (TBST)
four times, followed by incubation with horseradish
peroxidase-conjugated secondary antibodies for 1–2 h at room
temperature. Following washing with TBST, the target protein bands
were detected using an enhanced chemiluminescence detection kit
with a ChemiDOC™ MP imaging system (Bio-Rad, Hercules, CA,
USA).
qPCR
Total RNA was extracted from the cells at the end of
culture using TRIzol® (Invitrogen Life Technologies).
The primers of Fkn and GAPDH were designed by Beijing Sunbiotech
Co., Ltd. (Beijing, China). The reverse transcription reaction was
performed with 5 μg total RNA. For PCR amplification, cDNAs were
amplified using SYBR Green qPCR Mix (Takara, Dalian, China); PCR
protocols are summarized in Table
I. The amplification reaction for each sample was performed in
triplicate. Results are expressed as the ratio of Fkn to GAPDH
mRNA.
 | Table IPCR primer sequences and protocol. |
Table I
PCR primer sequences and protocol.
| Gene | Primer sequences | Length (bp) | PCR protocol |
|---|
| Fkn |
5′-ctcgtcccagagtgaggaag-3′
(sense)
5′-ctgctcctcaggcctacaac-3′ (antisense) | 106 | 95°C/15 sec, 60°C/60
sec, 45 cycles |
| GAPDH |
5′-agacagccgcatcttcttgt-3′
(sense)
5′-tgatggcaacaatgtccact-3′ (antisense) | 142 | 95°C/15 sec, 60°C/60
sec, 45 cycles |
Statistical analysis
SPSS 16.0 (SPSS Inc., Chicago, IL, USA) was used for
the statistical analysis. All data are expressed as the mean ±
standard deviation and were analyzed using the Student’s t-test for
two group comparisons or analysis of variance followed by the
Student-Newman-Keuls test for multiple groups. P<0.05 was
considered significant.
Results
AngII promotes RASMC proliferation and
Fkn gene expression
MTT assay showed that AngII could promote RASMC
proliferation in a time- and concentration-dependent manner
(P<0.05; Fig. 1A). Accompanied
by the enhanced proliferation of RASMCs, Fkn protein expression, as
shown by western blot analysis, was significantly upregulated
following the use of AngII, and the overexpression was also in a
concentration- and time-dependent manner (P<0.05; Fig. 1B). By contrast, control cells only
showed a baseline Fkn expression.
Fkn gene silencing suppresses
AngII-induced RASMC proliferation
Following Fkn siRNA transfection, the Fkn expression
at the mRNA and protein levels in the AngII-stimulated Fkn siRNA
group was suppressed in comparison with that in the
AngII-stimulated control siRNA group (P<0.05; Fig. 2A). Furthermore, MTT assay and flow
cytometry demonstrated that the Fkn siRNA transfection
significantly attenuated the AngII-induced cell proliferation
(P<0.05; Fig. 2B and C).
Neferine inhibits the proliferation of
both normal and AngII-stimulated RASMCs
MTT assay showed that neferine inhibited RASMC
proliferation in a concentration-dependent manner in comparison
with the control group (P<0.05). Neferine treatment at a
concentration of 5 μmol/l appeared to result in moderate
inhibition, and this concentration of neferine was used in the
following experiments (Fig. 3A).
MTT assay also showed that 5 μmol/l neferine attenuated the
proliferative effect of AngII at 1×10−6 M (neferine
pretreatment for 1 h followed by AngII treatment) (P<0.05;
Fig. 3B). This inhibitive effect
of neferine on the AngII-induced proliferation of RASMCs was
further confirmed by cell cycle analysis using flow cytometry
(P<0.05; Fig. 3C).
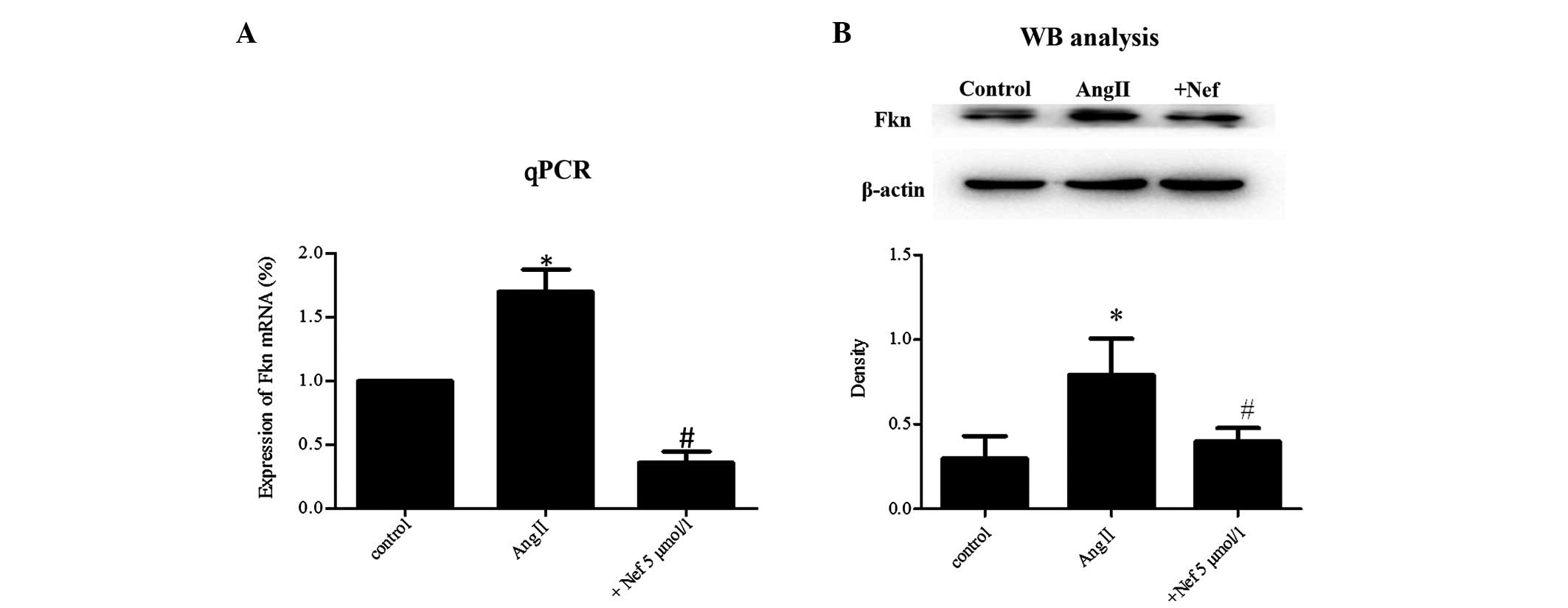
Neferine attenuates AngII-induced Fkn
expression in RASMCs
Fkn mRNA and protein expression in normal RASMCs was
limited, but, following exposure to AngII for 24 h, Fkn expression
was significantly upregulated (P<0.05). By contrast,
pretreatment with neferine (5 μmol/l) markedly attenuated the
enhanced Fkn expression at the mRNA and protein levels (P<0.05;
Fig. 4A and B).
Discussion
In the present study, the following observations
were noted: i) AngII stimulated Fkn expression in RASMCs; ii) Fkn
gene-knockdown attenuated the AngII-induced proliferation of RASMCs
and iii) neferine inhibited AngII-induced Fkn expression,
attenuating the proliferative effect of AngII on RASMCs.
Accumulating evidence has shown that Fkn plays an
important role in cardiovascular diseases (12,25,26).
Fkn is an atypical chemokine that exists in either a membrane-bound
form or as a soluble chemokine (9). The membrane-bound form acts as an
adhesion molecule and enables leucocyte adhesion via binding to
chemokine (C-X3-C motif) receptor 1 (CX3CR1), while the soluble
form functions as a chemoattractant for monocytes and T cells
(27). Fkn is expressed in
atherosclerotic plaques (28,29),
and an absence of Fkn or its receptor, CX3CR1, prevents
atherosclerotic lesion formation (30,31).
The proliferation of SMCs is involved in
atherosclerosis and hypertension, and AngII is an important
stimulus among the numerous involved (7,32).
AngII contributes to the vascular SMC growth, endothelial
dysfunction and vascular inflammation in hypertension. There is
little Fkn expression in endothelial cells or SMCs under normal
conditions; however, overexpression of Fkn has been observed when
cells are under a certain stimulation, such as interferon-γ and
tumor necrosis factor-α (9,23).
Although there has been a report of AngII inducing Fkn
overexpression in endothelial cells (16), the effect of AngII on Fkn
expression in SMCs has not been documented to date. In this study,
it was revealed that AngII upregulated Fkn expression in a time-
and concentration-dependent manner, and knockdown of Fkn caused a
significant decrease in cell proliferation in AngII-stimulated
RASMCs.
In this study, neferine was again found to be
capable of inhibiting AngII-induced RASMC proliferation, which was
consistent with a previous study (22), and the anti-proliferative effect of
neferine on AngII-stimulated RASMCs was revealed to be the result
of suppressed Fkn expression. Neferine markedly attenuated the
proliferative effect of AngII on the RASMCs by suppressing Fkn gene
expression.
In conclusion, the findings in this study show that
the Fkn gene plays an important role in the AngII-induced
proliferation of RASMCs. Through Fkn, AngII exerts its
proliferative effect on the cells, and by inhibiting Fkn
expression, neferine exerts its anti-proliferative effects on the
AngII-stimulated cells. Further studies are, however, required to
fully understand the mechanisms and underlying signaling pathways
of SMC proliferation.
Acknowledgements
This study was supported by a grant from the Hunan
Provincial Science and Technology Department, China (no.
12JJ5070).
References
|
1
|
Viiri LE, Full LE, Navin TJ, Bequm S,
Didangelos A, Astola N, Berge RK, Seppälä I, Shalhoub J, Frankin
IJ, Perretti M, Lehtimäki T, Davies AH, Wait R and Monaco C: Smooth
muscle cells in human atherosclerosis: proteomic profiling reveals
differences in expression of Annexin A1 and mitochondrial proteins
in carotid disease. J Mol Cell Cardiol. 54:65–72. 2013. View Article : Google Scholar
|
|
2
|
Savoia C, Burger D, Nishigaki N, Montezano
A and Touyz RM: Angiotensin II and the vascular phenotype in
hypertension. Expert Rev Mol Med. 13:e112011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu G, Hitomi H, Hosomi N, Lei B, Pelisch
N, Nakano D, Kiyomoto H, Ma H and Nishiyama A: Mechanical stretch
potentiates angiotensin II-induced proliferation in spontaneously
hypertensive rat vascular smooth muscle cells. Hypertens Res.
33:1250–1257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kasal DA and Schiffrin EL: Angiotensin II,
aldosterone, and anti-inflammatory lymphocytes: interplay and
therapeutic opportunities. Int J Hypertens.
2012:8297862012.PubMed/NCBI
|
|
5
|
Touyz RM and Schiffrin EL: Signal
transduction mechanisms mediating the physiological and
pathophysiological actions of angiotensin II in vascular smooth
muscle cells. Pharmacol Rev. 52:639–672. 2000.PubMed/NCBI
|
|
6
|
Eguchi S, Dempsey PJ, Frank GD, Motley ED
and Inagami T: Activation of MAPKs by angiotensin II in vascular
smooth muscle cells. Metalloprotease-dependent EGF receptor
activation is required for activation of ERK and p38 MAPK but not
for JNK. J Biol Chem. 276:7957–7962. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shi R, Hu C, Yuan Q, Yang T, Peng J, Li Y,
Bai Y, Cao Z, Cheng G and Zhang G: Involvement of vascular
peroxidase 1 in angiotensin II-induced vascular smooth muscle cell
proliferation. Cardiovasc Res. 91:27–36. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao Y, Liu J, Li L, Liu L and Wu L: Role
of Ras/PKCzeta/MEK/ERK1/2 signaling pathway in angiotensin
II-induced vascular smooth muscle cell proliferation. Regul Pept.
128:43–50. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bazan JF, Bacon KB, Hardiman G, Wang W,
Soo K, Rossi D, Greaves DR, Zlotnik A and Schall TJ: A new class of
membrane-bound chemokine with a CX3C motif. Nature. 385:640–644.
1997. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Balabanian K, Foussat A, Dorfmüller P,
Durand-Gasselin I, Capel F, Bouchet-Delbos L, Portier A,
Marfaing-Koka A, Krzysiek R, Rimaniol AC, Simonneau G, Emilie D and
Humbert M: CX(3)C chemokine fractalkine in pulmonary arterial
hypertension. Am J Respir Crit Care Med. 165:1419–1425. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schäfer A, Schulz C, Fraccarollo D, Tas P,
Leutke M, Eigenthaler M, Seidl S, Heider P, Ertl G, Massberg S and
Bauersachs J: The CX3C chemokine fractalkine induces vascular
dysfunction by generation of superoxide anions. Arterioscler Thromb
Vasc Biol. 27:55–62. 2007.PubMed/NCBI
|
|
12
|
Xuan W, Liao Y, Chen B, Huang Q, Xu D, Liu
Y, Bin J and Kitakaze M: Detrimental effect of fractalkine on
myocardial ischaemia and heart failure. Cardiovasc Res. 92:385–393.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sullivan JC, Pardieck JL, Doran D, Zhang
Y, She JX and Pollock JS: Greater fractalkine expression in
mesenteric arteries of female spontaneously hypertensive rats
compared with males. Am J Physiol Heart Circ Physiol.
296:H1080–H1088. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Perros F, Dorfmüller P, Souza R,
Durand-Gasselin I, Godot V, Capel F, Adnot S, Eddahibi S, Mazmanian
M, Fadel E, Hervé P, Simonneau G, Emilie D and Humbert M:
Fractalkine-induced smooth muscle cell proliferation in pulmonary
hypertension. Eur Respir J. 29:937–943. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chandrasekar B, Mummidi S, Perla RP,
Bysani S, Dulin NO, Liu F and Melby PC: Fractalkine (CX3CL1)
stimulated by nuclear factor kappaB (NF-kappaB)-dependent
inflammatory signals induces aortic smooth muscle cell
proliferation through an autocrine pathway. Biochem J. 373:547–558.
2003. View Article : Google Scholar
|
|
16
|
Rius C, Piqueras L, González-Navarro H,
Albertos F, Company C, López-Ginés C, Ludwig A, Blanes JI, Morcillo
EJ and Sanz MJ: Arterial and venous endothelia display differential
functional fractalkine (CX3CL1) expression by angiorensin-II.
Arterioscler Thromb Vasc Biol. 33:96–104. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao L, Wang X, Chang Q, Xu J, Huang Y,
Guo Q, Zhang S, Wang W, Chen X and Wang J: Neferine, a
bisbenzylisoquinline alkaloid attenuates bleomycin-induced
pulmonary fibrosis. Eur J Pharmcol. 627:304–312. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pan Y, Cai B, Wang K, Wang S, Zhou S, Yu
X, Xu B and Chen L: Neferine enhances insulin sensitivity in
insulin resistant rats. J Ethnopharmacol. 124:98–102. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lalitha G, Poornima P, Archanah A and
Padma VV: Protective effect of neferine against
isoproterenol-induced cardiac toxicity. Cardiovasc Toxicol.
13:168–179. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ho HH, Hsu LS, Chan KC, Chen HM, Wu CH and
Wang CJ: Extract from the leaf of nucifera reduced the development
of atherosclerosis via inhibition of vascular smooth muscle cell
proliferation and migration. Food Chem Toxicol. 48:159–168. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li XC, Tong GX, Zhang Y, Liu SX, Jin QH,
Chen HH and Chen P: Neferine inhibits angiotensin II-stimulated
proliferation in vascular smooth muscle cells through heme
oxygenase-1. Acta Pharmacol Sin. 31:679–686. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu S, Wang B, Li XZ, Qi LF and Liang YZ:
Preparative separation and purification of liensinine,
isoliensinine and neferine from seed embryo of Nelumbo
nucifera GAERTN using high-speed counter-current
chromatography. J Sep Sci. 32:2476–2481. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ludwig A, Berkhout T, Moores K, Groot P
and Chapman G: Fractalkine is expressed by smooth muscle cells in
response to IFN-gamma and TNF-alpha and is modulated by
metalloproteinase activity. J Immunol. 168:604–612. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lucas AD, Bursill C, Guzik TJ, Sadowski J,
Channon KM and Greaves DR: Smooth muscle cells in human
atherosclerotic plaques express the fractalkine receptor CX3CR1 and
undergo chemotaxis to the CX3C chemokine fractalkine (CX3CL1).
Circulation. 108:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ikejima H, Imanishi T, Tsujioka H,
Kashiwagi M, Kuroi A, Tanimoto T, Kitabata H, Ishibashi K, Komukai
K, Takeshita T and Akasaka T: Upregulation of fractalkine and its
receptor, CX3CR1, is associated with coronary plaque rupture in
patients with unstable angina pectoris. Circ J. 74:337–345. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Flierl U and Schäfer A: Fractalkine - a
local inflammatory marker aggravating platelet activation at the
vulnerable plaque. Thromb Haemost. 108:457–463. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Imai T, Hieshima K, Haskell C, Baba M,
Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ
and Yoshie O: Identification and molecular characterization of
fractalkine receptor CX3CR1, which mediates both leukocyte
migration and adhesion. Cell. 91:521–530. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Damås JK, Boullier A, Waehre T, Smith C,
Sandberg WJ, et al: Expression of fractalkine (CX3CL1) and its
receptor, CX3CR1, is elevated in coronary artery disease and is
reduced during statin therapy. Arterioscler Thromb Vasc Biol.
25:2567–2572. 2005.PubMed/NCBI
|
|
29
|
Stolla M, Pelisek J, von Brühl ML, Schäfer
A, Barocke V, Heider P, Lorenz M, Tirniceriu A, Steinhart A,
Bauersachs J, Bray PF, Massberg S and Schulz C: Fractalkine is
expressed in early and advanced atherosclerotic lesions and
supports monocyte recruitment via CX3CR1. PLoS One. 7:e435722012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Combadière C, Potteaux S, Gao JL, Esposito
B, Casanova S, et al: Decreased atherosclerotic lesion formation in
CX3CR1/apolipoprotein E double knockout mice. Circulation.
107:1009–1016. 2003.PubMed/NCBI
|
|
31
|
Lesnik P, Haskell CA and Charo IF:
Decreased atherosclerosis in CX3CR1−/− mice reveals a role for
fractalkine in atherogenesis. J Clin Invest. 111:333–340. 2003.
|
|
32
|
Cheng JF, Ni GH, Chen MF, Li YJ, Wang YJ,
Wang CL, Yuan Q, Shi RZ, Hu CP and Yang TL: Involvement of
profilin-1 in angiotensin II-induced vascular smooth muscle cell
proliferation. Vascul Pharmacol. 55:34–41. 2011. View Article : Google Scholar : PubMed/NCBI
|