Introduction
Acute ischemic stroke is one of the leading causes
of adult disability (1). The
excitatory neuronal toxicity caused by the accelerated release of
glutamate during brain ischemia is the key element leading to
neuronal death. The concentration of glutamate in the synaptic
cleft is regulated by membrane-bound excitatory amino-acid
transporters (EAATs) (1). To date,
five subtypes have been identified and termed EAAT1-5 in human
tissues. Glutamate/aspartate transporter, glutamate transporter-1
and excitatory amino-acid carrier 1 are the rodent homologs of
EAAT1, 2 and 3, respectively (3).
EAAT2 is primarily an astrocytic transporter and is highly
expressed throughout the central nervous system (CNS), removing
>90% of the total extracellular glutamate from the synaptic
cleft (4).
Tumor necrosis factor-α (TNF-α) is an important
inflammatory factor and its expression levels markedly increase in
the brain following ischemia (5).
For a long period of time, TNF-α was believed to induce neuronal
necrosis, resulting in brain injury (6), until Cheng et al (7) demonstrated that TNF-α (1–100 ng/ml)
pretreatment protected cultured neurons from glucose
deprivation-induced injury and excitatory amino-acid toxicity. By
knocking down the receptors for TNF-α in mice, Gary et al
(8) unexpectedly observed more
severe neuronal damage caused by focal brain ischemia, leading to
the hypothesis that TNF-α exhibits a protective role on ischemic
neurons. Since then, increasing evidence has shown that TNF-α plays
a dual role in ischemic neuronal injury by inducing necrosis and
protecting injured neurons (9–12).
Our previous study demonstrated that the permeability of the
blood-brain barrier to TNF-α transiently increased following injury
to the CNS (13). Notably, the
time-course of the TNF-α permeability was in accordance with that
of the functional recovery, which indicated that the optimal
concentration and timing of TNF-α administration contribute to
neuroprotection. Although different mechanisms have been proposed
to explain the dual role of TNF-α (10–12),
the association between TNF-α and EAATs on astrocytes remains
unclear. The aim of the present study was to determine whether
TNF-α regulates the expression level of EAAT2 in primary astrocytes
in culture and its role in brain ischemia.
Materials and methods
Animals and reagents
Adult male Sprague Dawley rats were obtained from
the Experimental Animal Centre of Zhejiang University (Hangzhou,
China). The experimental procedures were approved by the Animal
Ethics Committee of Zhejiang University and were carried out in
accordance with institutional guidelines. Recombinant rat TNF-α was
obtained from BioVision (Milpitas, CA, USA).
Primary hippocampal neuron culture
Rat hippocampal neurons were prepared as previously
described, with certain modifications (14). Briefly, the hippocampi of E18
embryos were dissected and dissociated in oxygenated Hank’s
balanced salt solution. Cells (1×106) were plated on
coverslips coated with poly-D-lysine (50 μg/ml; Sigma-Aldrich, St.
Louis, MO, USA) or on coverslips in Neurobasal® medium
supplemented with 2% B27 (Invitrogen Life Technologies, San Diego,
CA, USA) and 0.2 mM L-glutamine (Sigma-Aldrich_.
Cytosine-D-arabinoside (10 μM; Sigma-Aldrich) was added to the
cultures two days after plating to block the proliferation of
non-neuronal cells. One-half of the culture medium was changed
every four days. After culturing for 12 days, neurons were then
co-cultured with astrocytes.
Primary cortical astrocyte culture and
TNF-α treatment
Astrocytes in primary culture were prepared from the
cerebral cortices of newborn (0–12 h postnatal) rat pups as
previously described (15). In
brief, cells were trypsinized with 0.25% trypsin (Invitrogen Life
Technologies, Carlsbad, CA, USA) for 16 min, followed by
trituration with 10 mg/l DNAse (Sigma-Aldrich). Cells were plated
in poly-D-lysine-coated flasks and maintained in Dulbecco’s
Modified Eagle’s medium (DMEM; Invitrogen Life Technologies)
supplemented with 10% fetal bovine serum (PAA, Pasching, Austria).
The culture medium was changed every third day. After nine days,
the flasks were agitated for 12 h at 37°C to separate the microglia
from the more adherent mass of astrocytes. The adherent cells were
replated in dishes and cultured for another week prior to
experimentation. Immunostaining with the glial fibrillary acidic
protein (GFAP) antibody (Cell Signaling Technology, Inc., Danvers,
MA, USA) confirmed that the majority (>95%) of the cells
expressed GFAP (data not shown). To examine the dose-dependent
effect, recombinant rat TNF-α (1, 10, 20 and 50 ng/ml) was added
directly to the medium and the cells were lysed following 24-h
incubation. To explore the time-dependent effect, astrocytes were
treated with 10 ng/ml TNFα for 4, 8, 12, 24, 36 or 72 h and lysed
after treatment.
Co-culture of astrocytes and neurons
Co-cultures of astrocytes and neurons were prepared
as previously described, with certain modifications (16). Briefly, astrocytes in Petri dishes
were washed twice with phosphate-buffered saline (PBS) and
incubated in serum-free medium for 3 h. Glass coverslips with
hippocampal neurons were inverted over the separately-cultured
astrocytes so that the neurons were facing the astrocytes. The
cells were incubated together in Neurobasal medium containing B27
and L-glutamine for 48 h to induce direct contact. The cultures
were pretreated with 10 ng/ml TNF-α for 0–24 h prior to being
subjected to oxygen-glucose deprivation (OGD).
ODG
OGD was performed as previously described (17). Briefly, co-cultured cells were
washed with PBS and incubated in glucose-free DMEM. The cultures
were transferred to an anaerobic chamber filled with a gas mixture
of 95% N2 and 5% CO2 at 37°C for 2 h.
Exposure was terminated by removing the glucose-free DMEM and
adding Neurobasal medium. After 22 h of recovery in a normoxic
incubator, the coverslips were carefully transferred, face-up, to a
new 24-well plate containing Neurobasal medium and the cell
viability was assessed as described below. In each experiment,
cultures exposed to OGD were compared with normoxic controls
supplied with DMEM containing glucose and maintained in standard
incubation conditions.
MTT assay
Cell viability was measured using the MTT
(Sigma-Aldrich) assay. Cells were gently washed with fresh culture
medium, and DMEM and the serum-free medium containing MTT (5 mg/ml)
was added and incubated for an additional 4 h at 37°C. The
purple-blue MTT formazan precipitate was dissolved in 150 μl
dimethyl sulfoxide. Absorbance was measured at 490 nm.
TNF-α treatment in vivo
A total of 6 male rats were anesthetized by an
intraperitoneal injection of 2% sodium pentobarbital (40 mg/kg) and
mounted in a stereotaxic frame on a heating blanket. TNF-α was
administered according to previously described techniques (18), with certain modifications.
Intracerebroventricular (i.c.v.) injection was performed at the
following coordinates: 0.6 mm posterior, 4.5 mm ventral and 1.6 mm
lateral to the bregma. Recombinant rat TNF-α (100 ng/15 μl) was
injected into the left lateral ventricle over a period of 10 min. A
total of 3 rats in the control group received the same volume of
sterilized, artificial cerebrospinal fluid. The expression levels
of the EAATs in the cerebral cortex were determined by western blot
analysis 24 h after treatment with TNF-α.
Western blot assay
Cerebral cortical tissues or astrocytes were lysed
on ice in radioimmunoprecipitation assay buffer with a protease
inhibitor cocktail (Sigma-Aldrich) containing a mixture of several
protease inhibitors, including AEBSF at 104 mM, Aprotinin at 80 μM,
Bestatin at 4 mM, E-64 at 1.4 mM, Leupeptin at 2 mM and Pepstatin A
at 1.5 mM. Protein content was estimated using the bicinchoninic
acid assay method. Immunoblotting was performed as described
previously (19). Following
electrophoresis, protein samples were transferred to polyvinylidene
fluoride membranes. The membranes were incubated at 4°C overnight
with antibodies against EAAT1 (1:500; Wuhan Boster Biological
Technology, Ltd., Wuhan, China), EAAT2 (1:500; Wuhan Boster
Biological Technology, Ltd.), GFAP (1:2,000) or β-actin (1:1,000;
Cell Signaling Technology, Inc.) diluted in blocking solution.
Following 2 h of incubation with a horseradish peroxidase-labeled
secondary antibody (Jackson ImmunoResearch Laboratories, West
Grove, PA, USA) at room temperature, the blots were developed with
enhanced chemiluminescence reagents (Pierce, Rockford, IL, USA) and
exposed to X-ray film to obtain images. The intensity of each band
was quantified using ImageJ software (National Institutes of
Health, Bethesda, MD, USA) and normalized to β-actin.
Statistical analysis
Data are expressed as the mean ± standard deviation.
Comparisons between groups were analyzed using the Student’s t-test
or one-way analysis of variance, followed by Dunnett’s post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
TNF-α elevates EAAT2 expression in
vivo
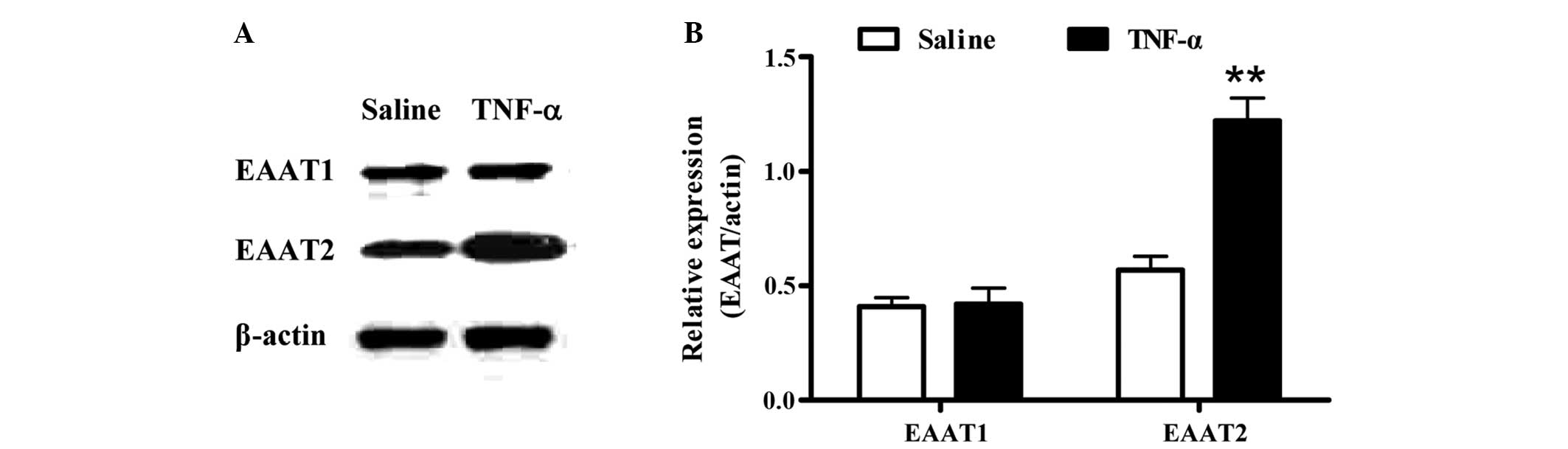
To investigate the effect of TNF-α on the expression
of EAATs in vivo, recombinant rat TNF-α was administered by
i.c.v. injection and the expression levels of the EAATs in the
cerebral cortical tissue were assessed by western blot analysis.
Compared with the EAAT2 levels in the saline control group, EAAT2
expression markedly increased (P<0.01) after TNF-α treatment for
24 h. However, no changes were observed in the expression level of
EAAT1 (Fig. 1).
TNF-α regulates EAAT2 expression in
astrocytes
To determine whether the expression levels of EAAT
proteins in astrocytes are regulated by TNF-α, the dose-dependent
effect of TNF-α (1, 10, 20 or 50 ng/ml) on primary cortical
astrocytes was analyzed (Fig. 2).
The optimal concentration of TNF-α (10 ng/ml) upregulated EAAT2
expression (P<0.01), whereas a high concentration of TNF-α (50
ng/ml) negatively regulated EAAT2 expression (P<0.05). Different
doses of TNF-α did not change the expression levels of EAAT1 or
GFAP, the latter representing astrocyte proliferation (Fig. 2).
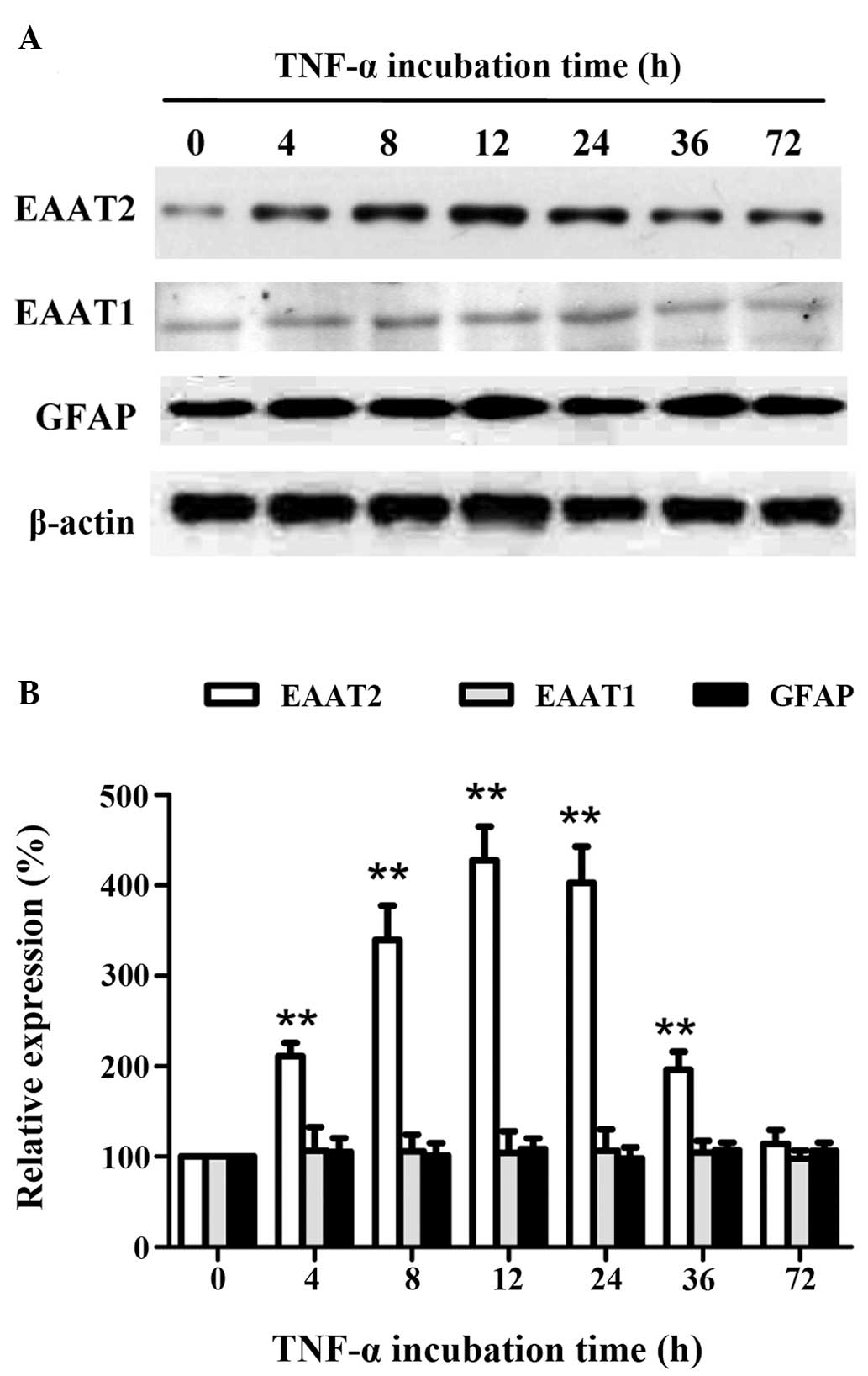
To further explore the association between
incubation time and the expression levels of EAATs in astrocytes,
the astrocytes were treated with the optimal concentration of TNF-α
(10 ng/ml) for different incubation periods (4, 8, 12, 24, 36 or 72
h). The EAAT2 protein expression showed a time-dependent increase,
followed by a time-dependent decrease, with a maximum level
(P<0.01) after 12 h (Fig. 3).
However, the expression levels of EAAT1 and GFAP did not change
with treatment. These results indicated that short-term treatment
with the optimal concentration of TNF-α promoted EAAT2 expression
in astrocytes, which was independent of astrocyte
proliferation.
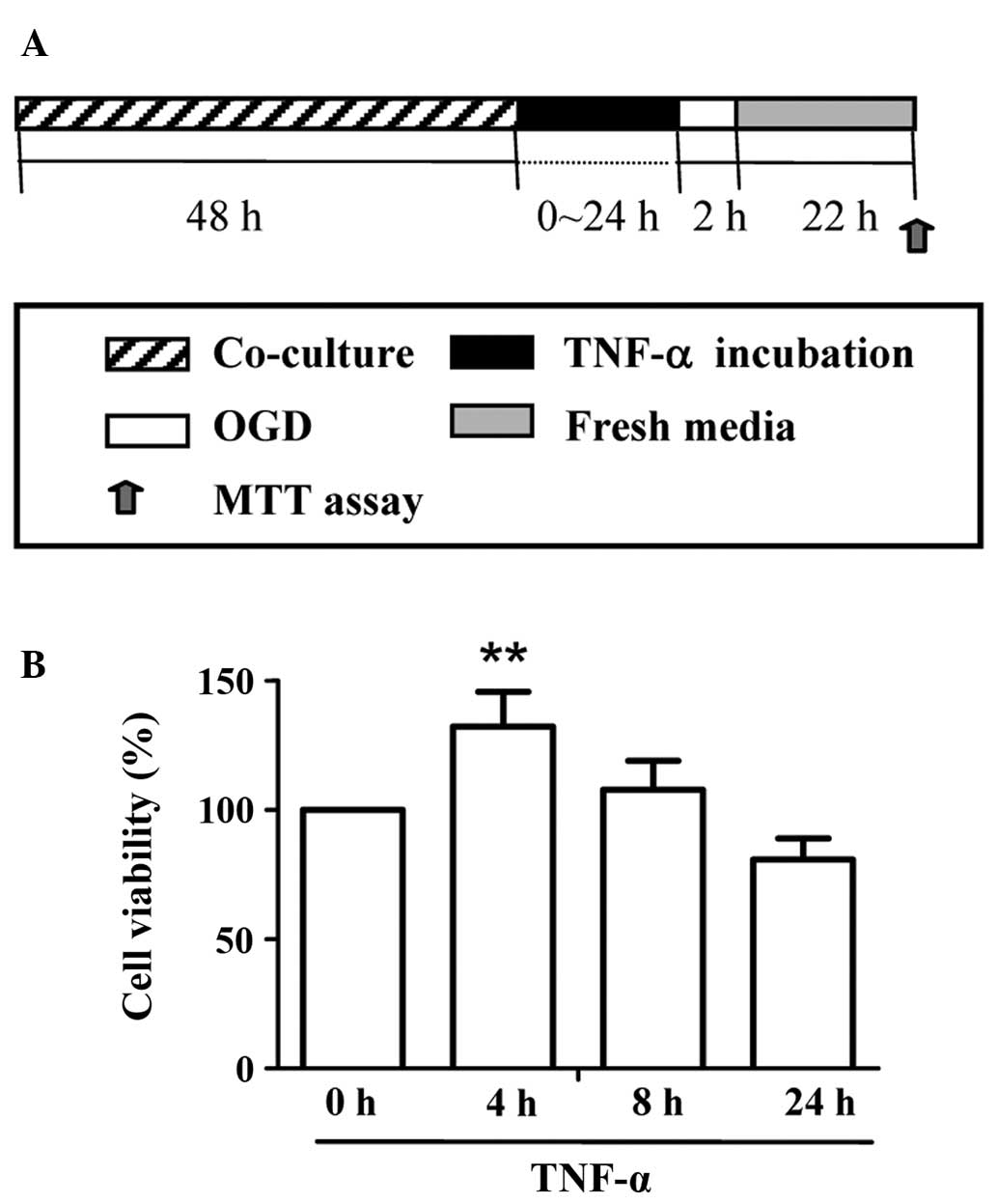
TNF-α treatment increases neuronal
viability following OGD injury
To determine whether TNF-α treatment is
neuroprotective against neuronal ischemic injury in vitro, a
co-culture system of astrocytes and neurons was established and
cell viability was assessed following OGD injury. After different
periods of incubation (0–24 h) with 10 ng/ml TNF-α, OGD was applied
for 2 h, followed by 22 h of reoxygenation (Fig. 4A). An MTT assay was performed on
the hippocampal neurons to reveal the cell viability following OGD
insult. The results demonstrated that only short-term (4 h) TNF-α
treatment led to a significantly higher cell viability value
compared with the other groups (P<0.01) (Fig. 4B).
Discussion
Clinical studies have demonstrated that a transient
ischemic attack (TIA) within a narrow time-window may enhance the
tolerance of the brain against a more severe ischemic insult
(20–22). Notably, elevation in the plasma
levels of TNF-α have been reported in patients for <72 h
following a TIA (23), leading to
the proposal that TNF-α pretreatment may mimic the neuroprotective
effect of ischemic preconditioning. Coinciding with this
hypothesis, it has been found that the elevation in TNF-α levels
induced by ischemic preconditioning and direct TNF-α pretreatment
contributes to the enhancement of cellular defense against the
second insult (10,15,24).
In the present study, in vivo TNF-α treatment markedly
decreased the infarct volume in the ischemic brain and improved the
functional recovery of rats receiving a subsequent middle cerebral
artery occlusion (data not shown).
Numerous studies have been performed to investigate
the mechanisms of ischemic tolerance caused by TNF-α exposure.
Glazner and Mattson found that TNF-α pretreatment reduced calcium
influx and upregulated the gene transcription of neuroprotective
factors (25). In addition,
Watters and O’Conner (26) and
Watters et al (27)
revealed that TNF-α pretreatment increased the resistance of
neurons to a subsequent insult from glutamate by attenuating
resting calcium activity and calcium-related responsiveness. In the
present study, the expression level of EAAT2, but not EAAT1, was
upregulated by TNF-α treatment in vivo and in vitro.
This was consistent with the findings of Davis and Patel (28), who demonstrated that the expression
level of EAAT2 was significantly increased in ischemic
preconditioning, suggesting that EAAT2 upregulation attenuates the
toxicity of the excitatory glutamate induced by a more severe
ischemic insult. This was further confirmed by the study of Weller
et al (29), in which it
was observed that astrocytes that had undergone transfection to
overexpress EAAT2 played a marked protective role against moderate
ischemia.
Consistent with the present results, Tilleux et
al (30) reported the
stimulatory effects of TNF-α on EAAT2 expression, although other
studies have found inhibitory effects. Su et al (31) revealed that the mRNA expression of
EAAT2 began to decline from 48 h with TNF-α (30 ng/ml) treatment.
Thus, the effect of TNF-α on EAAT2 expression remains
controversial. In the present study, TNF-α treatment promoted the
expression of EAAT2 in astrocytes in a concentration- and
time-dependent manner. The current results suggested that a lower
concentration (<20 ng/ml) of TNF-α applied as a short-term
(<36 h) treatment contributed to the induction of a
neuroprotective effect by elevating EAAT2 expression, while this
effect was lost or even reversed with increasing concentration and
incubation time. Although the underlying mechanism remains unclear,
this may explain why certain studies provide evidence for an
inhibitory influence of TNF-α on EAAT2 expression. Furthermore, it
was demonstrated in the present study that incubation with TNF-α
did not increase astrocyte proliferation. Therefore, the elevated
expression of EAAT2 in astrocytes was
proliferation-independent.
To further determine whether the elevated levels of
EAAT2 protein induced by TNF-α had a beneficial outcome in
vitro, the viability following OGD injury in a co-culture
system of astrocytes and neurons was assessed. Corresponding with
the in vivo results, it was revealed that TNF-α increased
the cell viability following OGD injury, suggesting that it has a
neuroprotective effect by promoting EAAT2 expression in
astrocytes.
The present study had several limitations. Firstly,
based on previous findings, TNF-α may have dual effects on the
expression and activity of EAATs. Thus, detection of the activity
of EAATs under these experimental conditions is required. Secondly,
it is necessary to demonstrate whether the protection is eradicated
following the blockade of glutamate uptake. Finally, an advanced
mechanistic study is also required. Therefore, these issues should
be addressed in future studies.
In conclusion, the present study revealed that an
optimal concentration and time-course of TNF-α elevates EAAT2
expression in astrocytes and has a beneficial effect on subsequent
ischemic insult.
Acknowledgements
This study was supported by the Natural Science
Foundation of Zhejiang Province (Y2100508) and the National Natural
Science Foundation of China (81000535).
References
|
1
|
Krishnamurthi RV, Feigin VL, Forouzanfar
MH, et al; GBD Stroke Experts Group. Global and regional burden of
first-ever ischaemic and haemorrhagic stroke during 1990-2010:
findings from the Global Burden of Disease Study 2010. Lancet Glob
Health. 1:e259–e281. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chao XD, Fei F and Fei Z: The role of
excitatory amino acid transporters in cerebral ischemia. Neurochem
Res. 35:1224–1230. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bunch L, Erichsen MN and Jensen AA:
Excitatory amino acid transporters as potential drug targets.
Expert Opin Ther Targets. 13:719–731. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maragakis NJ and Rothstein JD: Glutamate
transporters: animal models to neurologic disease. Neurobiol Dis.
15:461–473. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Botchkina GI, Meistrell ME III, Botchkina
IL and Tracey KJ: Expression of TNF and TNF receptors (p55 and p75)
in the rat brain after focal cerebral ischemia. Mol Med. 3:765–781.
1997.PubMed/NCBI
|
|
6
|
Feuerstein G, Wang X and Barone FC:
Cytokines in brain ischemia - the role of TNF alpha. Cell Mol
Neurobiol. 18:695–701. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cheng B, Christakos S and Mattson MP:
Tumor necrosis factors protect neurons against
metabolic-excitotoxic insults and promote maintenance of calcium
homeostasis. Neuron. 12:139–153. 1994. View Article : Google Scholar
|
|
8
|
Gary DS, Bruce-Keller AJ, Kindy MS and
Mattson MP: Ischemic and excitotoxic brain injury is enhanced in
mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood
Flow Metab. 18:1283–1287. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lambertsen KL, Clausen BH, Babcock AA, et
al: Microglia protect neurons against ischemia by synthesis of
tumor necrosis factor. J Neurosci. 29:1319–1330. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Saha RN, Liu X and Pahan K: Up-regulation
of BDNF in astrocytes by TNF-alpha: a case for the neuroprotective
role of cytokine. J Neuroimmune Pharmacol. 1:212–222. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nawashiro H, Martin D and Hallenbeck JM:
Inhibition of tumor necrosis factor and amelioration of brain
infarction in mice. J Cereb Blood Flow Metab. 17:229–232. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hallenbeck JM: The many faces of tumor
necrosis factor in stroke. Nat Med. 8:1363–1368. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pan W, Ding Y, Yu Y, et al: Stroke
upregulates TNFalpha transport across the blood-brain barrier. Exp
Neurol. 198:222–233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu P, Genc A, Zhang X, et al:
Heterogeneous expression and regulation of hippocampal
prostaglandin E2 receptors. J Neurosci Res. 81:817–826. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Saha RN, Ghosh A, Palencia CA, et al:
TNF-alpha preconditioning protects neurons via neuron-specific
up-regulation of CREB-binding protein. J Immunol. 183:2068–2078.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Roqué PJ, Guizzetti M, Giordano G and
Costa LG: Quantification of synaptic structure formation in
cocultures of astrocytes and hippocampal neurons. Methods Mol Biol.
758:361–390. 2011.PubMed/NCBI
|
|
17
|
Wang R, Zhang X, Zhang J, et al:
Oxygen-glucose deprivation induced glial scar-like change in
astrocytes. PLoS One. 7:e375742012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schmidt OI, Morganti-Kossmann MC, Heyde
CE, et al: Tumor necrosis factor-mediated inhibition of
interleukin-18 in the brain: a clinical and experimental study in
head-injured patients and in a murine model of closed head injury.
J Neuroinflammation. 1:132004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang XY, Chen ZH, Zhang RY, et al:
Construction of a eukaryotic expression vector pEGFP-C1-BMP-2 and
its effect on cell migration. J Zhejiang Univ Sci B. 13:356–363.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Durukan A and Tatlisumak T:
Preconditioning-induced ischemic tolerance: a window into
endogenous gearing for cerebroprotection. Exp Transl Stroke Med.
2:22010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dirnagl U, Becker K and Meisel A:
Preconditioning and tolerance against cerebral ischaemia: from
experimental strategies to clinical use. Lancet Neurol. 8:398–412.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schaller B: Ischemic preconditioning as
induction of ischemic tolerance after transient ischemic attacks in
human brain: its clinical relevance. Neurosci Lett. 377:206–211.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Castillo J, Moro MA, Blanco M, et al: The
release of tumor necrosis factor-alpha is associated with ischemic
tolerance in human stroke. Ann Neurol. 54:811–819. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang X, Li X, Erhardt JA, et al: Detection
of tumor necrosis factor-alpha mRNA induction in ischemic brain
tolerance by means of real-time polymerase chain reaction. J Cereb
Blood Flow Metab. 20:15–20. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Glazner GW and Mattson MP: Differential
effects of BDNF, ADNF9, and TNFalpha on levels of NMDA receptor
subunits, calcium homeostasis, and neuronal vulnerability to
excitotoxicity. Exp Neurol. 161:442–452. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Watters O and O’Connor JJ: A role for
tumor necrosis factor-α in ischemia and ischemic preconditioning. J
Neuroinflammation. 8:872011.
|
|
27
|
Watters O, Pickering M and O’Connor JJ:
Preconditioning effects of tumor necrosis factor-α and glutamate on
calcium dynamics in rat organotypic hippocampal cultures. J
Neuroimmunol. 234:27–39. 2011.
|
|
28
|
Davis DP and Patel PM: Ischemic
preconditioning in the brain. Curr Opin Anaesthesiol. 16:447–452.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Weller ML, Stone IM, Goss A, et al:
Selective overexpression of excitatory amino acid transporter 2
(EAAT2) in astrocytes enhances neuroprotection from moderate but
not severe hypoxia-ischemia. Neuroscience. 155:1204–1211. 2008.
View Article : Google Scholar
|
|
30
|
Tilleux S, Goursaud S and Hermans E:
Selective up-regulation of GLT-1 in cultured astrocytes exposed to
soluble mediators released by activated microglia. Neurochem Int.
55:35–40. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Su ZZ, Leszczyniecka M, Kang DC, et al:
Insights into glutamate transport regulation in human astrocytes:
cloning of the promoter for excitatory amino acid transporter 2
(EAAT2). Proc Natl Acad Sci USA. 100:1955–1960. 2003. View Article : Google Scholar : PubMed/NCBI
|