Introduction
Antimicrobial peptides (AMPs) are significant
components of the innate immune systems of numerous animal species,
where they act as an effective, largely non-discriminatory first
line of defense against invading pathogens (1). A variety of AMPs have been identified
from a variety of sources, ranging from plants and insects to lower
vertebrates and mammals, as described in previous studies (2,3).
Protegrin-1 (PG-1) is a β-hairpin AMP of 18 amino
acids (RGGRLCYCRRRFCVCVGR) that was originally isolated from
porcine leukocytes (4). PG-1 is
rich in cationic residues, such as arginine (Arg). The amphipathic
characteristic of this peptide enables it to interact with the
membranes of pathogens and kill the pathogen by releasing its
cellular contents (5). Studies
have revealed a number of the key steps in the action of protegrin,
including: Protegrin monomers dimerize in various types of lipid
environment; protegrin peptides interact strongly with lipid
bilayer membranes, particularly those that contain anionic lipids;
and protegrins form pores in lipid bilayers, which results in
uncontrolled ion transport and may be a key factor in bacterial
death (1). PG-1 is considered as a
potential pharmaceutical agent as it has shown a broad range of
antimicrobial activities against gram-positive and gram-negative
bacteria, including Escherichia coli (E. coli),
Pseudomonas aeruginosa and Neisseria gonorrhoeae
(5–7).
For pharmaceutical applications, a large quantity of
AMP is required. Chemical synthesis of AMPs is not economically
practical, particularly for the production of long peptides. Thus,
a biological expression system would be an improved alternative
method of production (2). Numerous
biological expression systems have been introduced for the
economical production of AMPs (8).
For the production of recombinant AMPs, E. coli-based
systems are well established (8,9) and
Pichia pastoris (P. pastoris) expression systems are
frequently used (10–14). However, each system has
limitations, including poor recovery yields, proteolysis of
products, low expression levels, toxicity of the product to host
cells, and occasionally an absence of the post-translational
modifications required for the biological activity of the AMPs
(2,8,15).
The aim of the present study was to express the
antimicrobial peptide PG-1 in P. pastoris X-33, isolate the
recombinant product and investigate its anticancer activity.
Materials and methods
Strains, vector, enzymes and other
reagents
E. coli DH5α (Shanghai Sangon Biological
Engineering Co. Ltd., Shanghai, China) was used for plasmid
amplification and P. pastoris X-33 was used for the
expression of the fusion protein. The pPICZα-A expression vector
and P. pastoris X-33 were purchased from Invitrogen Life
Technologies (Carlsbad, CA, USA). The restriction endonuclease, T4
DNA ligase and Taq DNA polymerase were purchased from Takara
Biotechnology Co., Ltd. (Dalian, China). Ni-chelating Sepharose
columns were purchased from Shanghai Sangon Biological Engineering
Technology and Services Co., Ltd. (Shanghai, China).
Cell culture
The HepG2 cell line was obtained from the Medical
College of Henan University of Science and Technology (Luoyang,
China). The cells were grown in Dulbecco’s modified Eagle’s medium
(DMEM; Shanghai Sangon Biological Engineering Co. Ltd.)
supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml
penicillin and 100 μg/ml streptomycin in a humidified 5%
CO2 atmosphere at 37°C.
Design and synthesis of the PG-1
nucleotide sequence
Based on the primary amino acid sequences of the
mature peptide and according to codon preference of P.
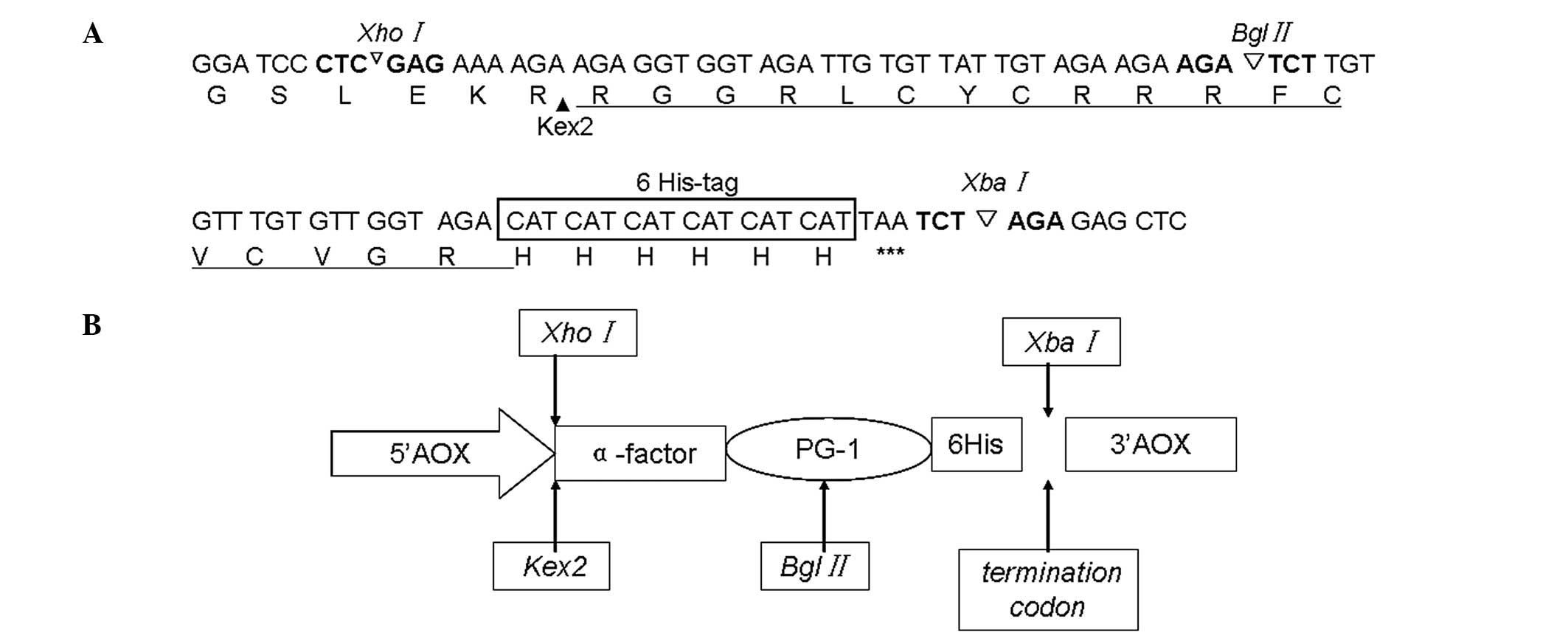
pastoris X-33, the gene sequence encoding PG-1 (Fig. 1A) was synthesized and used to
construct the plasmid pUC57-PG-1 (Shanghai Sangon Biological
Engineering Co. Ltd.). An XhoI restriction enzyme site was
introduced to allow in-frame cloning into the α-factor secretion
signal of the pPICZα-PG-1 shuttle vector. A sequence encoding the
Kex2 cleavage site (LEKR) and a 6His-tag were added upstream and
downstream of the PG-1 codon sequence, respectively. A termination
codon of PG-1 was introduced at the C-terminus, along with a
XbaI restriction enzyme site (Fig. 1B).
Construction of the expression
vectors
With the plasmid pUC57-PG-1 as the template, the
gene encoding PG-1 was amplified by polymerase chain reaction (PCR)
with the common primers pUC57+ (5′-ATCAGGCGCCATTCGCCATTC-3′) and
pUC57- (5′-CAGGTTCCCGACTGGAAAG-3′). The PCR mixture had a total
volume of 50 μl and comprised 5 μl 10X PCR buffer, 4 μl dNTP
mixture, 1 μl 20 μl/ml primer pUC57+, 1 μl 20 μl/ml primer pUC57-,
2 μl template, 1 μl Taq DNA polymerase and 36 μl ddH2O.
The PCR procedure involved 4 min predenaturation at 95°C; 30 sec
denaturation at 94°C, 30 sec annealing at 53°C and 30 sec extension
at 72°C for 30 cycles; followed by 3 min extension at 72°C. The
amplified products were verified by agarose gel electrophoresis.
The PCR products were and the pPICZα A plasmid were double digested
with XhoI and XbaI respectively, and ligated with T4 DNA
ligase to generate the fusion vector pPICZα-A. The plasmid was then
digested with the same restriction enzymes, to generate the fusion
vector pPICZα-A-PG-1. The ligation mixture was transformed into
E. coli DH5α, and recombinant E. coli cells were
selected on Zeocin-containing lysogeny broth plates (Invitrogen
Life Technologies). The recombinant plasmid pPICZα-A-PG-1 was
confirmed by restriction endonuclease digestion and DNA sequencing
analysis.
Transformation of P. pastoris and
selection of transformants
pPICZα-A-PG-1 was linearized with SacI and
transformed into competent cells of P. pastoris X-33
(Mut+) by electroporation (Gene Pulser Xcell™
Electroporation System; Bio-Rad, Hercules, CA, USA), according to
the manufacturer’s instructions (Pichia Expression kit;
Invitrogen Life Technologies). The empty pPICZα-A vector was
similarly linearized and transformed into P. pastoris X-33
as a negative control. All Zeocin-resistant colonies growing in
Yeast Extract Peptone Dextrose medium (1% yeast extract, 2%
peptone, 2% dextrose, 1 M sorbitol, 2% agar and 100 mg/ml Zeocin)
plates were plated in duplicate onto either minimal methanol with
histidine [MMH; 1.34% yeast nitrogen base (YNB), 0.05% biotin, 0.5%
methanol and 1.5% agar] or minimal dextrose with histidine (MDH;
1.34% YNB, 0.05% biotin, 1% dextrose and 1.5% agar) plates to
characterize the methanol-utilizing phenotype. Mut+
strains were obtained from the MDH plates and the inserts were
evaluated by PCR amplification of the yeast genomic DNA template,
using the common aldehyde oxidase 1 primers
(5′-GACTGGTTCCAATTGACAAGC and 5′-GCAAATGGCATTCTGACATCC). These
strains were subsequently used for the suspension culture.
Heterologous expression of recombinant
PG-1 in P. pastoris X-33
The positive P. pastoris transformants were
selected and inoculated in 10 ml buffered glycerol-complex medium
(1% yeast extract, 2% peptone, 100 mM potassium phosphate buffer pH
6.0, 1.34% YNB, 4×10−5% biotin and 1% glycerol) for 24 h
at 28°C. When the cell density reached ~0.6 at optical density
(OD)600 (725 UV Visible Spectrophotometer; Shanghai Precision &
Scientific Instrument Co., Ltd., Shanghai, China), the cells were
harvested by centrifugation at 3,000 × g for 2 min and resuspended
to an OD600 of 1.0 in buffered methanol-complex medium (1% yeast
extract, 2% peptone, 100 mM potassium phosphate buffer pH 6.0,
1.34% YNB, 4×10−5% biotin and 1.0% methanol). The cells
were then cultured for 4–5 days at 28°C in a flask while adding
methanol to a final concentration of 1% every 24 h. The cell
culture medium was harvested by centrifugation at 12,000 × g for 20
min. The presence of the fusion protein in the supernatant was
analyzed by Tricine-sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (Tricine-SDS-PAGE) with Coomassie Brilliant Blue
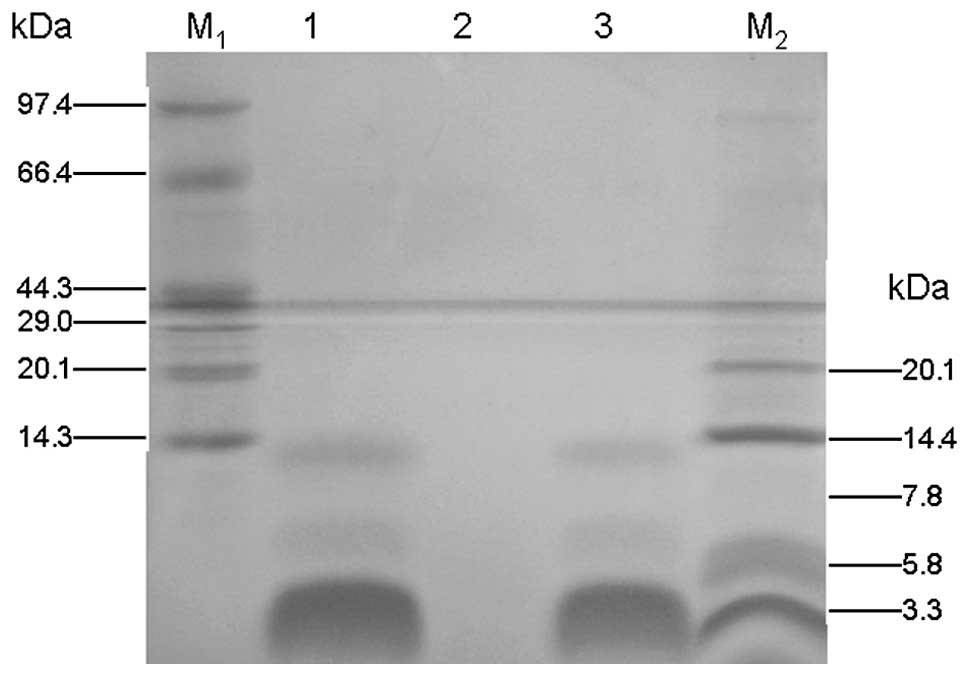
staining (16). As shown in
Fig. 2, a major protein
recombinant PG-1 band of ~2.9 kDa appeared following induction for
4 days. The protein concentration was determined using Bio-Rad
Protein Assay Dye Reagent with bovine serum albumin (BSA) as
standard (17).
Purification of peptide PG-1 and
concentration determination
The expression supernatant was applied to a
Ni-chelating Sepharose column (1.6×10 cm) pre-equilibrated with
phosphate-buffered saline to purify the PG-1 fusion protein.
Subsequently, the column was washed with 20 bed volumes of the same
buffer to remove contaminating proteins. The protein was eluted
with a linear gradient from 0.1 to 1.0 M imidazole in 1X Ni-NTA
elution buffer (50 mM NaH2PO4 pH 8.0 and 300
mM NaCl). The peak amount of PG-1 eluted was determined by analysis
of the fractions with 15% Tricine-SDS-PAGE, and the fractions
containing PG-1 were collected. The collected fractions were
dialyzed in Milli-Q water (Millipore, Billerica, MA, USA). The
protein concentration of the purified PG-1 was determined using
Bio-Rad Protein Assay Dye Reagent with BSA as standard.
Subsequently, the purified peptide was lyophilized and stored at
−20°C until use.
In vitro assay for cytotoxic
activity
The cytotoxicity of the purified PG-1 fusion protein
was determined by a tetrazolium (MTT) assay (18). The HepG2 cells (3×103
cells/well) were plated in 100 μl DMEM per well in 96-well plates
(Costar Corning, Corning, NY, USA). Following overnight incubation,
purified PG-1 was added in various concentrations (10, 20, 40, 60
and 160 μg/ml) to the wells, with six wells for each concentration.
After treatment with purified PG-1 for 1, 2, 3, 4 and 5 days, 20 μl
5 mg/ml MTT (pH 4.7) was added to each well and the cells were
cultivated for a further 4 h. The supernatant fluid was then
removed, 100 μl DMSO was added to each well and the samples were
agitated for 15 min. The absorbance at 490 nm was measured with a
microplate reader (Bio-Rad) using wells without cells as blanks.
Three independent experiments were performed for each set of
conditions.
Results
Construction of the recombinant plasmid
pPICZα-A-PG-1
Using degenerate primers, the PG-1 gene was
amplified by PCR and verified by agarose gel electrophoresis. The
PCR product was ligated into the pPICZα-A expression vector
together with an α-mating factor secretion signal sequence at the
N-terminus and a 6His-tag at the C-terminus of the PG-1 peptide.
The recombinant plasmid pPICZα-A-PG-1 was successfully constructed
and verified by restriction enzyme analysis and DNA sequencing.
Recombinant pPICZα-A-PG-1 expression and
purification
The recombinant plasmid pPICZα-A-PG-1 was linearized
with SacI and transformed into competent cells of P.
pastoris X-33. The pPICZα-A-PG-1 transformants of P.
pastoris were grown in flasks at 28°C and, after culture for
120 h, the cell culture medium was harvested by centrifugation. The
supernatant from the flask culture was analyzed by
Tricine-SDS-PAGE. As shown in Fig.
2, a major band at ~2.9 kDa was observed after a 96-h
induction. The protein concentration in the supernatent was
15.6mg/100ml, as measured using Bio-Rad Protein Assay Dye Reagent
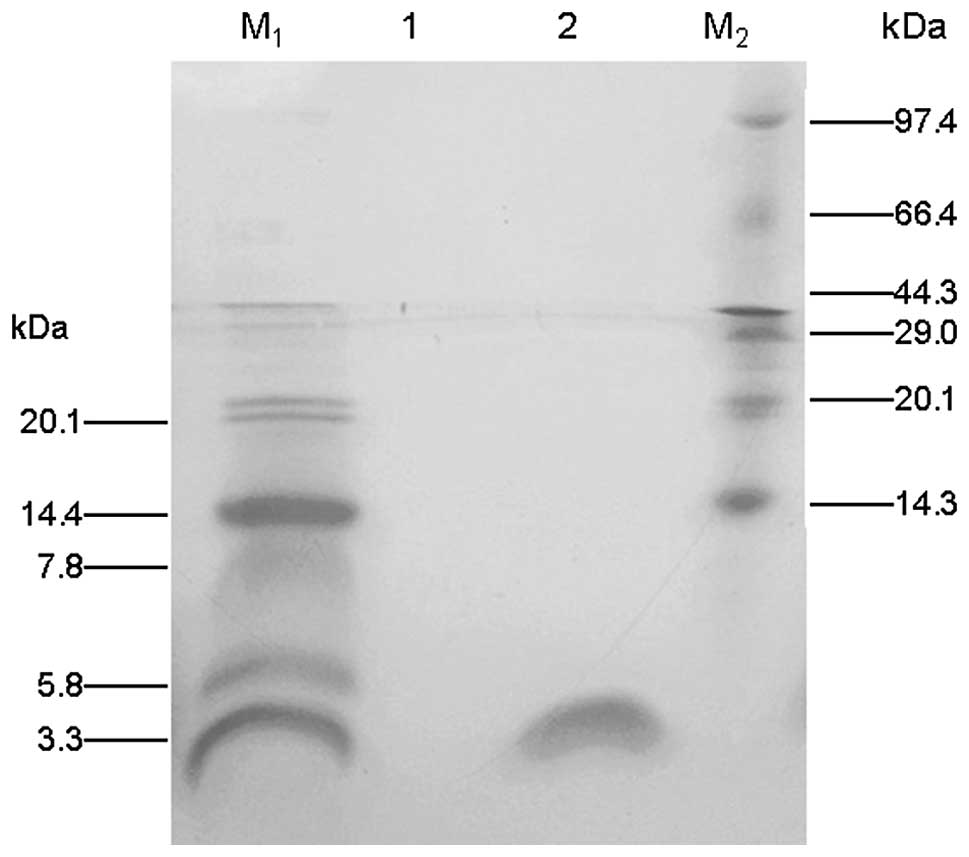
Using an Ni-NTA column, the fusion protein was eluted at 250 mM
imidazole. The eluted fractions were collected and dialyzed, and
the results of the Tricine-SDS-PAGE (Fig. 3) indicated that the recombinant
PG-1 fusion protein (2.9 kDa) had been obtained with high-purity.
Approximately 20 mg PG-1/6His was purified from 500 ml culture
medium.
Cytotoxic activity of PG-1 against HepG2
cells
The results of the in vitro assay of the
cytotoxic activity of the PG-1 fusion protein against HepG2 cells
are shown in Fig. 4. The
percentage of growth inhibition of the HepG2 cells by PG-1/6His at
various concentrations was determined by the number of viable
treated cells in comparison with the number of viable cells of the
untreated controls. The results showed that PG-1/6His had a dose-
and time-dependent inhibitory effect on cell growth.
Discussion
PG-1
(H3N+-RGGRLCYCRRRFCVCVGR-CONH2),
with a high content of positively charged Arg and cysteine (Cys)
residues, has a β-hairpin structure that is stabilized by disulfide
bonds linking Cys-6 and Cys-15, and Cys-8 and Cys-13 (19,20).
Its rigid structure separates the hydrophobic and hydrophilic
residues of the peptide, resulting in PG-1 having an amphipathic
nature that is common to numerous other types of AMP (1,21).
PG-1 is highly cationic (charge of 7+) at physiological pH, which
is essential for its ability to bind strongly to bacterial cell
membranes. It is the amphipathic characteristic of PG-1 that
enables it to interact with the membranes of pathogens (4). Protegrin monomers dimerize in various
types of lipid environment (1,4,19,21,22).
However, the characteristics of the protegrin peptide biologically
expressed in the present study, and whether fusion to 6His affects
its structure and function are unknown. These factors require
investigation in further studies.
The methylotrophic yeast P. pastoris has been
utilized widely as a heterologous gene expression system. Although
PG-1 has been successfully expressed in E. coli (8), the expression levels that have been
achieved are lower than those obtained for P.
pastoris-derived PG-1 in the present study. Furthermore,
certain recombinant AMPs produced in P. pastoris have
stronger activity than those produced in E. coli, including
crab AMP scygonadin (12), plant
defensin corn 1 (23), human
secretory leukocyte protease inhibitor (24) and non-specific lipid-transfer
proteins (25).
In order to facilitate the purification of PG-1 in
the present study, a 6His-tag was introduced at the C-terminus of
PG-1. A previous study has observed that Mdcec/6His has the same
levels of activity against bacteria as those of Mdcec, and has
slightly increased activity levels against fungi (11). P. pastoris-derived
scygonadin/6His has been effectively expressed with higher activity
levels against bacteria than those of scygonadin (12). In the present study, the expressed
PG-1/6His demonstrated strong dose- and time-dependent anticancer
activity against HepG2 cells.
Acknowledgements
This study was supported by grants from Henan
University of Science and Technology Youth Science Fund Project
(2010QN0015) and Ph.D. Programs Foundation (09001279).
References
|
1
|
Bolintineanu DS and Kaznessis YN:
Computational studies of protegrin antimicrobial peptides: a
review. Peptides. 32:188–201. 2011. View Article : Google Scholar :
|
|
2
|
Wang L, Lai C, Wu Q, Liu J, Zhou M, Ren Z,
Sun D, Chen S and Xu A: Production and characterization of a novel
antimicrobial peptide HKABF by Pichia pastoris. Process Biochem.
43:1124–1131. 2008. View Article : Google Scholar
|
|
3
|
Chen H, Xu Z, Peng L, Fang X, Yin X, Xu N
and Cen P: Recent advances in the research and development of human
defensins. Peptides. 27:931–940. 2006. View Article : Google Scholar
|
|
4
|
Rui H, Lee J and Im W: Comparative
molecular dynamics simulation studies of protegrin-1 monomer and
dimer in two different lipid bilayers. Biophys J. 97:787–795. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Panchal RG, Smart ML, Bowser DN, Williams
DA and Petrou S: Pore-forming proteins and their application in
biotechnology. Curr Pharm Biotechnol. 3:99–115. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kokryakov VN, Harwig SS, Panyutich EA,
Shevchenko AA, Aleshina GM, Shamova OV, et al: Protegrins:
leukocyte antimicrobial peptides that combine features of
corticostatic defensins and tachyplesins. FEBS Lett. 327:231–236.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qu XD, Harwig SS, Shafer WM and Lehrer RI:
Protegrin structure and activity against Neisseria gonorrhoeae.
Infect Immun. 65:636–639. 1997.PubMed/NCBI
|
|
8
|
Fan F, Wu Y and Liu J: Expression and
purification of two different antimicrobial peptides, PR-39 and
Protegrin-1 in Escherichia coli. Protein Expr Purif. 73:147–151.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Feng X, Liu C, Guo J, Song X, Li J, Xu W
and Li Z: Recombinant expression, purification, and antimicrobial
activity of a novel hybrid antimicrobial peptide LFT33. Appl
Microbiol Biotechnol. 95:1191–1198. 2012. View Article : Google Scholar
|
|
10
|
Moers AP, Wolbert EJ, de Wolf FA and
Werten MW: Secreted production of self-assembling peptides in
Pichia pastoris by fusion to an artificial highly hydrophilic
protein. J Biotechnol. 146:66–73. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jin F, Xu X, Zhang W and Gu D: Expression
and characterization of a housefly cecropin gene in the
methylotrophic yeast, Pichia pastoris. Protein Expr Purif.
49:39–46. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Peng H, Liu HP, Chen B, Hao H and Wang KJ:
Optimized production of scygonadin in Pichia pastoris and analysis
of its antimicrobial and antiviral activities. Protein Expr Purif.
82:37–44. 2012. View Article : Google Scholar
|
|
13
|
Zhang J, Yang Y, Teng D, Tian Z, Wang S
and Wang J: Expression of plectasin in Pichia pastoris and its
characterization as a new antimicrobial peptide against
Staphyloccocus and Streptococcus. Protein Expr Purif. 78:189–196.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Niu M, Li X, Wei J, Cao R, Zhou B and Chen
P: The molecular design of a recombinant antimicrobial peptide CP
and its in vitro activity. Protein Expr Purif. 57:95–100. 2008.
View Article : Google Scholar
|
|
15
|
Lee JH, Kim JH, Hwang SW, Lee WJ, Yoon HK,
Lee HS and Hong SS: High-level expression of antimicrobial peptide
mediated by a fusion partner reinforcing formation of inclusion
bodies. Biochem Biophys Res Commun. 277:575–580. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schägger H and von Jagow G: Tricine-sodium
dodecyl sulfate-polyacrylamide gel electrophoresis for the
separation of proteins in the range from 1 to 100 kDa. Anal
Biochem. 166:368–379. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lehrer RI, Rosenman M, Harwig SS, Jackson
R and Eisenhauer P: Ultrasensitive assays for endogenous
antimicrobial polypeptides. J Immunol Methods. 137:167–173. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Selvakumaran M, Pisarcik DA, Bao R, Yeung
AT and Hamilton TC: Enhanced cisplatin cytotoxicity by disturbing
the nucleotide excision repair pathway in ovarian cancer cell
lines. Cancer Res. 63:1311–1316. 2003.PubMed/NCBI
|
|
19
|
Capone R, Mustata M, Jang H, Arce FT,
Nussinov R and Lal R: Antimicrobial protegrin-1 forms ion channels:
molecular dynamic simulation, atomic force microscopy, and
electrical conductance studies. Biophys J. 98:2644–2652. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Langham AA, Khandelia H, Schuster B,
Waring AJ, Lehrer RI and Kaznessis YN: Correlation between
simulated physicochemical properties and hemolycity of
protegrin-like antimicrobial peptides: predicting experimental
toxicity. Peptides. 29:1085–1093. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jang H, Ma B, Lal R and Nussinov R: Models
of toxic beta-sheet channels of protegrin-1 suggest a common
subunit organization motif shared with toxic alzheimer beta-amyloid
ion channels. Biophys J. 95:4631–4642. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bolintineanu D, Hazrati E, Davis HT,
Lehrer RI and Kaznessis YN: Antimicrobial mechanism of pore-forming
protegrin peptides: 100 pores to kill E. coli. Peptides. 31:1–8.
2010. View Article : Google Scholar :
|
|
23
|
Kant P, Liu WZ and Pauls KP: PDC1, a corn
defensin peptide expressed in Escherichia coli and Pichia pastoris
inhibits growth of Fusarium graminearum. Peptides. 30:1593–1599.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Z, Moy A, Sohal K, Dam C, Kuo P,
Whittaker J, Whittaker M, Düzgünes N, Konopka K, Franz AH,
Lin-Cereghino J and Lin-Cereghino GP: Expression and
characterization of recombinant human secretory leukocyte protease
inhibitor (SLPI) protein from Pichia pastoris. Protein Expr Purif.
67:175–181. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pokoj S, Lauer I, Fötisch K, Himly M, Mari
A, Enrique E, del Miguel-Moncin MM, Lidholm J, Vieths S and
Scheurer S: Pichia pastoris is superior to E. coli for the
production of recombinant allergenic non-specific lipid-transfer
proteins. Protein Expr Purif. 69:68–75. 2010. View Article : Google Scholar
|