Introduction
Lower back pain (LBP) is a common and remitting
problem that cannot be cured but is relieved by current treatments.
Multiple studies have demonstrated that 80% of adults will have at
least one episode of back pain during adulthood (1,2). One of
the main causes of LBP is thought to be degeneration of the
intervertebral disc (IVD) (3).
However, current treatments for IVD degeneration and LBP are aimed
at relieving symptoms; they are not curative and offer little hope
of restoring the IVD to its original function (4). To date, there is no approved
conservative therapy to prevent or inhibit IVD degeneration.
Therefore, elucidating the mechanisms of IVD degeneration will be
necessary for the development of agents to prevent and treat IVD
degeneration.
The IVD is a composite tissue, composed of the
nucleus pulposus (NP), annulus fibrosis, and cartilaginous end
plate. Human IVD degeneration is characterized by changes in
architecture and biochemical composition, which alter the disc's
ability to bear weight (5). Studies
characterizing the extracellular matrix (ECM) and the inflammatory
environment of IVD tissue isolated from surgical patients found
that degeneration was associated with a loss of proteoglycan (PG)
content, an increase in degenerative fibrillation, decreased water
content and upregulation of degradative enzymes (6–10). Via
an upregulation of molecules such as proinflammatory cytokines and
catabolic growth factors, homeostasis of the ECM shifts toward a
degenerative, catabolic state with subsequent breakdown of ECM
components, including the collagen fibrils surrounding and
restraining large, hydrated aggregates of PG, principally aggrecan
(11–14). Elevated levels of molecular mediators
of inflammation have been described in pathological disc tissue,
and have been shown to increase in correlation with the grade of
degeneration (15,16). Similar results have been observed for
interleukin-1β (IL-1β) and tumor necrosis factor α (TNFα), both of
which have established roles in regulating nitric oxide (NO) and
prostaglandin production, metalloproteinase expression, and
apoptosis, all of which are changes that may contribute to the
progressive pathology of the IVD (17). Therefore, it is crucial to identify
and inhibit the key points of the signal pathway responsible for
producing proinflammatory cytokines and matrix metalloproteases for
the prevention and treatment of IVD degeneration.
The mammalian Toll-like receptors (TLRs),
germline-encoded receptors expressed by cells of the innate immune
system, are stimulated by structural motifs referred to as
pathogen-associated molecular patterns (PAMPs), which are
characteristically expressed by bacteria, viruses and fungi
(18,19). Notably, TLR interactions trigger the
expression of proinflammatory cytokines, as well as the functional
maturation of antigen presenting cells of the innate immune system
(19,20). TLR4 signals the presence of
lipopolysaccharide (LPS) on the cell membrane of gram-negative
bacteria and activates an inflammatory response (21). When the majority of TLRs are
stimulated, they interact with an adapter protein referred to as
myeloid differentiation primary response gene 88 (MyD88), which
couples the TLR to downstream signaling kinases, eventually
culminating in the activation (by translocation from the cytoplasm
to the nucleus) of the transcription factor nuclear factor κB
(NF-κB) (22). However,
MyD88-independent signaling pathways for TLR3 and TLR4 are also
known to exist (23). Therefore,
TLR4 is able to affect signal transduction in two different ways,
either through MyD88 or a TIR domain-containing adapter that
induces interferon-beta (TRIF), while TLR3 can signal only through
TRIF (24). Although the
MyD88-dependent and -independent pathways utilize distinct adapter
proteins, both signaling pathways involve the activation and
nuclear translocation of NF-kB, leading to the expression of
numerous proinflammatory cytokines. To date, it has been confirmed
that the TLR4 gene is expressed in IVD NP cells, and that it plays
an important role in the molecular mechanism of IVD degeneration
(6). However, the signal
transduction pathway that activates the TLR4 in NP cells in IVD
degeneration remains unknown, as does the MyD88-dependent or
-independent nature of this signal transduction pathway.
The aims of the current study were to determine
whether the TLR4 signal pathway in IVD degeneration was
MyD88-dependent or -independent, as well as to assess the
consequences associated with activation of TLR4 in IVD NP cells.
Achievement of these aims may provide direct evidence in support of
the hypothesis that the MyD88-dependent TLR4 signal pathway is the
target pathway underlying IVD degeneration, as well as providing a
theoretical basis for researching the molecular mechanisms
underlying IVD degeneration.
Materials and methods
Isolation and culture of IVD NP
cells
Animal experiments were approved by the ethics
review board of Guangzhou Medical University (Guangzhou, China) and
were performed in accordance with the guidelines on animal use of
Guangzhou Medical University. NP cells were isolated from
4-week-old female Wistar rat (weight, 200 g; Laboratory Animal
Center, Sun Yat-sen University) lumbar discs using methods reported
by Hiyama et al (25).
Briefly, the rats were euthanized by injection with an overdose of
pentobarbital sodium (100 mg/kg; Nembutal; Amresco LLC, Cleveland,
OH, USA). The spinal column was then removed under aseptic
conditions, and the lumbar IVDs were separated under microscopy.
The obtained NP tissue was allowed to digest in a mixture of 0.01%
trypsin (Sigma-Aldrich, St. Louis, MO, USA) at 37°C for 15 min. The
isolated cells (1×108 L−1) were maintained in
Dulbecco's modified Eagle's medium/Nutrient Mixture F-12 (DMEM/F12,
1:1) and 10% fetal bovine serum supplemented with 100 U/ml
penicillin and 100 µg/ml streptomycin (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) at 37°C in a humidified
atmosphere of 5% CO2. When confluent, the NP cells were
harvested and subcultured in 10-cm dishes.
Morphological observation
For the observation of morphology, 6-well culture
plates with an additional coverglass in each well were used. The
primary or P1 NP chondrocytes that adhered to the coverglasses were
used for observation of morphological changes under an inverted
phase contrast microscope (IX51; Olympus Corporation, Tokyo,
Japan). For hematoxylin and eosin (HE) staining, the coverglasses
were washed with phosphate-buffered saline (PBS) prior to fixation
in 4% paraformaldehyde for 30 min, followed by consecutive staining
in HE. For Oil Red O staining (Sigma-Aldrich), the coverglasses
were washed with PBS and fixed as for HE staining, stained with Oil
Red O for 30 min, and counterstained with hematoxylin for another 5
min. For toluidine blue staining, the coverglasses were washed and
fixed as for the HE staining, and were immersed for 2 h in a 1%
toluidine blue solution (KeyGen Biotech Co., Ltd., Nanjing, China)
prior to rinsing in 95% ethanol. For immunohistochemistry staining
of collagen II, the endogenous peroxidase was blocked by 3%
H2O2 in methanol, and then the coverglasses
were incubated for 30 min with anti-human type II collagen antibody
(ab34712; Abcam, Cambridge, MA, USA) at a 1:50 dilution. A
secondary antibody linked with avidin-biotin-peroxidase (SV0002;
Abcam) and 3,3′-diaminobenzidine substrate solutions was used to
visualize the immunoreactivity, followed by counterstaining in
hematoxylin. Negative control was processed without the anti-rat
type II collagen antibody (26,27).
Co-culture of NP cells and LPS
A 6-well co-cultured system was used. LPS was
suspended in sterile dH2O by sonication, diluted in
serum-free media, and re-sonicated immediately prior to use. Cells
were rinsed and cultured in serum-free DMEM/F12 (control) ± LPS (10
µg/ml) for 1, 3, 6, 9 or 12 h (n=3–6 wells per time phase point)
prior to use of the reverse transcription-quantitative polymerase
chain reaction (RT-qPCR) to determine mRNA (TLR4, MyD88, TNFα and
IL-1β) expression levels, and ensure a best time phase point. In
addition, cells were rinsed and cultured in serum-free DMEM/F12
(control) ± LPS (10 µg/ml) for 24, 48 and 72 h (n=3–6 wells per
time phase point), prior to western blot and enzyme-linked
immunosorbent assay (ELISA) analyses to determine protein (TLR4,
MyD88, TNFα and IL-1β) expression levels, and ensure a best time
phase point. In additional experiments, cells were rinsed and
cultured in serum-free DMEM/F12 (control) ± LPS (0.1, 1, 10 and 100
µg/ml, n=3–6 wells per dose) for different lengths of time (as
determined by the results of the qPCR experiment), prior to use of
qPCR to determine the mRNA (TLR4, MyD88, TNFα and IL-1β) expression
levels, and ensure a best dose of LPS. In other experiments, cells
were rinsed and cultured in serum-free DMEM/F12 (control) ± LPS
(0.1, 1, 10 and 100 µg/ml, n=3–6 wells per dose) for different
lengths of time (determined by the results of the western blot and
ELISA analyses), prior to the use of western blot and ELISA
analyses to determine the protein (TLR4, MyD88, TNFα and IL-1β)
expression levels, and ensure a best dose of LPS.
RT-qPCR
Total RNA was isolated from cell cultures at various
time phase points using RNAiso Plus reagent (Takara Bio, Inc.,
Tokyo, Japan), prior to elution from the column, RNA was treated
with RNase-free DNase I to remove genomic DNA. Absorbances at 260
and 280 nm were measured for RNA quantification and quality
control. All RNA samples exhibited high quality RNA and were
subsequently reverse transcribed to cDNA using the PrimeScript™ RT
reagent kit (Perfect Real Time) according to the manufacturer's
instructions (Takara Bio, Inc.). Subsequently, qPCR was conducted
to determine the levels of mRNA expression using an ABI Prism 7000
sequence detection system (Applied Biosystems; Thermo Fisher
Scientific, Inc., Foster City, CA, USA) in triplicate in 96-well
plates in a final volume of 20 µl under standard conditions. qPCR
was conducted on cDNA samples using the SYBR Green method with
SYBR® Premix Ex Taq™ (Tli RNaseH Plus; Takara Bio,
Inc.). Reaction mixes contained 10 µl 2X SYBR Green mastermix, 1 µl
(6 µM) forward primer, 1 µl (6 µM) reverse primer, 6 µl water and 2
µl (5 ng/µl) cDNA. qPCR was performed as follows: Initial
denaturation at 95°C for 30 sec for activation of AmpliTaq Cold DNA
polymerase (Applied Biosystems; Thermo Fisher Scientific, Inc.),
followed by 40 cycles of denaturation at 95°C for 5 sec, annealing
at 60°C for 30 sec, and extension at 95°C for 15 sec. Forward and
reverse primer sequences are listed in Table I and were synthesized by Takara Bio,
Inc. To normalize each sample, a control gene (GAPDH) was used, and
the arbitrary intensity threshold of amplification was computed.
The 2-ΔΔCq method was used to calculate the relative
expression of each target gene, as described previously (28).
 | Table I.Primers for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primers for reverse
transcription-quantitative polymerase chain reaction.
| Target | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| GAPDH |
ATGGGAAGCTGGTCATCAAC |
GTGGTTCACACCCATCACAA |
| TLR4 |
GAGGACTGGGTGAGAAACGA |
AGATACACCAACGGCTCTGG |
| MyD88 |
GAGATCCGCGAGTTTGAGAC |
CTGTTTCTGCTGGTTGCGTA |
| TNFα |
CATCTGCTGGTACCACCAGTT |
TGAGCACAGAAAGCATGATC |
| IL-1β |
GGGTTCCATGGAGAAGTCAAC |
CACCTCTCAAGCAGAGCACAG |
Western blot analysis
Following treatment, NP cells were immediately
placed on ice and washed with cold PBS. Proteins were prepared
using the CellLytic NuCLEAR extraction kit (Sigma-Aldrich). Protein
quantification was performed using a microplate bicinchoninic acid
protein assay kit (Pierce Biotechnology, Thermo Fisher Scientific,
Inc., Rockford, IL, USA). All wash buffers and the final
resuspension buffer included a 1X protease inhibitor cocktail
(Pierce Biotechnology), NaF (5 mM) and Na3VO4
(200 mM). Nuclear or total cell proteins (50 µg) were separated by
10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and
were subsequently electroblotted onto nitrocellulose membranes
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Membranes were
blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline
with Tween 20 (TBST: 50 mM Tris, pH 7.6, 150 mM NaCl, 0.1% Tween
20) and were incubated overnight at 4°C in 5% BSA in TBST with an
anti-β-catenin antibody (1:1,000; 9582; Cell Signaling Technology,
Inc., Danvers, MA, USA) for 1 h, followed by washing three times
with TBS and incubation with a peroxidase-conjugated goat
anti-rabbit secondary antibody (1:5,000; 111-035-003; Jackson
Immunoresearch, Baltimore, MD, USA) for 2 h at room temperature.
Immunolabeling was detected using enhanced chemiluminescence
reagents (Amersham Biosciences; GE Healthcare, Little Chalfont,
UK.
ELISA
The concentrations of TNFα and IL-1β in the NP cells
were assayed using an ELISA kit (Invitrogen; Thermo Fisher
Scientific, Inc., Carlsbad, CA, USA) according to the
manufacturer's instructions.
Statistical analysis
Experiments were repeated three times in biological
replicates to obtain mean values. Data are presented as the mean ±
standard deviation. Differences among groups were assessed using a
one-way analysis of variance. Least significant difference t-tests
were used when a single control group was compared with all other
groups. Statistical significance (P<0.001) as compared to
control group is denoted with an asterisk (*). All statistical
analyses were conducted using SPSS 13.0 software (SPSS, Inc.,
Chicago, IL, USA).
Results
Characterization of IVD NP cells
The third passage of NP cells appeared round or
multi-angular with activity. HE staining appeared homogeneous with
blue nuclei and pink cytoplasm; no cells size increases were
observed. No inhomogeneous or poor light refraction in the
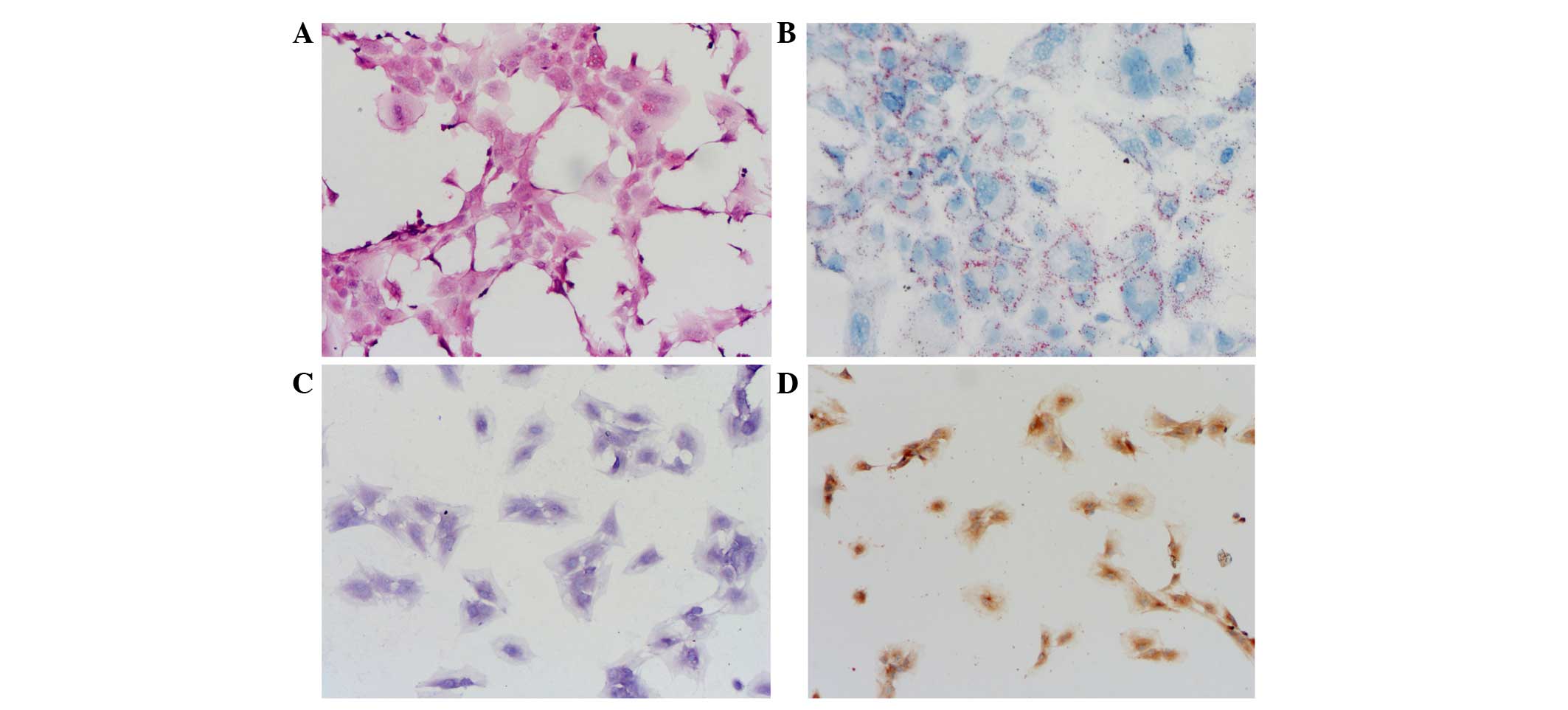
cytoplasm was detected (Fig. 1A).
Oil Red O staining (Fig. 1B),
toluidine blue staining (Fig. 1C)
and type II collagen immunohistochemistry staining (Fig. 1D) were all positive and maintained a
good cell phenotype. These results indicated that the third passage
of NP cells were active and homogeneous, thus were fit for
researching the molecular mechanism of IVD degeneration.
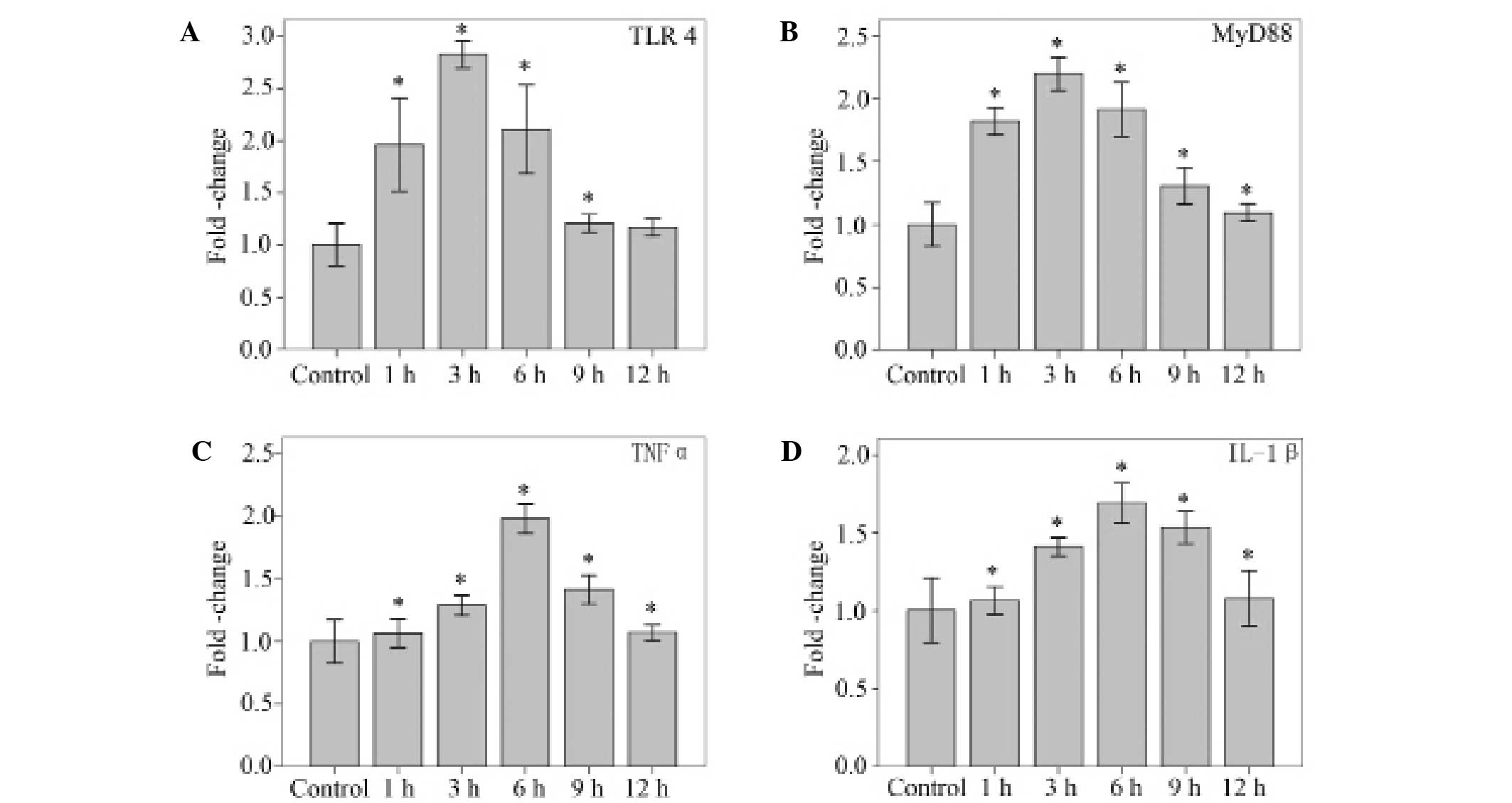
Time phase-dependent association
between LPS and the TLR4 signal pathway (TLR4, MyD88, TNFα and
IL-1β)
Using qPCR, mRNA levels for TLR4, MyD88, and the
proinflammatory cytokines TNFα and IL-1β were detected in NP cells
stimulated with 10 µg/ml LPS. LPS (10 µg/ml) significantly
increased the mRNA levels of TLR4 at 1, 3, 6 and 9 h (P<0.001,
P<0.001, P<0.001 and P=0.040, respectively), and MyD88 at 1,
3, 6, 9 and 12 h (all P<0.001). The mRNA levels of TNFα and
IL-1β increased significantly at 1, 3, 6, 9 and 12 h (all
P<0.001). The peak TLR4, MyD88, TNFα and IL-1β responses were
observed at 3, 3, 6 and 6 h, respectively (Fig. 2).
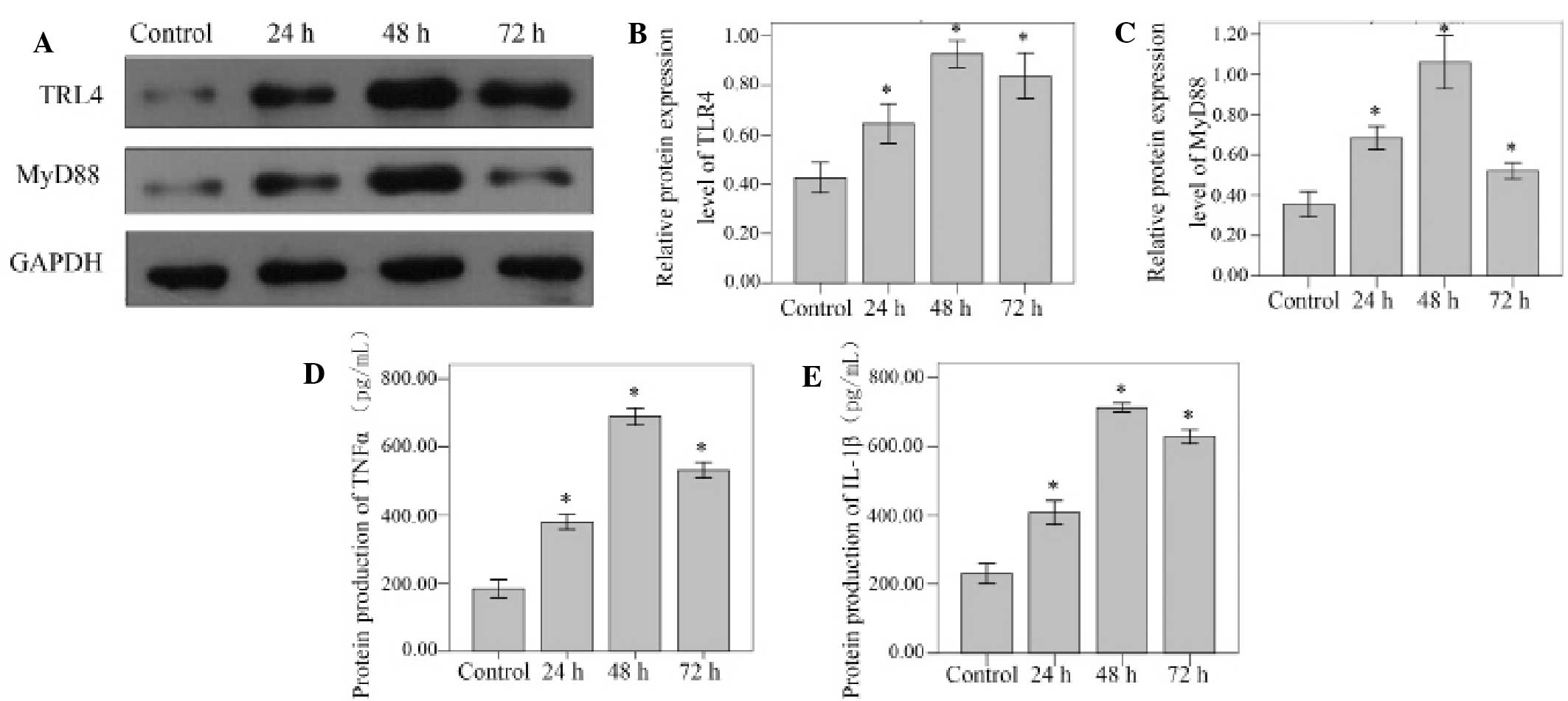
Western blot analysis was used to detect the TLR4
and MyD88 protein levels. Compared with unstimulated control cells,
the TLR4 and MyD88 protein levels increased significantly at 24, 48
and 72 h (all P<0.001). Peak protein expression levels were
observed at the 48 h time point for TLR4 and MyD88. (Fig. 3A-C).
The levels of TNFα and IL-1β protein were confirmed
via ELISA analysis of the cell supernatants. TNFα and IL-1β protein
levels standardized against GAPDH increased significantly at 24, 48
and 72 h in the NP cells stimulated by 10 µg/ml LPS (all
P<0.001). Levels of both proteins reached a peak at the 48 h
time point (Fig. 3D and E).
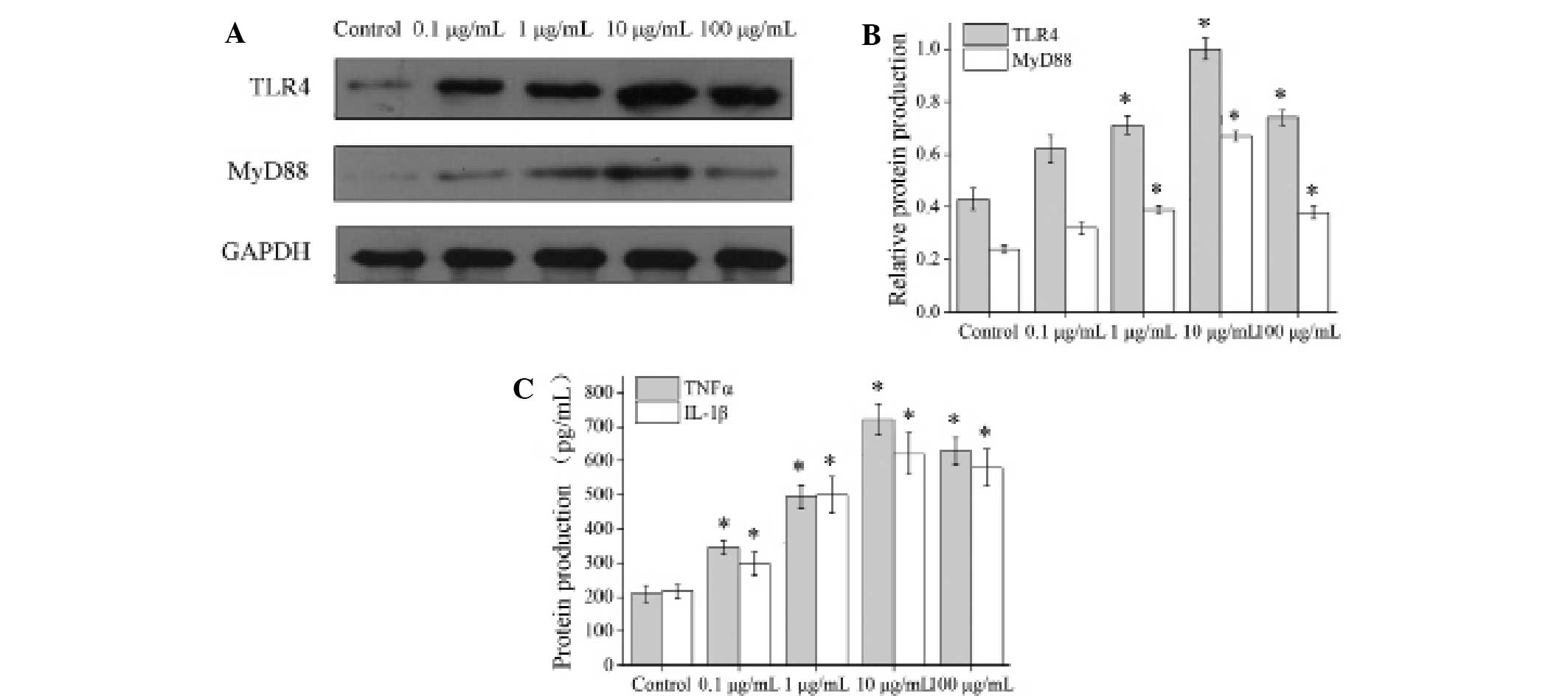
Dose-dependent association between LPS
and the TLR4 signal pathway (TLR4, MyD88, TNFα and IL-1β)
The NP cells stimulated by 0.1, 1, 10 and 100 µg/ml
LPS were assessed using qPCR. The mRNA levels of TLR4 and MyD88
were detected by qPCR after 3 h, and the mRNA levels of TNFα and
IL-1β were detected after 6 h. The results indicated that the mRNA
levels of TLR4, MyD88, TNFα and IL-1s increased significantly in
the cells stimulated by 0.1, 1, 10 and 100 µg/ml LPS as compared to
a control group, and reached a peak in the 10 µg/ml LPS group (all
P<0.001; Fig. 4).
Using western blot and ELISA analyses, TLR4, MyD88,
TNFα and IL-1β proteins were detected in NP cells stimulated with
LPS (0.1, 1, 10 and 100 µg/ml) for 48 h. Western blot analysis was
used to detect TLR4 and MyD88 protein levels (Fig. 5A), and ELISA analysis was used to
confirm the presence of TNFα and IL-1β proteins. The results
indicated that levels of all four target proteins were increased
significantly in the LPS stimulated cells, most notably in the 10
µg/ml LPS group (TLR4, all P<0.001; MyD88, P=0.019, P<0.001,
P<0.001 and P<0.001, respectively; TNFα and IL-1β, all
P<0.001) (Fig. 5).
Discussion
The aims of the current study were to investigate
whether TLR4 was expressed in IVD NP cells, and if so, whether its
signaling was MyD88-dependent. Additionally, the response of NP
cells to stimulation with a TLR4 ligand (LPS) was evaluated in
vitro. To accomplish these goals, the presence of a functional
TLR4 signal pathway in the IVD of rats was confirmed, and LPS was
used to stimulate NP cells in order to assess the time
phase-dependent association between LPS and the TLR4 signal
pathway. The present results demonstrated that NP cells
constitutively expressed TLR4 that could be activated by LPS at
different time phase points. Subsequently, we used various
concentrations of LPS to stimulate NP cells in order to ascertain
the nature of the dose-dependent association between LPS and the
TLR4 signal pathway. Indeed, TLR4 expression was modulated by
stimulation with LPS in a dose-dependent manner via the
MyD88-dependent signal pathway, resulting in upregulation of a
coordinated set of proinflammatory mediators in vitro.
Currently, the treatment of symptomatic IVD
degeneration consists of either conservative measures, such as the
application of analgesics and physiotherapy or surgery in cases
where conservative measures prove unhelpful (29). These approaches have not been shown
to slow the degeneration process, and consequently, relapses or
other adverse sequelae of discectomy, dynamic stabilization
techniques, total disc replacement or fusion surgery may be
expected (30–33). As IVD degeneration has a high
prevalence and is associated with major socioeconomic costs, the
current study sought to elucidate causative mechanisms underlying
IVD degeneration in order to identify targets for its prevention
and therapy.
Recent studies have partially elucidated the
mechanisms of the LPS/TLR4 signaling pathway, and this
understanding may be applied to model the regulation of additional
TLR4 signaling pathways (34,35). As
improper regulation of LPS/TLR4 signaling has the potential to
induce massive inflammation and cause acute sepsis or chronic
inflammatory disorders, it is crucial to investigate this pathway
further and evaluate novel targets to counteract these conditions
(36,37).
Consistent with the known response of TLR4
activation in the immune system, the present results indicated that
the expression of TNFα and IL-1β in NP cells was significantly
upregulated in response to various concentrations of LPS at various
time points, confirming that TLR4 activation can trigger an
inflammatory cascade in IVD NP cells. These findings demonstrated
that LPS was capable of upregulating a coordinated set of
inflammatory cytokines via the MyD88-dependent TLR4 signal pathway,
and that the response to inflammatory stimulation was time phase-
and dose-dependent.
In addition to the PAMPs, such as LPS from
Gram-negative bacteria, a fusion protein from the respiratory
syncytial virus, and the envelope protein from a mouse mammary
tumor virus, TLR4 signaling may also be initiated by endogenous
molecules that interact either directly or indirectly with TLR4,
such as heat-shock proteins, hyaluronic acid and β-defensin 2
(34). LPS is among the most
extensively studied immunostimulatory components of bacteria, and
can induce systemic inflammation and sepsis if excessive signals
occur (38). Furthermore, LPS is an
important structural component of the outer membrane of
Gram-negative bacteria, consisting of three parts; lipid A, core
oligosaccharide and an O side chain. Lipid A is the main PAMP of
LPS (34). Using the C3H/HeJ mouse
strain, known to have a defective response to LPS, Beutler's group
demonstrated that TLR4 was an important sensor for LPS (39). The active upregulation of TLR4 in IVD
NP cells following LPS stimulation suggests that TLR4 may
participate in these responses.
The ability of TLR4 activation by LPS to provoke the
secretion of multiple cytokines may provide an opportunity to study
broad aspects of the physiological inflammatory process in the IVD.
Results from the current study suggest that in vivo
inflammatory stimulation can induce degenerative changes in the
IVD, without the use of physical destruction of disc integrity as
an injury stimulus (6). Commonly
used animal models of degeneration, where a stab or laceration
lesion of the disc is performed, and reproduce morphological
changes of IVD degeneration in general (40–43).
These animal models have also been associated with transient
increases in the expression or secretion of proinflammatory
cytokines (44,45), results that differ from what is
observed in human degenerative disc diseases. Clinical disc
degeneration in humans is associated with chronically elevated
levels of multiple proinflammatory cytokines, indicative of the
crucial roles that inflammatory mediators play in degenerative
etiology. The findings of the current study indicate that altering
the stimulus of degeneration, from physical disruption to an
inflammatory stimulant, can also initiate a degenerative process in
the IVD in vivo. This suggests that inflammatory insults
alone may be able to initiate degeneration of the IVD.
The current study had several limitations. For
example, LPS was the only agent used to interfere with the studied
signal pathway. Additionally, we did not study any aspect of other
TLR4 signal pathways, including the MyD88-independent pathway. In a
planned study, we will interfere with different location points
(e.g., MyD88 and TRAF6) along the TLR4 signal pathway using siRNA,
and will also study changes to the ECM following activation of the
TLR4 signal pathway.
In conclusion, the present findings confirm the
activity of the MyD88-dependent TLR4 signal pathway in IVD NP cells
in IVD degeneration, and that this signal pathway was stimulated by
LPS in vitro. Activation of this pathway was time phase- and
dose-dependent. When activated, this signal pathway led to the
release of inflammatory factors that participated in IVD
degeneration. Although these findings cannot be used directly to
prevent and treat IVD degeneration, they suggest a potential
therapeutic benefit of inhibition of the MyD88-dependent TLR4
signal pathway in degenerative disc disease. Therefore, the results
of the current study provide a foundation for further
investigations into therapeutic agents designed to prevent or treat
IVD degeneration.
Acknowledgements
This study was supported by a grants from the
Medical Scientific Research Foundation of Guangdong Province (grant
no. B2013284), the Scientific Research Foundation of Guangzhou
Medical University (grant no. 2012C61). and the Natural Science
Foundation of Guangdong Province (grant no. 2016A030313607).
References
|
1
|
Deyo RA, Cherkin D, Conrad D and Volinn E:
Cost, controversy, crisis: Low back pain and the health of the
public. Annu Rev Public Health. 12:141–156. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kabbara A and Hayek SM: Intradiscal
electrothermal therapy (IDET) for the treatment of discogenic pain.
Tech Reg Anesth Pain Manag. 13:102–108. 2009. View Article : Google Scholar
|
|
3
|
Cheung KM, Karppinen J, Chan D, Ho DW,
Song YQ, Sham P, Cheah KS, Leong JC and Luk KD: Prevalence and
pattern of lumbar magnetic resonance imaging changes in a
population study of one thousand forty-three individuals. Spine
(Phila Pa 1976). 34:934–940. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ireland D: Molecular mechanisms involved
in intervertebral disc degeneration and potential new treatment
strategies. Biosci Horiz. 2:83–89. 2009. View Article : Google Scholar
|
|
5
|
Maidhof R, Alipui DO, Rafiuddin A, Levine
M, Grande DA and Chahine NO: Emerging trends in biological therapy
for intervertebral disc degeneration. Discov Med. 14:401–411.
2012.PubMed/NCBI
|
|
6
|
Rajan NE, Bloom O, Maidhof R, Stetson N,
Sherry B, Levine M and Chahine NO: Toll-like receptor 4 (TLR4)
expression and stimulation in a model of intervertebral disc
inflammation and degeneration. Spine (Phila Pa 1976). 38:1343–1351.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Antoniou J, Steffen T, Nelson F,
Winterbottom N, Hollander AP, Poole RA, Aebi M and Alini M: The
human lumbar intervertebral disc: Evidence for changes in the
biosynthesis and denaturation of the extracellular matrix with
growth, maturation, ageing, and degeneration. J Clin Invest.
98:996–1003. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roughley PJ, Alini M and Antoniou J: The
role of proteoglycans in aging, degeneration and repair of the
intervertebral disc. Biochem Soc Trans. 30:869–874. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Le Maitre CL, Freemont AJ and Hoyland JA:
Localization of degradative enzymes and their inhibitors in the
degenerate human intervertebral disc. J Pathol. 204:47–54. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pockert AJ, Richardson SM, Le Maitre CL,
Lyon M, Deakin JA, Buttle DJ, Freemont AJ and Hoyland JA: Modified
expression of the ADAMTS enzymes and tissue inhibitor of
metalloproteinases 3 during human intervertebral disc degeneration.
Arthritis Rheum. 60:482–491. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goldring MB: The role of the chondrocyte
in osteoarthritis. Arthritis Rheum. 43:1916–1926. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee S, Moon CS, Sul D, Lee J, Bae M, Hong
Y, Lee M, Choi S, Derby R, Kim BJ, et al: Comparison of growth
factor and cytokine expression in patients with degenerated disc
disease and herniated nucleus pulposus. Clin Biochem. 42:1504–1511.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Masuda K, Imai Y, Okuma M, Muehleman C,
Nakagawa K, Akeda K, Thonar E, Andersson G and An HS: Osteogenic
protein-1 injection into a degenerated disc induces the restoration
of disc height and structural changes in the rabbit anular puncture
model. Spine. 31:742–754. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ellman MB, Kim JS, An HS, Chen D, KC R, An
J, Dittakavi T, van Wijnen AJ, Cs-Szabo G, Li X, et al: Toll-like
receptor adaptor signaling molecule MyD88 on intervertebral disk
homeostasis: in vitro, ex vivo studies. Gene. 505:283–290. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Weiler C, Nerlich AG, Bachmeier BE and
Boos N: Expression and distribution of tumor necrosis factor alpha
in human lumbar intervertebral discs: A study in surgical specimen
and autopsy controls. Spine (Phila Pa 1976). 30:44–53; discussion
54. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Le Maitre CL, Hoyland JA and Freemont AJ:
Catabolic cytokine expression in degenerate and herniated human
intervertebral discs: IL-1beta and TNFalpha expression profile.
Arthritis Res Ther. 9:R772007. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tukhvatulin AI, Logunov DY, Shcherbinin
DN, Shmarov MM, Naroditsky BS, Gudkov AV and Gintsburg AL:
Toll-like receptors and their adapter molecules. Biochemistry
(Mosc). 75:1098–1114. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Janeway CJ and Medzhitov R: Innate immune
recognition. Annu Rev Immunol. 20:197–216. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee HK and Iwasaki A: Innate control of
adaptive immunity: Dendritic cells and beyond. Semin Immunol.
19:48–55. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takeda K, Kaisho T and Akira S: Toll-like
receptors. Annu Rev Immunol. 21:335–376. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Beutler B: Inferences, questions and
possibilities in Toll-like receptor signalling. Nature.
430:257–263. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Youn HS, Lee JY, Fitzgerald KA, Young HA,
Akira S and Hwang DH: Specific inhibition of MyD88-independent
signaling pathways of TLR3 and TLR4 by resveratrol: Molecular
targets are TBK1 and RIP1 in TRIF complex. J Immunol.
175:3339–3346. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Akira S, Takeda K and Kaisho T: Toll-like
receptors: Critical proteins linking innate and acquired immunity.
Nat Immunol. 2:675–680. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hiyama A, Sakai D, Risbud MV, Tanaka M,
Arai F, Abe K and Mochida J: Enhancement of intervertebral disc
cell senescence by WNT/β-catenin signaling-induced matrix
metalloproteinase expression. Arthritis Rheum. 62:3036–3047. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang F, Wu XT, Zhuang SY, Wang YT, Hong X,
Zhu L and Bao JP: Ex vivo observation of human nucleus pulposus
chondrocytes isolated from degenerated intervertebral discs. Asian
Spine J. 5:73–81. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mao M, Lei H, Liu Q, Chen Y, Zhao L, Li Q,
Luo S, Zuo Z, He Q, Huang W, et al: Effects of miR-33a-5P on
ABCA1/G1-mediated cholesterol efflux under inflammatory stress in
THP-1 macrophages. PLoS One. 9:e1097222014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
McGirt MJ, Ambrossi GL, Datoo G, Sciubba
DM, Witham TF, Wolinsky JP, Gokaslan ZL and Bydon A: Recurrent disc
herniation and long-term back pain after primary lumbar discectomy:
Review of outcomes reported for limited versus aggressive disc
removal. Neurosurgery. 64:338–344; discussion 344–345. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Glassman SD, Carreon LY, Djurasovic M,
Dimar JR, Johnson JR, Puno RM and Campbell MJ: Lumbar fusion
outcomes stratified by specific diagnostic indication. Spine J.
9:13–21. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mirza SK and Deyo RA: Systematic review of
randomized trials comparing lumbar fusion surgery to nonoperative
care for treatment of chronic back pain. Spine. 32:816–823. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Galbusera F, Bellini CM, Zweig T, Ferguson
S, Raimondi MT, Lamartina C, Brayda-Bruno M and Fornari M: Design
concepts in lumbar total disc arthroplasty. Eur Spine J.
17:1635–1650. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bothmann M, Kast E, Boldt GJ and Oberle J:
Dynesys fixation for lumbar spine degeneration. Neurosurg Rev.
31:189–196. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lu YC, Yeh WC and Ohashi PS: LPS/TLR4
signal transduction pathway. Cytokine. 42:145–151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Barton GM and Medzhitov R: Toll-like
receptor signaling pathways. Science. 300:1524–1525. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
O'Neill LA: Targeting signal transduction
as a strategy to treat inflammatory diseases. Nat Rev Drug Discov.
5:549–563. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kanzler H, Barrat FJ, Hessel EM and
Coffman RL: Therapeutic targeting of innate immunity with Toll-like
receptor agonists and antagonists. Nat Med. 13:552–559. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Netea MG, van Deuren M, Kullberg BJ,
Cavaillon JM and Van der Meer JW: Does the shape of lipid A
determine the interaction of LPS with Toll-like receptors? Trends
Immunol. 23:135–139. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Beutler B: Tlr4: Central component of the
sole mammalian LPS sensor. Curr Opin Immunol. 12:20–26. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Masuda K, Aota Y, Muehleman C, Imai Y,
Okuma M, Thonar EJ, Andersson GB and An HS: A novel rabbit model of
mild, reproducible disc degeneration by an anulus needle puncture:
Correlation between the degree of disc injury and radiological and
histological appearances of disc degeneration. Spine (Phila Pa
1976). 30:5–14. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sobajima S, Kompel JF, Kim JS, Wallach CJ,
Robertson DD, Vogt MT, Kang JD and Gilbertson LG: A slowly
progressive and reproducible animal model of intervertebral disc
degeneration characterized by MRI, X-ray and histology. Spine
(Phila Pa 1976). 30:15–24. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kim KS, Yoon ST, Li J, Park JS and Hutton
WC: Disc degeneration in the rabbit: A biochemical and radiological
comparison between four disc injury models. Spine (Phila Pa 1976).
30:33–37. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rousseau MA, Ulrich JA, Bass EC, Rodriguez
AG, Liu JJ and Lotz JC: Stab incision for inducing intervertebral
disc degeneration in the rat. Spine (Phila Pa 1976). 32:17–24.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sobajima S, Shimer AL, Chadderdon RC,
Kompel JF, Kim JS, Gilbertson LG and Kang JD: Quantitative analysis
of gene expression in a rabbit model of intervertebral disc
degeneration by real-time polymerase chain reaction. Spine J.
5:14–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ulrich JA, Liebenberg EC, Thuillier DU and
Lotz JC: ISSLS prize winner: Repeated disc injury causes persistent
inflammation. Spine (Phila Pa 1976). 32:2812–2819. 2007. View Article : Google Scholar : PubMed/NCBI
|