Introduction
Parkinsons disease (PD), which is the second most
common neurodegenerative disease among the aging human population
after Alzheimer's disease (AD), is thought to be caused by genetic
factors and environmental agents, including
1-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine (MPTP) (1). MPTP was initially discovered to be a
dopaminergic neurotoxin in the early 1980s after it contaminated a
source of synthetic heroin (2),
although it is predominantly used to produce animal models of PD.
MPTP is converted to its metabolite 1-methyl-4-phenylpyridinium ion
(MPP+) by monoamine oxidase B (MAO-B), which is predominantly
localized within astrocytes (3).
Previous studies have reported a role for autophagy in PD, and a
defect in autophagy, particularly in mitochondrial autophagy
(mitophagy), has emerged as a potential novel pathogenic mechanism
underlying the development of PD (4–7).
Previous studies have suggested that MPP+ may initiate dopaminergic
neuronal cell death, and induce autophagy in SH-SY5Y cells, SK-N-SH
cells, neuronal PC12 cells and the rat brain (8–12).
Astrocytes have numerous essential functions in the
healthy central nervous system (CNS), including responding to
oxidative stress-induced injury (13). Various neurological disease states,
including PD, AD and ischemic injury, have previously been
associated with varying degrees of astrocyte activation or
astrogliosis (14,15). Autophagy has been shown to be
activated in rat cortical astrocytes following permanent middle
cerebral artery occlusion (pMCAO), and in primary astrocytes
following oxygen and glucose deprivation (OGD) injury, in which
autophagy markedly decreased cell survival following OGD and focal
cerebral ischemia (16). Astrocytes
express NF-E2-related factor (Nrf2), which binds to the antioxidant
response element (ARE) in order to induce the expression of
antioxidant enzymes. Overexpression of Nrf2 in astrocytes has been
reported to protect against 6-hydroxydopamine damage in mice
(17), thus suggesting a potential
therapeutic strategy for the treatment of PD. Chen et al
(18) demonstrated that Nrf2
expression in astrocytes was able to protect against MPTP, and
suggested that modulation of the Nrf2-ARE pathway may be considered
a promising target for therapeutics aimed at reducing or preventing
neuronal death in patients with PD. These results supported the
hypothesis that astrocytes may have a neuroprotective role in
PD.
Autophagy is a cellular homeostatic process that
involves the sequestration of cytoplasmic material by lysosomes for
bulk degradation. Previous studies have suggested that autophagy
may have an important role in the pathogenic process of PD
(19–22). However, whether activation of
autophagy exerts beneficial or detrimental effects in PD is
currently unclear, since both protective and destructive effects
have previously been reported (22,23).
Lithium has been shown to induce autophagy by inhibiting inositol
monophosphatase, which in turn leads to depletion of free inositol
and decreased levels of inositol 1,4,5-trisphosphate (24,25).
Previous studies have detected therapeutic and protective effects
of lithium compounds in various models of neuronal disease,
including brain ischemia, AD, affective bipolar disease and
kainate-induced neuronal cell death (26–28).
Astrocytes have important functions and may be
beneficial to neurons under certain conditions. In addition, MPP+
may trigger oxidative stress, which may subsequently induce
autophagy. However, to the best of our knowledge, no previous study
has investigated whether MPP+ is able to induce autophagy in
astrocytes and the underlying mechanisms. Therefore, the present
study aimed to investigate whether MPP+ was able to induce
autophagy in astrocytes and its function. Furthermore, the ability
of lithium to protect astrocytes treated with MPP+, and its
potential underlying mechanisms, were analyzed.
Materials and methods
Ethics statement
The present study was approved by the Institutional
Animal Ethical Committee of Sun Yat-sen University (Guangzhou,
China), in accordance with recommendations in the Guide for the
Care and Use of Laboratory Animals (National Institutes of Health,
Bethesda, MA, USA).
Primary astrocyte culture
A total of 100 specific-pathogen-free neonatal male
C57BL/6 mice (Guangzhou University of Chinese Medicine, Guangzhou,
China), aged 1-day-old, were maintained at 25°C. Following
sacrifice via an overdose of 10% chloral hydrate (0.03 ml;
Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) via
intraperitoneal injection and disinfection with 75% alcohol,
astrocyte-enriched cultures were prepared from the cerebral cortex.
The meninges were removed from dissected cerebral cortexes and
tissues were cut into ~1 mm3 sections, which were
subsequently digested using 0.25% trypsin (Gibco; Thermo Fisher
Scientific, Inc. Waltham, MA, USA) at 37°C for 15 min. Digestion
was terminated using Dulbecco's modified Eagle's medium/nutrient
F12 (DMEM/F12) supplemented with 10% fetal bovine serum (FBS) and
penicillin/streptomycin (50 U/ml; 50 µg/ml) (all Gibco; Thermo
Fisher Scientific, Inc.). Following centrifugation at 112 × g for 5
min, the astrocytes were gently forced through a sterile 70 µm
Nitex mesh, after which they were resuspended in DMEM/F12
containing 10% heat-inactivated FBS, 2 mM L-glutamine, 50 U/ml
penicillin and 50 mg/ml streptomycin (all Gibco; Thermo Fisher
Scientific, Inc.). Subsequently, astrocytes (1×106
cells/ml) were seeded into a poly-lysine-coated flask, which was
stored in a humidified atmosphere containing 5% CO2 and
95% air at 37°C. The culture medium was replaced after 24 h, and
was subsequently replaced every 2–3 days. Upon reaching confluence
(typically 12–14 days later), microglia were detached from the
astrocytes by agitation at 260 rpm for 16 h. Astrocytes were
subsequently detached using trypsin-ethylenediaminetetraacetic acid
solution (Gibco; Thermo Fisher Scientific, Inc.), and were seeded
in the same culture medium. Following three or more consecutive
passages, cells were seeded into 96-well plates (105
cells/well) or dishes for further experimentation. The purity of
the astrocytes was determined using glial fibrillary acidic protein
(GFAP) immunocytochemistry using rabbit anti-GFAP polyclonal
antibody (1:5,000; ab7260; Abcam, Cambridge, UK), which indicated
that 98% of the cultured cells were GFAP-positive, using a
microscope (Bx51; Olympus Corporation, Tokyo, Japan).
Cell treatment
In order to measure the toxicity of MPP+, the cells
were divided into seven groups, including one control group and six
groups treated with MPP+, which were treated with 50, 100, 200,
400, 800 or 1,200 µM MPP+ (Sigma-Aldrich, St. Louis, MO, USA),
respectively. In order to measure the induction of autophagy in the
astrocytes, the cells were divided into several groups, including
the control group (treated with FBS), the starvation group
(incubated in DMEM/F12 without FBS or MPP+; positive control), MPP+
groups (0.2, 0.4 and 0.8mM MPP+, respectively), and MPP+ and
inhibitor groups, which were treated with 10mM Pepstain A lysosomal
inhibitor (Sigma-Aldrich). Furthermore, in order to explore the
function of the induced autophagy, the cells were pretreated with
autophagy inhibitors, 10 mM 3-methyladenine (3-MA), 0.01 mM
chloroquine (CQ), 10 mM lysosomal inhibitor Pepstain A and 10 mM
lithium (all Sigma-Aldrich) for 1 h, after which MPP+ was added for
an additional 24 h.
Cell viability assay
Cell viability was analyzed using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich) assay, as outlined in a previous study (29). Briefly, 24 h following treatment with
the various concentrations of MPP+, astrocytes in the 96-well
plates were washed with phosphate-buffered saline, after which the
cells were incubated with MTT (5 mg/ml) at 37°C for 4 h.
Subsequently, the supernatants were removed, 100 µl dimethyl
sulfoxide was added to each well, and the plates were agitated on a
microplate shaker in order to dissolve the blue MTT-formazan.
Absorbance was measured at 570 nm using a ELx800 microplate reader
(BioTek Instruments, Inc., Winooski, VT, USA). Cell viability was
expressed as the ratio of the signal obtained from the treated
cultures to the control cultures.
Western blotting
Cells were harvested and lysed using the Mammalian
Cell Extraction kit (BioVision, Inc., Milpitas, CA, USA). The
resulting lysates were subjected to the Bradford Protein assay
(Pierce Biotechnology, Inc., Rockford, IL, USA), in order to
determine protein concentrations and to ensure equal protein
loading. Protein samples (30 µg) were separated by 8, 10 or 12%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and were
subsequently transferred onto polyvinylidene difluoride membranes
(EMD Millipore, Billerica, MA, USA). The membranes were blocked
using Tris-buffered saline Tween 20 [50 mM Tris-HCl, 154 mM NaCl,
0.1% Tween 20 (pH 7.5)], containing 5% nonfat dry milk for 1 h, and
were subsequently probed with rabbit anti-microtubule-associated
protein light chain-3 (LC3; 1:5,000; NB100-2220; Novus Biologicals,
LLC, Littleton, CO, USA) polyclonal antibody or anti-phosphorylated
(p)AKT (1:5,000; ab81283; Abcam) monoclonal antibody overnight at
4°C, according to the manufacturer's protocol. Following primary
antibody incubation, the membranes were washed and incubated with
either horseradish peroxidase-conjugated anti-mouse (1:1,000; 7076)
or anti-rabbit immunoglobulin G (1:1,000; 7074; both Cell Signaling
Technology, Inc., Billerica, MA, USA) for 1 h at room temperature.
Chemiluminescence reactions were conducted according to the
manufacturer's protocol (EMD Millipore, Bedford, MA, USA). The
intensity (INT × area) of each band was measured and analyzed using
the ChemiDoc XRS+ Imaging system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Data are presented as the percentage of the
vehicle control, and β-actin (1:4,000; ab-32092; Novus Biologicals,
LLC) was used as an internal control.
Statistical analysis
Data were analyzed using SPSS 13.0 for Windows
(SPSS, Inc., Chicago, IL, USA) and presented as the mean ± standard
deviation. All experiments were repeated at least three times.
Statistical analysis was conducted using one-way analysis of
variance, followed by Student's t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
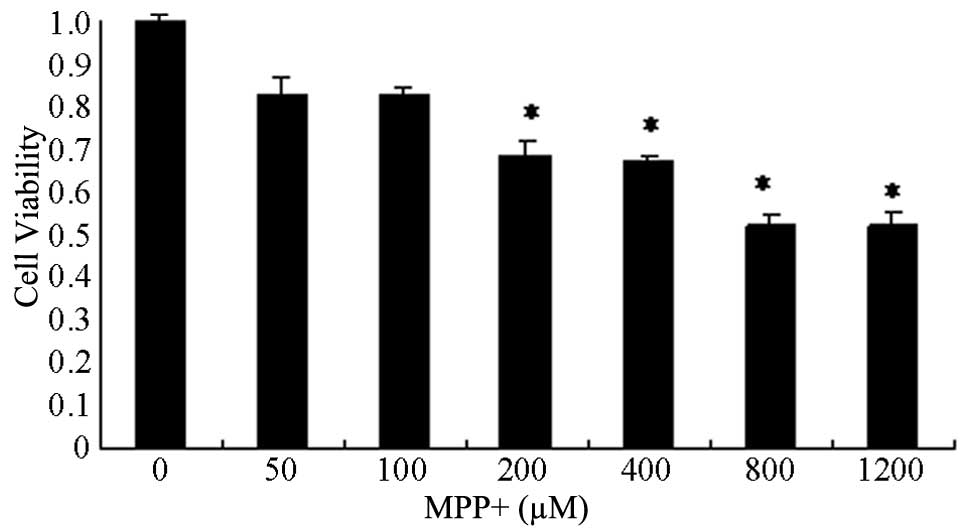
Toxicity of MPP+
Primary astrocyte cells were treated with increasing
concentrations of MPP+, ranging from 0 to 1,200 µM, for 24 h, and
the cell viability was determined in order to identify the optimal
concentration of MPP+ (Fig. 1). MPP+
decreased cell viability in a concentration-dependent manner. The
viability of cells treated with >200 µM MPP+ was significantly
decreased, particularly at 800 and 1,200 µM MPP+ (P<0.05;
Fig 1). The median lethal dose of
MPP+ was ~430 µM. Therefore, 400 and 800 µM MPP+ concentrations
were selected for further experimentation.
Induction of autophagy in MPP+-treated
primary astrocytes
In order to investigate autophagy induction in the
control cells, as well as those treated with various concentrations
of MPP+, total protein was extracted from the astrocytes and
western blotting using anti-LC3 primary antibodies was conducted.
In the primary astrocytes treated with >200 µM MPP+, the protein
expression levels of LC3 II were significantly increased in a
concentration-dependent manner (P<0.05; Fig. 2A and B), as compared with the control
cells. However, it was unclear whether the increase in LC3 II
protein expression levels was associated with autophagy induction
or lysosomal dysfunction; therefore, the cells were pretreated with
lysosomal inhibitors. The protein expression levels of LC3 II in
the MPP+-treated cells pretreated with lysosomal inhibitors were
significantly increased in a concentration-dependent manner
(P<0.05; Fig. 2C and D), as
compared with the control cells; thus suggesting that LC3 II
protein expression levels increased in MPP+-treated astrocytes due
to the induction of autophagy and not the impairment of
lysosomes.
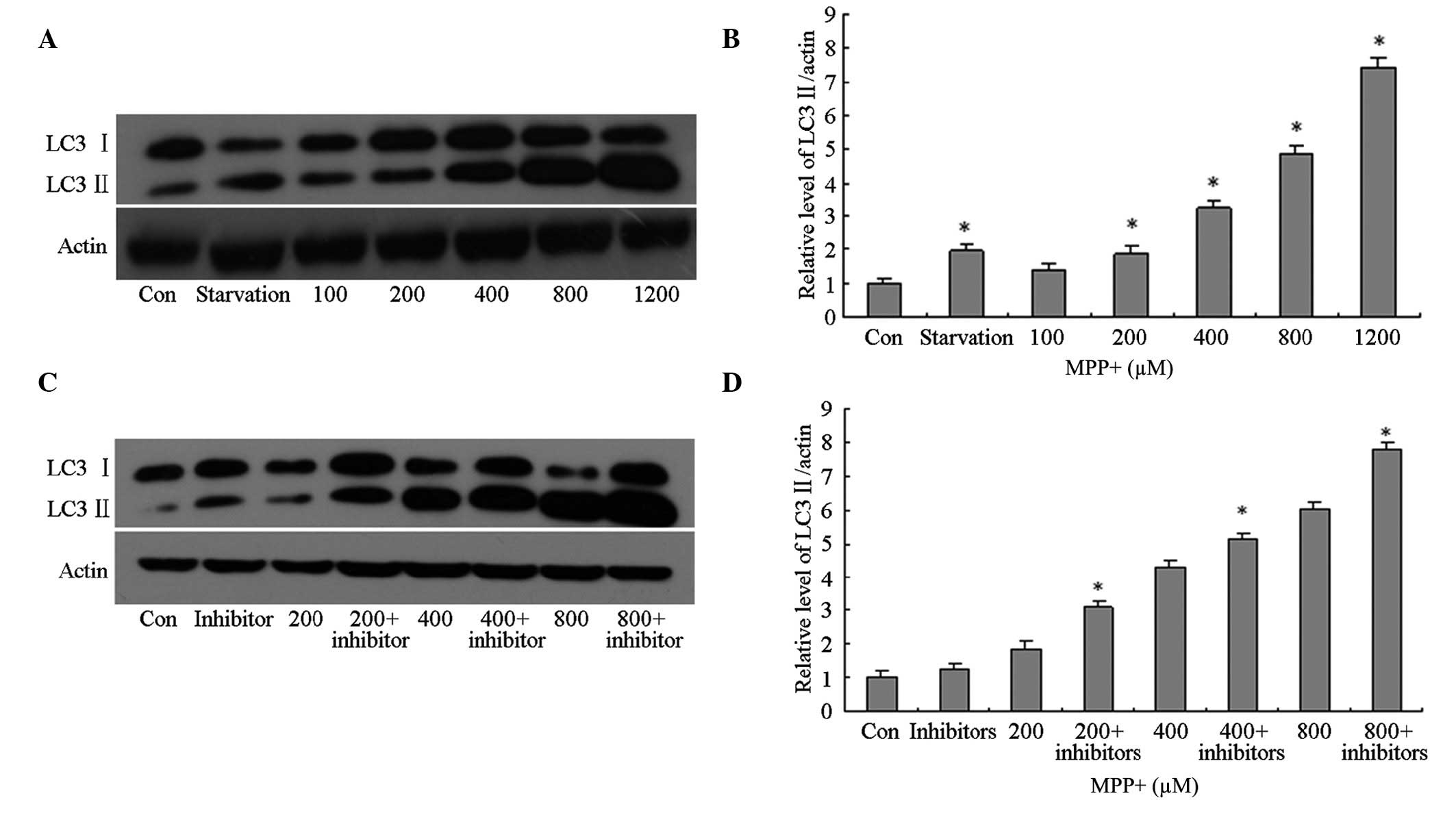 | Figure 2.Effects of MPP+ on the protein
expression levels of LC3 II in primary astrocytes. (A and B) The
cells were treated with FBS (Con), without FBS (starvation) or MPP+
(100, 200, 400, 800 or 1,200 µM) for 24 h. Autophagy was determined
by detecting the protein expression levels of LC3 II using western
blotting. Protein concentrations were quantified by measuring the
band density. (C and D) The cells were treated with FBS (Con),
without FBS (starvation) or MPP+ (200, 400 or 800 µM), and a
lysosomal inhibitor for 24 h. The protein expression levels of LC3
II were detected using western blotting. Protein concentrations
were quantified by measuring the band density. Data are presented
as the mean ± standard error of the mean of triplicate experiments.
*P<0.05 vs. the control group. MPP+,
1-methyl-4-phenylpyridinium; LC3, microtubule-associated protein
light chain-3; FBS, fetal bovine serum; Con, control group. |
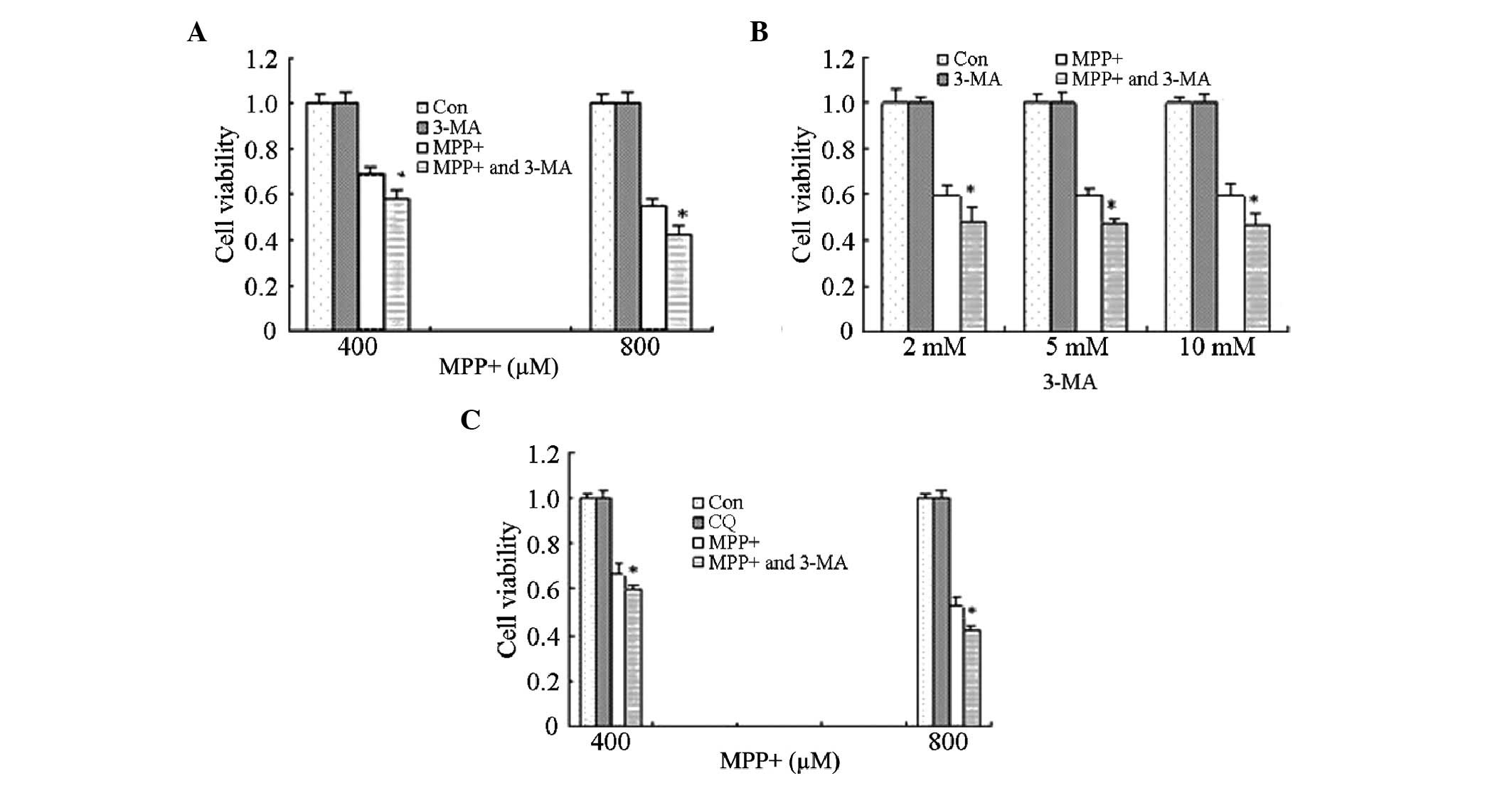
Effects of autophagy inhibitors on the
viability of astrocytes treated with MPP+
In order to investigate the effects of MPP+-induced
autophagy, astrocytes were treated with 10 mM 3-MA for 1 h, prior
to treatment with MPP+ for 24 h. Cell viability was significantly
decreased in the cells treated with MPP+ and 3-MA, as compared with
the cells treated with MPP+ alone (P<0.05; Fig. 3A). These results suggest that
MPP+-induced autophagy exerts potential protective effects on
astrocytes.
However, previous studies reported that high doses
of 3-MA were lethal to cells, and therefore the effects of three
different doses of 3-MA (2, 5 and 10 mM) were analyzed in the
present study. Treatment with 3-MA (2, 5 or 10 mM) alone did not
affect cell viability; however, cell viability was significantly
decreased following treatment of astrocytes with all three
concentrations of 3-MA in combination with MPP+, as compared with
MPP+ treatment alone (P<0.05; Fig.
3B). These results suggest that the ability of 3-MA to decrease
cell viability is not associated with the dose but with other
mechanisms, such as the inhibition of autophagy.
Astrocytes were also treated with CQ in order to
corroborate the effects of autophagy inhibitors on the cell
viability of MPP+-treated astrocytes. Consistent with the results
for 3-MA, astrocytes pretreated with CQ for 1 h exhibited decreased
cell viability, as compared with the MPP+ treatment alone
(P<0.05; Fig. 3C). These results
suggest that MPP+-induced autophagy exerts protective effects.
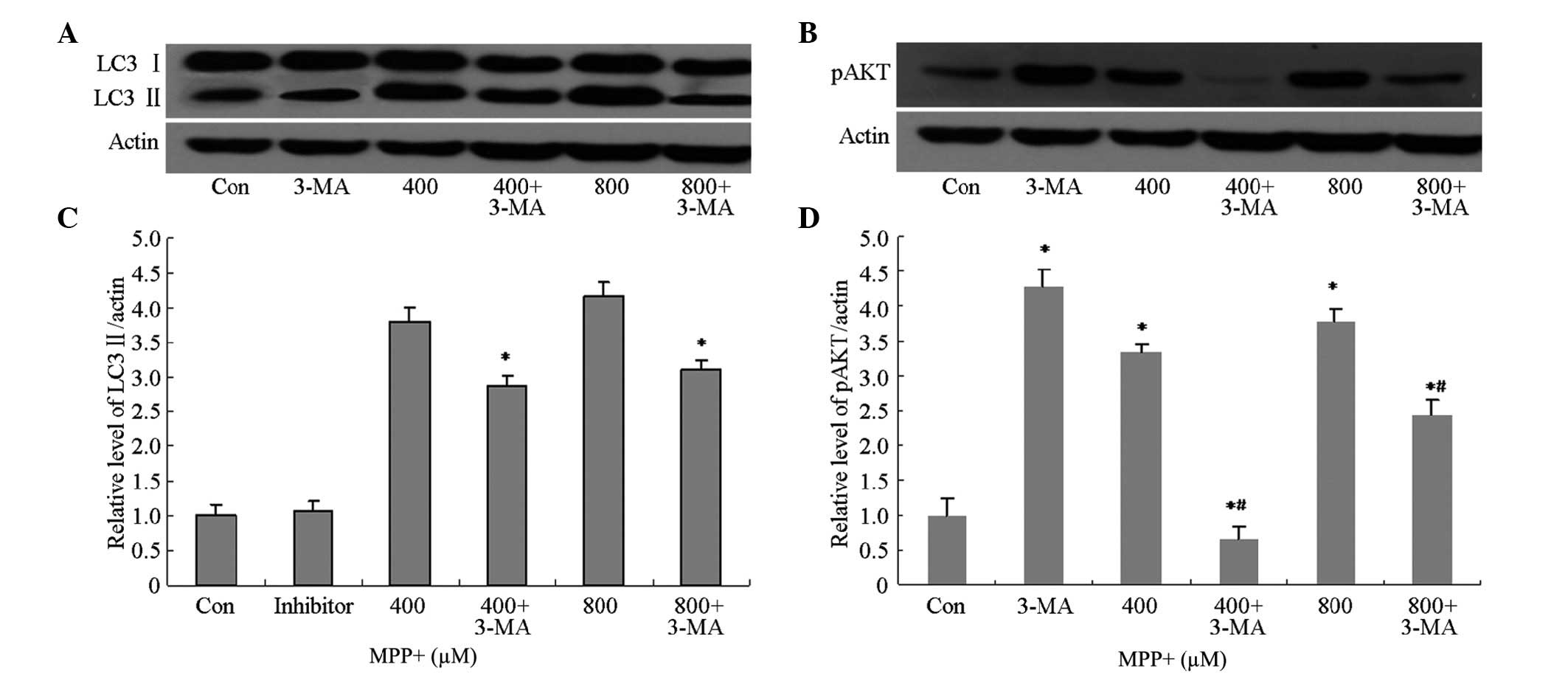
Potential mechanisms underlying the
effects of 3-MA on MPP+-treated cells
3-MA is a selective inhibitor of class III
phosphatidylinositol kinase (30,31), and
has been accepted as a specific inhibitor of autophagy. In order to
investigate whether 3-MA was able to inhibit MPP+-induced
autophagy, the protein expression levels of autophagy-associated
proteins, including LC3 II and pAkt, were analyzed by western
blotting. The protein expression levels of LC3 II and pAkt in
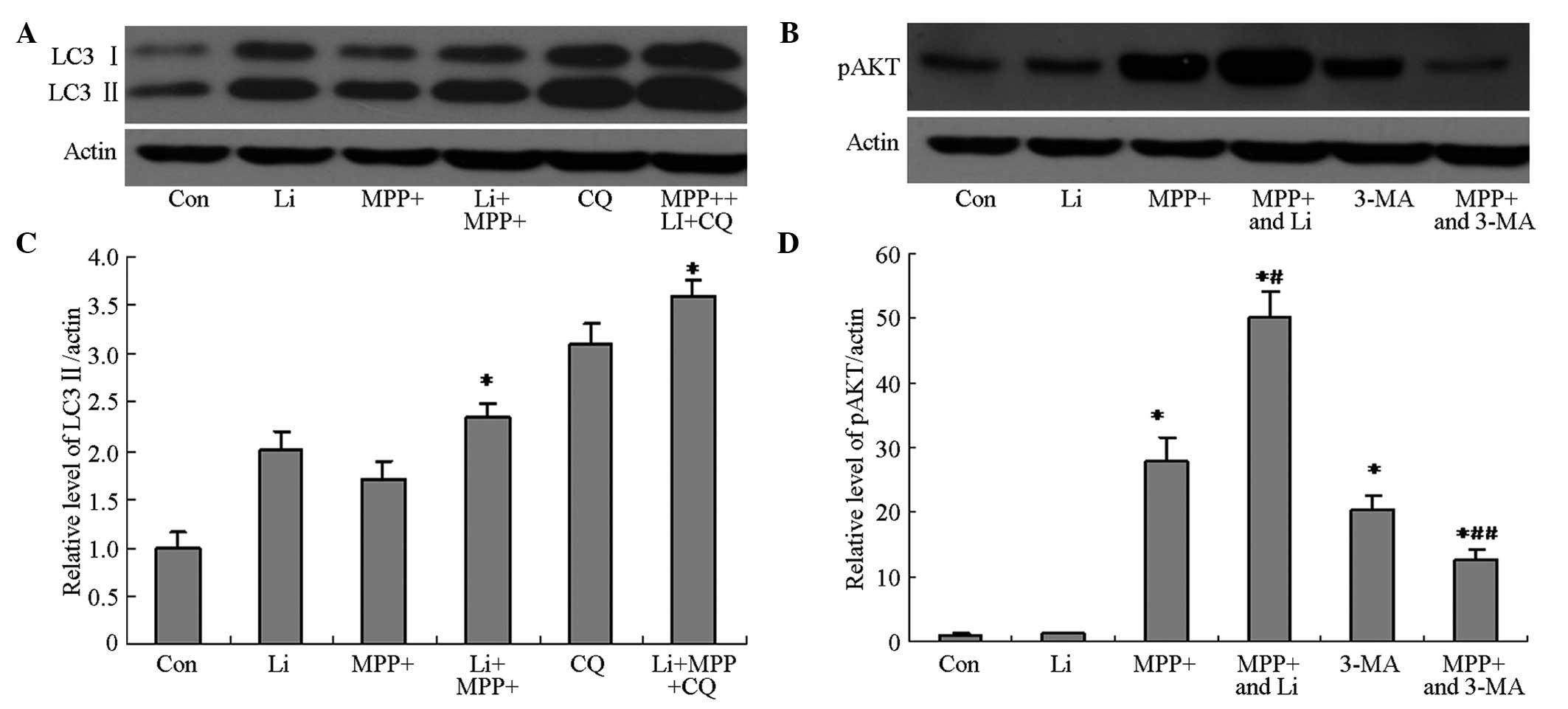
astrocytes pretreated with 3-MA were markedly decreased (Fig. 4A and B), and were shown to be
significantly decreased by quantification of optical density (OD)
values (P<0.05; Fig. 4C and D).
These results suggest that 3-MA is able to inhibit MPP+-induced
autophagy via inhibition of the phosphoinositide 3-kinase
(PI3K)/AKT pathway.
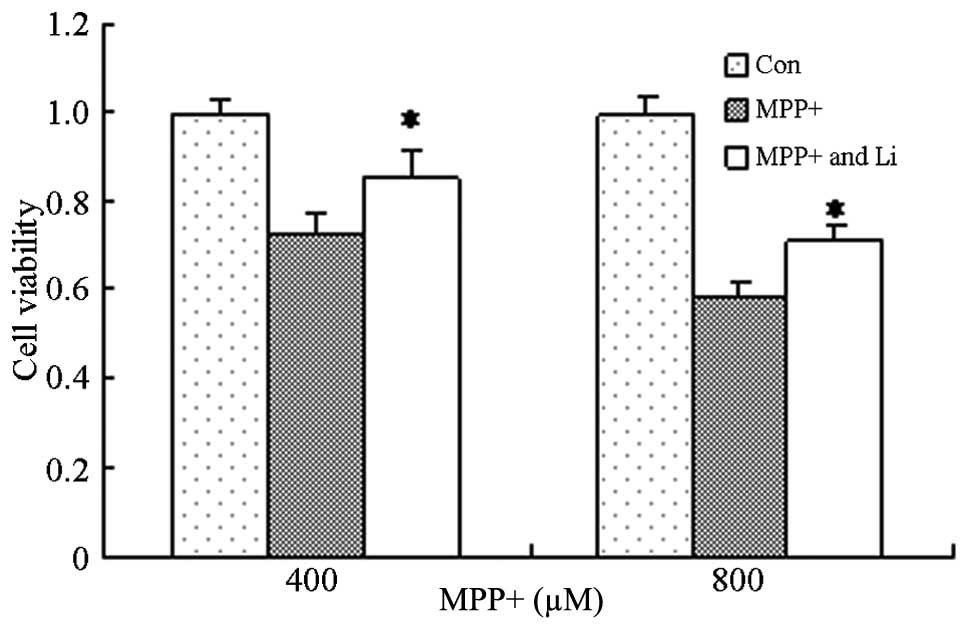
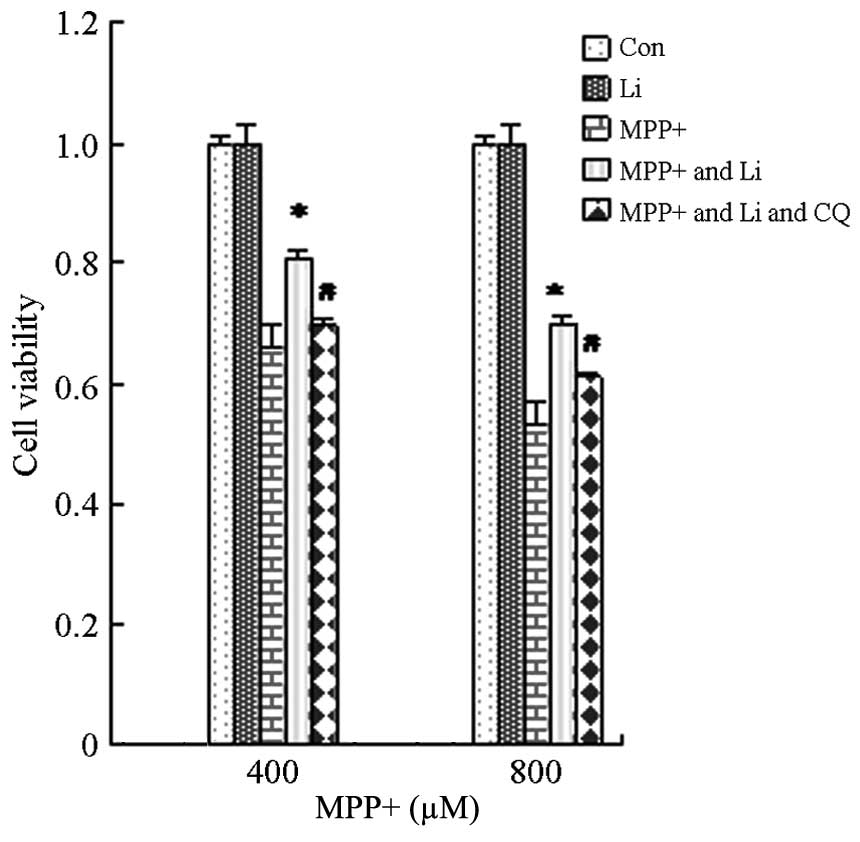
Protective effects of lithium on
astrocytes treated with MPP+
Lithium exerts protective effects in numerous
diseases, including AD and Huntington's disease, and its underlying
mechanism has been shown to involve the inhibition of glycogen
synthase kinase-3β, activation of the PI3K/Akt pathway, and the
induction of autophagy (24,32). In order to investigate the effects of
lithium on astrocytes treated with MPP+, astrocytes were pretreated
with lithium for 1 h and cell viability was determined. The
viability of cells pretreated with lithium significantly increased,
as compared with the cells treated with MPP+ alone (P<0.05;
Fig. 5). These results suggest that
lithium is able to protect astrocytes against the effects MPP+.
Potential mechanisms underlying the
protective effects of lithium
The ability of lithium to induce autophagy in
MPP+-treated astrocytes was investigated, in order to elucidate the
mechanism underlying the protective effects of lithium. The protein
expression levels of LC3 II and pAkt in cells pretreated with
lithium were markedly increased, as compared with the cells treated
with MPP+ alone (Fig. 6A and B), and
were shown to be significantly increased by quantification of OD
values (P<0.05; Fig. 6C and D).
Furthermore, pretreatment with CQ, as well as lithium, markedly
attenuated the lithium-induced upregulation of LC3 II and pAkt
protein expression (Fig. 6), and
viability (Fig. 7) of cells treated
with MPP+. These results suggest that the protective effects of
lithium may be associated with its ability to induce autophagy in
astrocytes treated with MPP+ via activation of the PI3K/Akt
pathway.
Discussion
Glial cells have an active role in normal brain
functioning, and their interactions with neuronal cells are
important for maintaining homeostasis in the extracellular
microenvironment (33–35). The function of glial cells is
particularly important following a CNS insult, such as ischemia.
Following injury, astrocytes are responsible for the removal of
accumulated excitotoxic neurotransmitters and ions, including
glutamate, lactate, hydrogen, and potassium ions, which have
previously been associated with brain injury (36). High levels of glutamine synthetase
have been detected in astrocytes, which, alongside a specific
glutamate transporter, acts to remove and detoxify extracellular
glutamate and generate the neuronal substrate glutamine, as part of
the glutamate/glutamine cycle (37,38).
Astrocytes store glycogen and are able to provide lactate as an
alternative aerobic substrate for neuronal energy production during
recovery, and also maintain the osmotic environment (39–41).
Astrocytes produce various cytokines and growth factors, which
function in the CNS as mediators of immune and inflammatory
responses, and may exert neurotoxic and neuroprotective effects
(42). High levels of reduced
glutathione, which is an important antioxidant in the CNS, have
previously been detected in astrocytes, but not in neurons
(43). Astrocytes support and
protect themselves, and other cellular elements with which they are
in intimate contact, and also actively participate in the
destruction of brain tissue following ischemia; thus suggesting
that a disturbance of astroglial activities may induce neuronal
dysfunction.
The present study hypothesized that the MPP+
neurotoxin may induce autophagy in primary astrocytes, and aimed to
investigate the effects of the induced autophagy. In addition, the
ability of lithium treatment to enhance autophagy and exert
protective effects on astrocytes, as well as the potential
underlying mechanisms, was investigated. The results of the present
study suggested that MPP+ was able to induce autophagy in
astrocytes, likely via the induction of oxidative stress.
In the present study, astrocytes pretreated with
autophagy inhibitors, including 3-MA and CQ, exhibited decreased
cell viability, as compared with cells treated with MPP+ alone,
thus suggesting that autophagy induced by MPP+ exerts protective
effects on astrocytes. These results were inconsistent with Zhu
et al (9), who demonstrated
that neither 3-MA nor wortmannin (WT) were able to inhibit the
increase in autophagic vacuoles (AV)/late AVs induced by MPP+. In
addition, 3-MA (5 mmol/l) and WT (50–5 mol/l) had no significant
effects on basal SH-SY5Y cell viability.
Previous studies have demonstrated that lithium may
induce autophagy (24,25). Lithium compounds have been shown to
exert therapeutic and protective effects in various models of
neuronal disease, including brain ischemia and AD. In addition,
LiCl was demonstrated to attenuate a reduction in the cell
viability of PC12 cells induced by treatment with MPP+ via the
induction of autophagy (44). In the
present study, the cell viability of astrocytes pretreated with
lithium for 1 h were significantly increased, as compared with the
cells treated with MPP+ only. These results were consistent with
our hypothesis and the results from Youdim and Arraf (44); however, they were inconsistent with
previous studies, which were unable to demonstrate cytoprotective
effects for lithium in astroglial cells and cerebellar granule
neurons (45,46). However, this inconsistency may be
attributed to the analysis of different cell types among the
studies. In addition, the results of the present study suggested
that the protective effects of lithium on astrocytes treated with
MPP+ were associated with the induction of autophagy and activation
of the PI3K/AKT pathway, which was consistent with previous studies
(24,47).
In the present study, MPP+ was able to induce
autophagy in astrocytes, which was shown to exert protective
effects on the cells. In addition, lithium was able to protect
astrocytes from the toxic effects of MPP+ by enhancing the rate of
autophagy; thus supporting the hypothesis that induced autophagy in
astrocytes treated with MPP+ may exert protective effects. The
results of the present study may help to elucidate the important
role of astrocytes in the pathophysiology of PD, and direct the
development of a novel strategy to treat patients with PD. Future
studies should endeavor to elucidate the effects of induced
autophagy in astrocytes on neurons.
Acknowledgements
The present study was supported by grants from the
National Basic Research Program of China (grant no. 2011CB504100),
the National Natural Science Foundation of China (grant no.
30770766), the Guangdong Natural Science Foundation (grant no.
10151008901000187) and the Guangdong Science and Technology
Foundation (grant nos. 2008B080703021, 2006B36004021 and
2006B60501023).
References
|
1
|
Dauer W and Przedborski S: Parkinson's
disease: Mechanisms and models. Neuron. 39:889–909. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Langston JW, Ballard P, Tetrud JW and
Irwin I: Chronic Parkinsonism in humans due to a product of
meperidine-analog synthesis. Science. 219:979–980. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vives BC, Zhou C, Huang Y, Cui M, de Vries
RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al:
PINK1-dependent recruitment of Parkin to mitochondria in mitophagy.
Proc Natl Acad Sci USA. 107:378–383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Narendra DP, Jin SM, Tanaka A, Suen DF,
Gautier CA, Shen J, Cookson MR and Youle RJ: PINK1 is selectively
stabilized on impaired mitochondria to activate Parkin. PLoS Biol.
8:e10002982010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Geisler S, Holmström KM, Skujat D, Fiesel
FC, Rothfuss OC, Kahle PJ and Springer W: PINK1/Parkin-mediated
mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol.
12:119–131. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kawajiri S, Saiki S, Sato S, Sato F,
Hatano T, Eguchi H and Hattori N: PINK1 is recruited to
mitochondria with parkin and associates with LC3 in mitophagy. FEBS
Lett. 584:1073–1079. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Levitt P, Pintar JE and Breakefield XO:
Immunocytochemical demonstration of monoamine oxidase B in brain
astrocytes and serotonergic neurons. Proc Natl Acad Sci USA.
79:6385–6389. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zigmond MJ and Stricker EM: Animal models
of parkinsonism using selective neurotoxins: Clinical and basic
implications. Int Rev Neurobiol. 31:1–79. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu JH, Horbinski C, Guo F, Watkins S,
Uchiyama Y and Chu CT: Regulation of autophagy by extracellular
signal-regulated protein kinases during
1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol.
170:75–86. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nopparat C, Porter JE, Ebadi M and
Govitrapong P: 1-methyl-4-phenylpyridinium-induced cell death via
autophagy through a Bcl-2/Beclin 1 complex-dependent pathway.
Neurochem Res. 39:225–232. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rodríguez-Blanco J, Martín V,
García-Santos G, Herrera F, Casado-Zapico S, Antolín I and
Rodriguez C: Cooperative action of JNK and AKT/mTOR in
1-methyl-4-phenylpyridinium-induced autophagy of neuronal PC12
cells. J Neurosci Res. 90:1850–1860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hung KC, Huang HJ, Lin MW, Lei YP and Lin
AM: Roles of autophagy in MPP+ -induced neurotoxicity in vivo: The
involvement of mitochondria and α-synuclein aggregation. PLoS One.
9:e910742014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pekny M and Nilsson M: Astrocyte
activation and reactive gliosis. Glia. 50:427–434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hirsch EC, Breidert T, Rousselet E, Hunot
S, Hartmann A and Michel PP: The role of glial reaction and
inflammation in Parkinson's disease. Ann N Y Acad Sci. 991:214–228.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Teismann P and Schulz JB: Cellular
pathology of Parkinson's disease: Astrocytes, microglia and
inflammation. Cell Tissue Res. 318:149–161. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qin AP, Liu CF, Qin YY, Hong LZ, Xu M,
Yang L, Liu J, Qin ZH and Zhang HL: Autophagy was activated in
injured astrocytes and mildly decreased cell survival following
glucose and oxygen deprivation and focal cerebral ischemia.
Autophagy. 6:738–753. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jakel RJ, Townsend JA, Kraft AD and
Johnson JA: Nrf2-mediated protection against 6-hydroxy-dopamine.
Brain Res. 1144:197–201. 2007. View Article : Google Scholar
|
|
18
|
Chen PC, Vargas MR, Pani AK, Smeyne RJ,
Johnson DA, Kan YW and Johnson JA: Nrf2-mediated neuroprotection in
the MPTP mouse model of Parkinson's disease: Critical role for the
astrocyte. Proc Natl Acad Sci USA. 106:2933–2938. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Plowey ED, Cherra SJ III, Liu YJ and Chu
CT: Role of autophagy in G2019S-LRRK2-associated neurite shortening
in differentiated SH-SY5Y cells. J Neurochem. 105:1048–1056. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Matsuda N, Sato S, Shiba K, Okatsu K,
Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al:
PINK1 stabilized by mitochondrial depolarization recruits Parkin to
damaged mitochondria and activates latent Parkin for mitophagy. J
Cell Biol. 189:211–221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tong Y, Yamaguchi H, Giaime E, Boyle S,
Kopan R, Kelleher RJ III and Shen J: Loss of leucine-rich repeat
kinase 2 causes impairment of protein degradation pathways,
accumulation of alpha-synuclein and apoptotic cell death in aged
mice. Proc Natl Acad Sci USA. 107:9879–9884. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alegre-Abarrategui J, Christian H, Lufino
MM, Mutihac R, Venda LL, Ansorge O and Wade-Martins R: LRRK2
regulates autophagic activity and localizes to specific membrane
microdomains in a novel human genomic reporter cellular model. Hum
Mol Genet. 18:4022–4034. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pan T, Rawal P, Wu Y, Xie W, Jankovic J
and Le W: Rapamycin protects against rotenone-induced apoptosis
through autophagy induction. Neuroscience. 164:541–551. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sarkar S, Floto RA, Berger Z, Imarisio S,
Cordenier A, Pasco M, Cook LJ and Rubinsztein DC: Lithium induces
autophagy by inhibiting inositol monophosphatase. J Cell Biol.
170:1101–1111. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sarkar S and Rubinsztein DC: Inositol and
IP3 levels regulate autophagy: Biology and therapeutic
speculations. Autophagy. 2:132–134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chuang DM: The antiapoptotic actions of
mood stabilizers: molecular mechanisms and therapeutic potentials.
Ann NY Acad Sci. 1053:195–204. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cappuccio I, Calderone A, Busceti CL,
Biagioni F, Pontarelli F, Bruno V, Storto M, Terstappen GT,
Gaviraghi G, Fornai F, et al: Induction of Dickkopf-1, a negative
modulator of the Wnt pathway, is required for the development of
ischemic neuronal death. J Neurosci. 25:2647–2657. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Busceti CL, Biagioni F, Aronica E, Riozzi
B, Storto M, Battaglia G, Giorgi FS, Gradini R, Fornai F,
Caricasole A, et al: Induction of the Wnt inhibitor, Dickkopf-1, is
associated with neurodegeneration related to temporal lobe
epilepsy. Epilepsia. 48:694–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Loo DT and Rilleman JR: Measurement of
cell death. Methods Cell Biol. 57:251–264. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Seglen PO and Gordon PB: 3-Methyladenine:
Specific inhibitor of autophagic/lysosomal protein degradation in
isolated rat hepatocytes. Proc Natl Acad Sci USA. 79:1889–1892.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schu PV, Takegawa K, Fry MJ, Stack JH,
Waterfield MD and Emr SD: Phosphatidylinositol 3-kinase encoded by
yeast VPS34 gene essential for protein sorting. Science. 260:88–91.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sarkar S, Krishna G, Imarisio SA, Saiki S,
O'Kane CJ and Rubinsztein DC: A rational mechanism for combination
treatment of Huntington's disease using lithium and rapamycin. Hum
Mol Genet. 17:170–178. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Norenberg MD: Astrocyte responses to CNS
injury. J Neuropathol Exp Neurol. 53:213–220. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Norenberg MD: Active and passive roles of
astrocytes in neurologic disease: Commentary on forum position
paper. Neurotoxicology. 19:23–26. 1998.PubMed/NCBI
|
|
35
|
Ridet JL, Malhotra SK, Privat A and Gage
FH: Reactive astrocytes: Cellular and molecular cues to biological
function. Trends Neurosci. 20:570–577. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Amédée T, Robert A and Coles JA: Potassium
homeostasis and glial energy metabolism. Glia. 21:46–55. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sonnewald U, Westergaard N and Schousboe
A: Glutamate transport and metabolism in astrocytes. Glia.
21:56–63. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tansey FA, Farooq M and Cammer W:
Glutamine synthetase in oligodendrocytes and astrocytes: New
biochemical and immunocytochemical evidence. J Neurochem.
56:266–272. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schurr A, Payne RS, Miller JJ and Rigor
BM: Brain lactate is an obligatory aerobic energy substrate for
functional recovery after hypoxia: Further in vitro validation. J
Neurochem. 69:423–426. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gerhart DZ, Enerson BE, Zhdankina OY,
Leino RL and Drewes LR: Expression of monocarboxylate transporter
MCT1 by brain endothelium and glia in adult and suckling rats. Am J
Physiol. 273(1)1E207–E213. 1997.PubMed/NCBI
|
|
41
|
Koehler-Stec EM, Simpson IA, Vannucci SJ,
Landschulz KT and Landschulz WH: Monocarboxylate transporter
expression in mouse brain. Am J Physiol. 275(3)1E516–E524.
1998.PubMed/NCBI
|
|
42
|
Aschner M: Astrocytic functions and
physiological reactions to injury: The potential to induce and/or
exacerbate neuronal dysfunction-a forum position paper.
Neurotoxicology. 19:7–17. 1998.PubMed/NCBI
|
|
43
|
Raps SP, Lai JC, Hertz L and Cooper AJ:
Glutathione is present in high concentrations in cultured
astrocytes but not in cultured neurons. Brain Res. 493:398–401.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Youdim MB and Arraf Z: Prevention of MPTP
(N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) dopaminergic
neurotoxicity in mice by chronic lithium: Involvements of Bcl-2 and
Bax. Neuropharmacology. 46:1130–1140. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lai JS, Zhao C, Warsh JJ and Li PP:
Cytoprotection by lithium and valproate varies between cell types
and cellular stresses. Eur J Pharmacol. 539:18–26. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yeste M, Alvira1 D, Verdaguer E, Tajes M,
Folch J, Rimbau V, Pallàs M and Camins A: Evaluation of acute
antiapoptotic effects of Li+ in neuronal cell cultures. J Neural
Transm. 114:405–416. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
King TD, Bijur GN and Jope RS: Caspase-3
activation induced by inhibition of mitochondrial complex I is
facilitated by glycogen synthase kinase-3beta and attenuated by
lithium. Brain Res. 919:106–114. 2001. View Article : Google Scholar : PubMed/NCBI
|