Introduction
It is widely recognized that re-establishing the
blood flow to ischemic myocardial tissue has become the most viable
therapeutic approach for the treatment of ischemic heart disease
(1). However, subsequent
ischemia/reperfusion (I/R) injury may reduce the therapeutic
benefit (2). Indeed, a wide range of
pathological processes contribute to myocardial I/R injury
(3), and it has been widely accepted
that inflammation is important in myocardial I/R injury (4). There is comprehensive experimental and
clinical evidence that anti-inflammatory actions attenuate I/R
injury (5,6). It is therefore necessary that an
effective therapeutic target against inflammation is identified to
attenuate I/R injury.
microRNAs (miRNAs) are a class of high abundance,
evolutionarily conserved, non-coding, small single-stranded RNAs
that negatively regulate gene expression by binding to the 3′
untranslated region of target mRNAs (7). miRNAs regulate genes involved in a
diverse range of biological processes by this mechanism, including
development, differentiation, inflammation, stress responses,
angiogenesis, adhesion, proliferation and apoptosis (8,9). In
addition, miRNAs are also recognized as critical regulators of
cardiovascular diseases (10). A
previous study revealed that miRNAs also have an important role in
myocardial I/R injury (11).
Recently, one specific miRNA, miR-22 was observed to have decreased
in rat hearts after I/R (12). These
data indicate that miR-22 may also be involved in the pathological
progression of myocardial I/R injury. Furthermore, our previously
published data also demonstrated that adenovirus-mediated miR-22
overexpression may protect against myocardial I/R injury via
anti-apoptosis in rats by targeting cAMP response element-binding
protein binding protein (CBP) (12).
However, the specific function of miR-22 in myocardial I/R injury
is far from fully elucidated.
p38-mitogen-activated protein kinase (p38-MAPK) and
CBP are general transcriptional co-activators, which are capable of
phosphorylating various sequence-specific transcription factors,
including activator protein (AP)-1 and p53, in order to regulate
their downstream gene expression (13–15).
AP-1 is a regulator of cytokine expression and a significant
modulator in inflammatory diseases, including rheumatoid arthritis,
psoriasis, psoriatic arthritis and myocardial I/R injury (16). c-Jun is considered to be a dominant
component of the AP-1 complex (c-Jun-AP-1) (17). p38 MAPK and CBP have been identified
as potential miR-22 targets using Target-Scan bioinformatics
software. A previous unpublished study performed in our laboratory
also identified that the levels of p38 MAPK, CBP and c-Jun-AP-1
were upregulated in rat hearts following I/R. However, it is
remains unknown whether miR-22 is able to protect against
myocardial I/R injury through anti-inflammation in rats via the
suppression of p38 MAPK, CBP and c-Jun-AP-1.
In the present study, adenoviral overexpression of
miR-22 was used to investigate another cardioprotective signaling
mechanism of miR-22 in myocardial I/R injury. The data demonstrated
that miR-22 was able to efficiently attenuate I/R injury by
regulating p38 MAPK, CBP and c-Jun-AP-1 expression and inhibiting
inflammation.
Materials and methods
Animals
A total of 40 adult male Sprague-Dawley rats
(weight, 220–250 g) were purchased from China Three Gorges
University (CTGU; Yichang, China). Rats received a standard diet
and free access to water, and were maintained at 23±3°C with a 12 h
light/dark cycle. The procedures for experiments and animal care
were approved by the Animal Care and Use Committee of CTGU, and
conformed to the Guide for the Care and Use of Laboratory Animals
outlined by the National Institutes of Health (NIH publication no.
80–23).
Construction of miR-22 expression
adenoviral vector
Rno-miR-22 precursor DNA (MI0000851) was synthesized
by Genechem, Co., Ltd., (Shanghai, China). The adenovirus
expressing miR-22 (Ad-miR-22) or control adenovirus expressing
scramble miRNA (Ad-Scramble) were generated using the AdMax system
(Microbix Biosystems, Ontario, Canada) according to the
manufacturer's instructions. These resulting adenoviruses were
subsequently packaged and amplified in HEK293 cells (Sunbio, Inc.,
Shanghai, China), and purified using cesium chloride banding
(12). Viral titer was routinely
concentrated to ~1×109 plaque-forming unit (PFU) as
determined by the plaque assay.
In vivo gene transfer and
establishment of a myocardial I/R model
Rats were subjected to adenovirus-mediated gene
transfer and a subsequent myocardial I/R surgical procedure. The
myocardial ischemia reperfusion model was performed as explained in
a previous study (12). Briefly,
rats were anesthetized with pentobarbital (30 mg/kg) via
intraperitoneal injection ventilated with oxygen using a small
animal breathing machine. After gently opening the chest through
the fourth and fifth ribs and safely exposing the heart, a 100 µl
solution of Ad-miR-22 (1×109 PFU), Ad-Scramble
(1×109 PFU) or saline was respectively injected into six
separate sites of the left ventricular anterior wall using a
26-gauge needle. Following the adenovirus or saline delivery, the
chest was closed and the rat was allowed to recover from
anesthesia. Four days after the rats were re-anesthetized with
pentobarbital (30 mg/kg) and the chest was re-opened allowing 100%
oxygen ventilation via a small animal breathing machine. The thorax
was reopened through the original intercostal space and the left
anterior descending coronary artery (LAD) was identified. LAD was
ligated using a 6-0 silk suture. Additionally, a medical latex tube
(socket inner diameter, 1.5 mm) was placed between the ligature and
the LAD. Myocardial ischemia was induced by tightening the ligature
around the latex tube. Successfully surgical myocardial ischemia
was detected on the basis of S-T segment elevation on an
electrocardiogram. After 30 min, the suture was withdrawn to
restore normal circulation for a 4 h period of reperfusion.
Following 4 h of reperfusion, the hearts of the rats and blood
samples were harvested for further investigation. Sham-operated
rats underwent similar procedures without the occlusion of LAD and
myocardial ischemic reperfusion.
Biochemical studies
Blood serum samples were collected to measure the
expression levels of two specific marker enzymes, CK (03010702011)
and LDH (03010703011), using commercial kits (Beijing Kemeidongya
Biotechnology Ltd., Beijing, China), according to the
manufacturer's instructions. The results were presented as U/l.
Detection of myocardial infarct area
(IA)/area at risk (AAR)
Evans blue/triphenyltetrazolium chloride (TTC)
double-staining was performed to determine the IA of the myocardium
as previously described (12). In
brief, following 4 h reperfusion, LAD was immediately re-occluded
and 1 ml 2.0% Evans blue (Sigma-Aldrich, St. Louis, MO, USA) was
intravenously administered to discriminate between the viable
non-ischemic area and the zone at risk. Stained hearts were quickly
removed, frozen and sliced to yield five sections (~2-mm thick),
which were subsequently incubated in 1.5% TTC (Sigma-Aldrich) for
15 min at 37°C. The viable myocardium at risk of ischemic was
stained red with TTC, whereas the IA without staining was pale
white. Therefore, the AAR (red plus white) and IA (white) from each
slice were delineated and assessed using Image-Pro Plus 5.0
software (Media Cybernetic, Rockville, MD, USA). The percentage of
IA/AAR was calculated.
Histological examination
Formalin-fixed, paraffin-embedded sections of
myocardial tissues were stained with hematoxylin and eosin and
examined under a light microscope (magnification, ×400).
Total RNA extraction and reverse
transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA from the cardiac muscle samples was
extracted using 1 ml TRIzol reagent (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) per 100 mg of tissue using a glass
homogenizer. For miRNA detection, 4.0 µg RNA was reverse
transcribed using a commercial cDNA synthesis kit (Fermentas;
Thermo Fisher Scientific, Inc.) at 42°C for 1 h. A mirVana RT-qPCR
miRNA detection kit (Thermo Fisher Scientific, Inc.) was used to
measure miR-22 expression levels. Amplification and detection via
qPCR were performed in a total reaction volume of 20 µl consisting
of SYBR Premix Ex Taq II (10 µl), 50X ROX Reference Dye II
(0.4 µl), forward primer (0.8 µl), reverse primer (0.8 µl),
DNA-template (2 µl) and nuclease-free water (6 µl), using an ABI
Prism 7500 system (Thermo Fisher Scientific, Inc.), and U6 was used
as an internal control. The relative level of miR-22 was calculated
based on the 2−ΔΔCq method (18). qPCR was run as follows: 50°C for 2
min, 95°C for 10 min, and 40 cycles of 95°C for 30 sec and 60°C for
30 sec. The following sequence-specific primers were used to
amplify the gene products: miR-22, forward,
5′-TGCGCAGTTCTTCAGTGGCAAG-3′ and reverse,
5′-CCAGTGCAGGGTCCGAGGTATT-3′; and U6, forward,
5′-CGCTTCGGCAGCACATATAC-3′ and reverse, 5′-AAATATGGAACGCTTCACGA-3′.
Samples were examined in triplicate.
Western blot analysis
To determine the protein levels of p38 MAPK, CBP,
c-Jun-AP-1, phospho (p)-c-Jun-AP-1 and GAPDH in the myocardial
tissue, protein was extracted from the AAR of the heart and western
blot analysis was performed as previously described (12). Briefly, the extracted proteins (50
µg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel
and transferred onto nitrocellulose membranes. Non-specific binding
was blocked with 5% non-fat dry milk for 2 h at room temperature.
The membrane was subsequently rinsed and incubated with anti-p38
MAPK (sc-535; 1:500), anti-CBP (sc-632; 1:500), anti-c-Jun-AP-1
(sc-44; 1:1,000) and anti-p-c-Jun-AP-1 (sc-101721; 1:1,000) primary
antibodies overnight at 4°C. Following washing three times with
TBST, the membranes were incubated with peroxidase-conjugated
secondary antibodies (1:50,000; BA1054; Boster Biological
Technology, Ltd., Wuhan, China) for 2 h at room temperature. Bands
were visualized using an enhanced chemiluminescence detection kit
(Pierce Biotechnology, Inc., Rockford, IL, USA). The expression
level of GAPDH served as a loading control and was used to
normalize the densities of the different samples. ImageJ 6.0
software (National Institutes of Health, Bethesda, MA, USA) was
used to quantify the optical density of each band.
ELISA
The ELISA method was used to determine the TNF-α and
IL-6 levels in cardiac muscle samples according to the
manufacturer's instructions. TNF-α (F16960) and IL-6 (F15870) ELISA
kits from Xitang Company (Shanghai, China) were used.
Statistical analysis
All the values were presented as the mean ± standard
error of the mean. Differences between groups were analyzed for
significance using one-way analysis of variance and
Student-Newman-Keuls-q test using SPSS software (version
14.0; SPSS Inc., Chicago, IL, USA). P<0.05 was used to indicate
a statistically significant difference.
Results
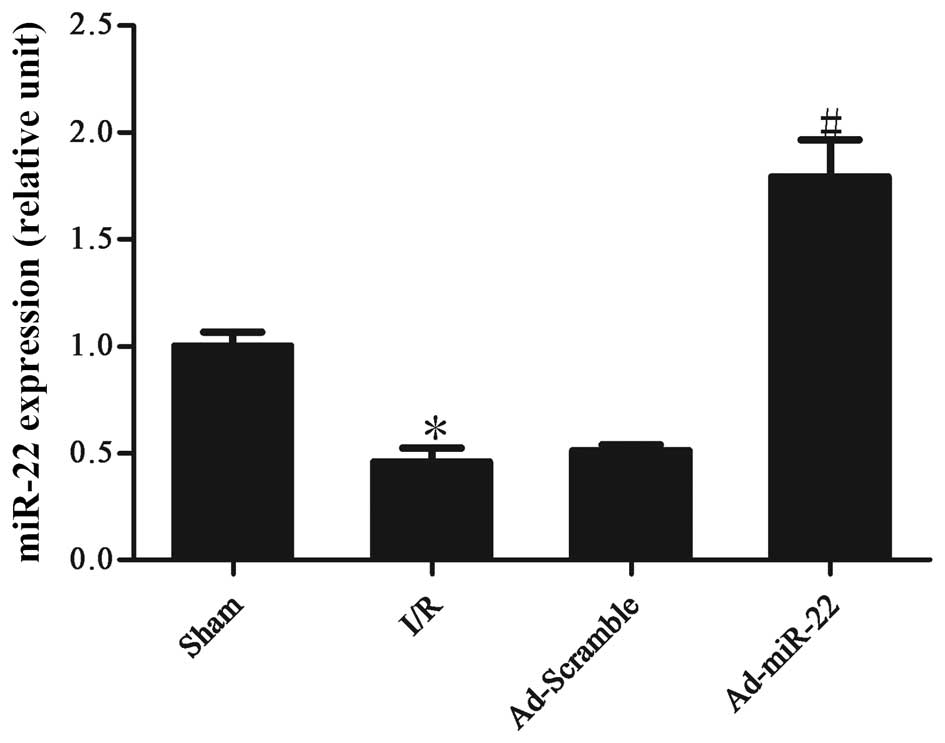
Myocardial I/R induces the
downregulation of miR-22 expression levels
Following 4-h reperfusion the levels of miR-22
expressed by the I/R myocardium were significantly reduced in
comparison to the non-ischemic sham control (I/R vs. sham group;
P<0.05; Fig. 1). Following
transfection to induce adenoviral overexpression of miR-22 into the
I/R myocardium for 4 days, miR-22 expression was significantly
increased (Ad-miR-22 vs. I/R group; P<0.05). However,
Ad-Scramble had no apparent effect on the expression level of
miR-22 compared with the I/R group (Ad-Scramble vs. I/R group;
P>0.05).
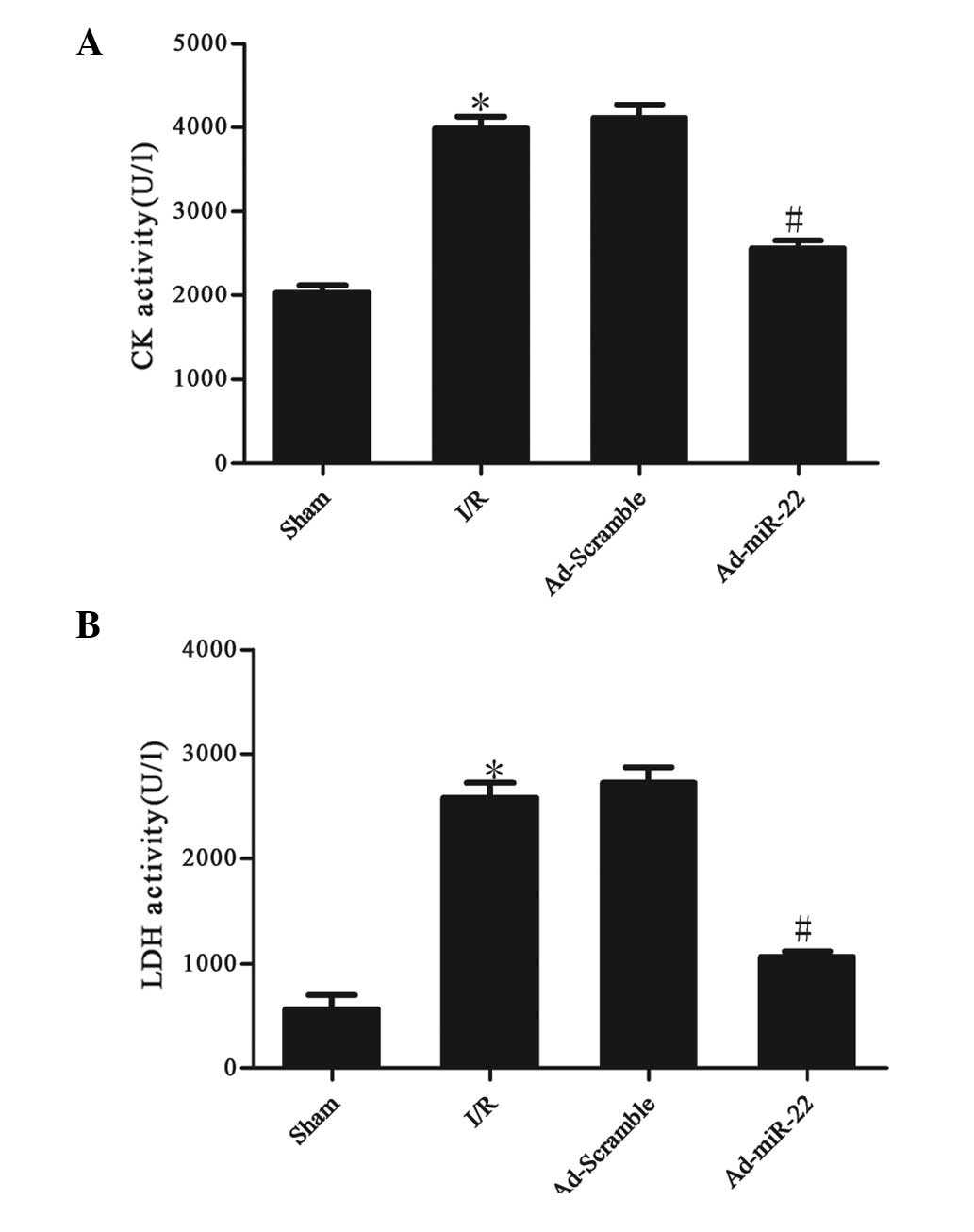
miR-22 reduces serum marker enzyme
levels
The activities of CK and LDH in the serum were used
to monitor the damage to the myocardium. Serum CK and LDH activity
significantly increased after 4 h of reperfusion in the I/R group
compared with the sham group (I/R vs. sham group; P<0.05;
Fig. 2). However, after Ad-miR-22
transfection, the CK and LDH levels in the Ad-miR-22 group
significantly decreased in comparison to the I/R group (Ad-miR-22
vs. I/R group; P<0.05). Ad-Scramble did not affect I/R-induced
increased leakage of CK and LDH from the myocardium (Ad-Scramble
group vs. I/R group; P>0.05).
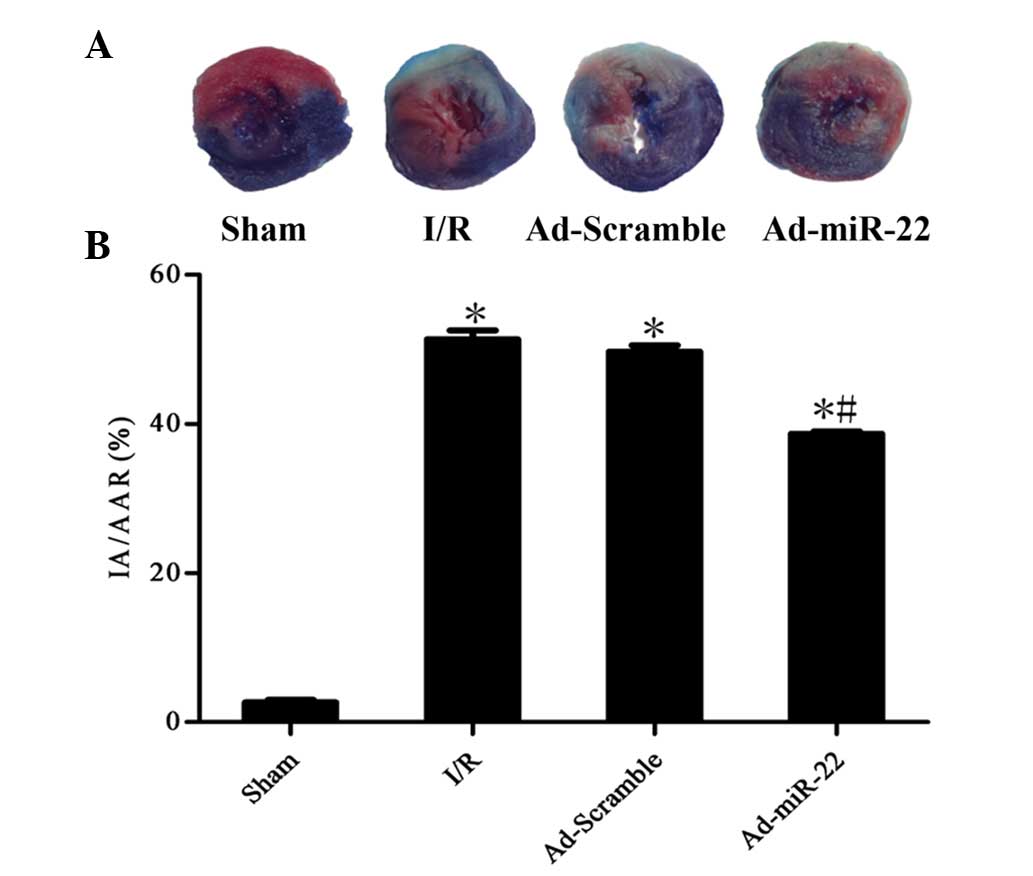
Upregulation of miR-22 decreases
infarct size
To quantitatively analyze the damage and potential
prognosis after reperfusion, IA/AAR was introduced as described
previously. Infarct size was measured as 51.4±2.6% of the IA/AAR in
the I/R group. Furthermore, overexpression of miR-22 using a
recombinant adenoviral vector exerted cardioprotective effects on
the development of myocardial I/R injury by significantly reducing
IR/AAR (Ad-miR-22 vs. I/R group; P<0.05; Fig. 3). Transfection of Ad-Scramble had no
significant effect on the damage and prognosis after myocardial I/R
in comparison with the I/R group (Ad-Scramble vs. I/R group;
P>0.05). This demonstrates that overexpression of miR-22 may be
used as a cardioprotective agent against myocardial I/R injury.
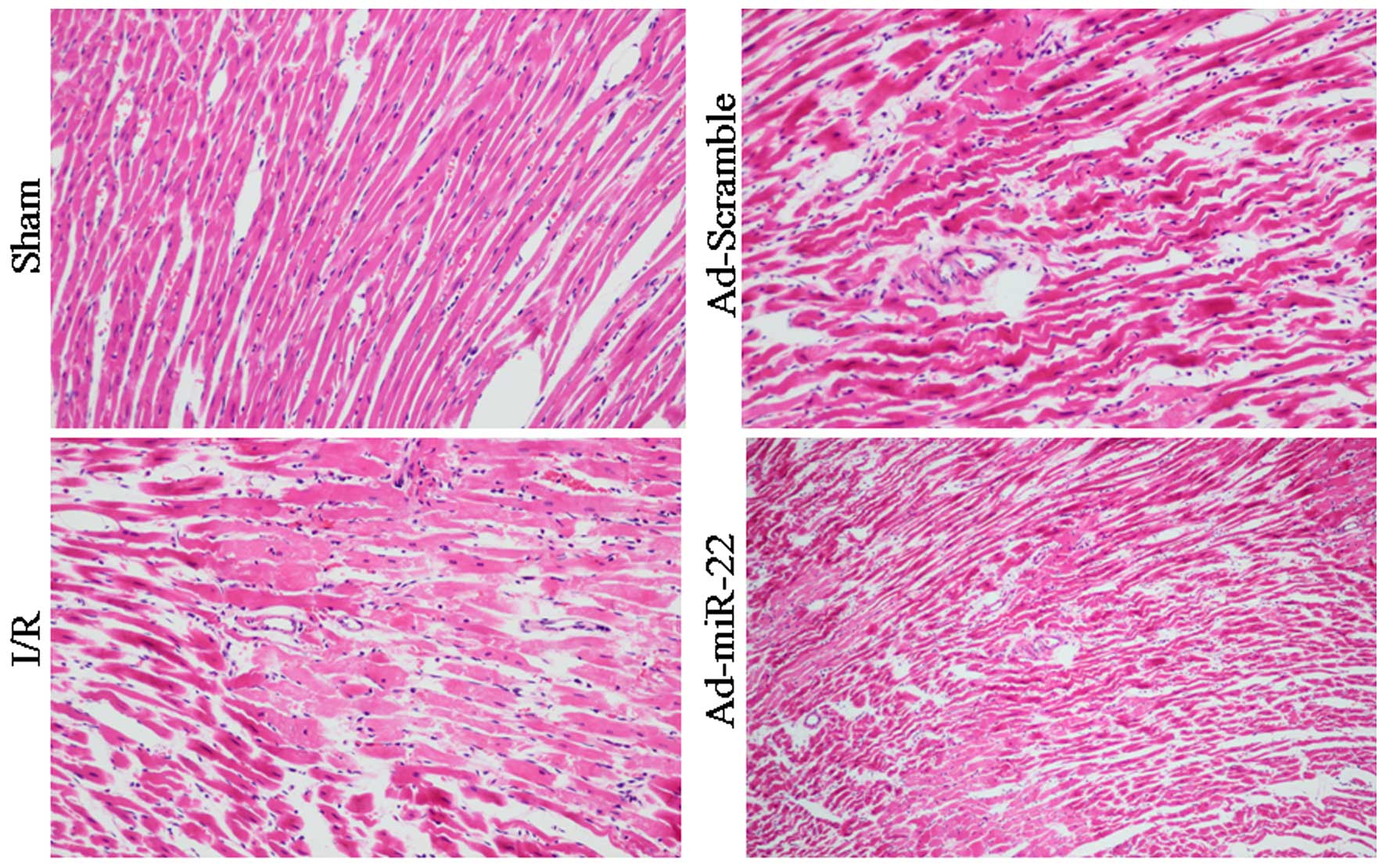
Light microscopy evaluation
Myocardial fibers of the Sham group were arranged
regularly without any apparent degeneration or necrosis. However,
disorganized myocardial fibers, edema, and ruptured and lysed cells
were observed in the I/R group. Furthermore, delivery of miR-22
partially rescued myocardium injury and inflammatory cell
infiltration. In addition, transfection with Ad-Scramble had no
notable effect on the morphological changes, compared with the I/R
group (Fig. 4).
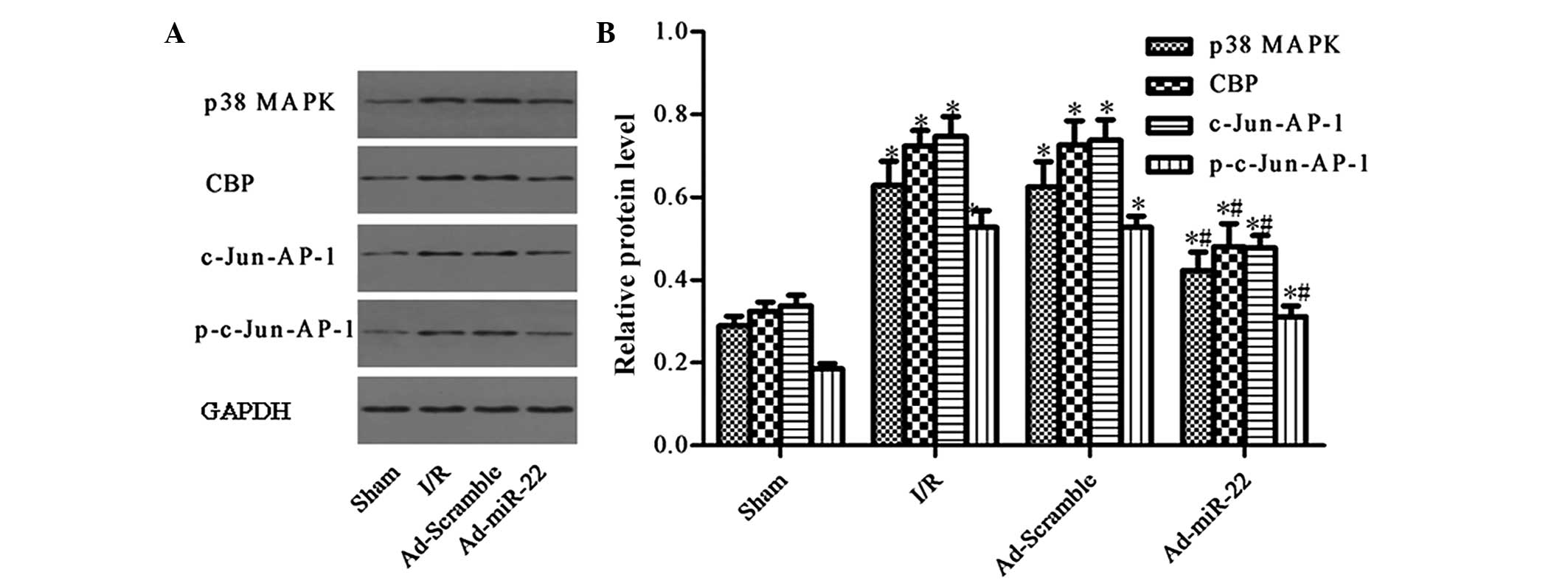
Upregulation of miR-22 suppresses the
expression of the p38 MAPK/CBP/c-Jun-AP-1 signaling pathway
p38 MAPK and CBP were demonstrated to be predicted
target genes of miR-22 by TargetScan (12)(genes.mit.edu/tscan/targetscan2003.html; Table I). c-Jun is a dominant component of
the c-Jun-AP-1 transcription factor, and p-c-Jun-AP-1 is the active
form of c-Jun-AP-1 (17). Compared
to the Sham group, the aforementioned protein levels were both
significantly upregulated in the I/R injury group (I/R vs. sham
group; P<0.05; Fig. 5).
Furthermore, delivery of miR-22 into the myocardium significantly
reduced the levels of p38 MAPK, CBP, c-Jun-AP-1 and p-c-Jun-AP-1 by
33.12, 32.90 and 38.50%, respectively (Ad-miR-22 vs. I/R group;
P<0.05). In addition, adenoviral transfection of Ad-Scramble had
no significant effect on the proteins mentioned above compared with
the I/R group (Ad-Scramble vs. I/R group; P>0.05). Therefore,
overexpression of miR-22 may specifically suppress the expression
of the p38 MAPK/CBP/c-Jun-AP-1 signaling pathway.
 | Table I.p38 MAPK and CBP are both miR-22
predicted targets using the TargetScan software. |
Table I.
p38 MAPK and CBP are both miR-22
predicted targets using the TargetScan software.
| Position | Predicted
consequential pairing of target region (top) and miRNA
(bottom) | Seed match | Sitetype
contribution | 3′ pairing
contribution | Local AU
contribution | Position
contribution | Context score | Context score
percentile | Conserved branch
length | PCT |
|---|
| 1211–1217 of
CREBBP | 5′
AUUGCAGUGGGUAUUGGCAGCUG | 7merm-8 | −0.161 | 0.005 | −0.019 | 0.073 | −0.10 | 41 | 0.515 | <0.1 |
| 3′ UTR mo-miR-22 | 3′
UGUCAAGAAGUUGACCGUCGAA |
|
|
|
|
|
|
|
|
|
| 1283–1289 of
CREBBP | 5′
CACAGAGAGUGAGGGGGCAGCUC | 7merm-8 | −0.161 | 0.005 | 0.092 | 0.077 | 0.01 | 3 | 0.086 | <0.1 |
| 3′ UTR mo-miR-22 | 3′
UGUCAAGAAGUUGACCGUCGAA |
|
|
|
|
|
|
|
|
|
| 1285–1291 of
MAPK14 | 5′
GGCCCCCCCGCCCCCGGCAGCUU | 7merm-8 | −0.161 | 0.067 | 0.114 | 0.030 | 0.05 | 0 | 1.796 | 0.59 |
| 3′ UTR
mo-miR-22 | 3′
UGUCAAGAAGUUGACCGUCGAA |
|
|
|
|
|
|
|
|
|
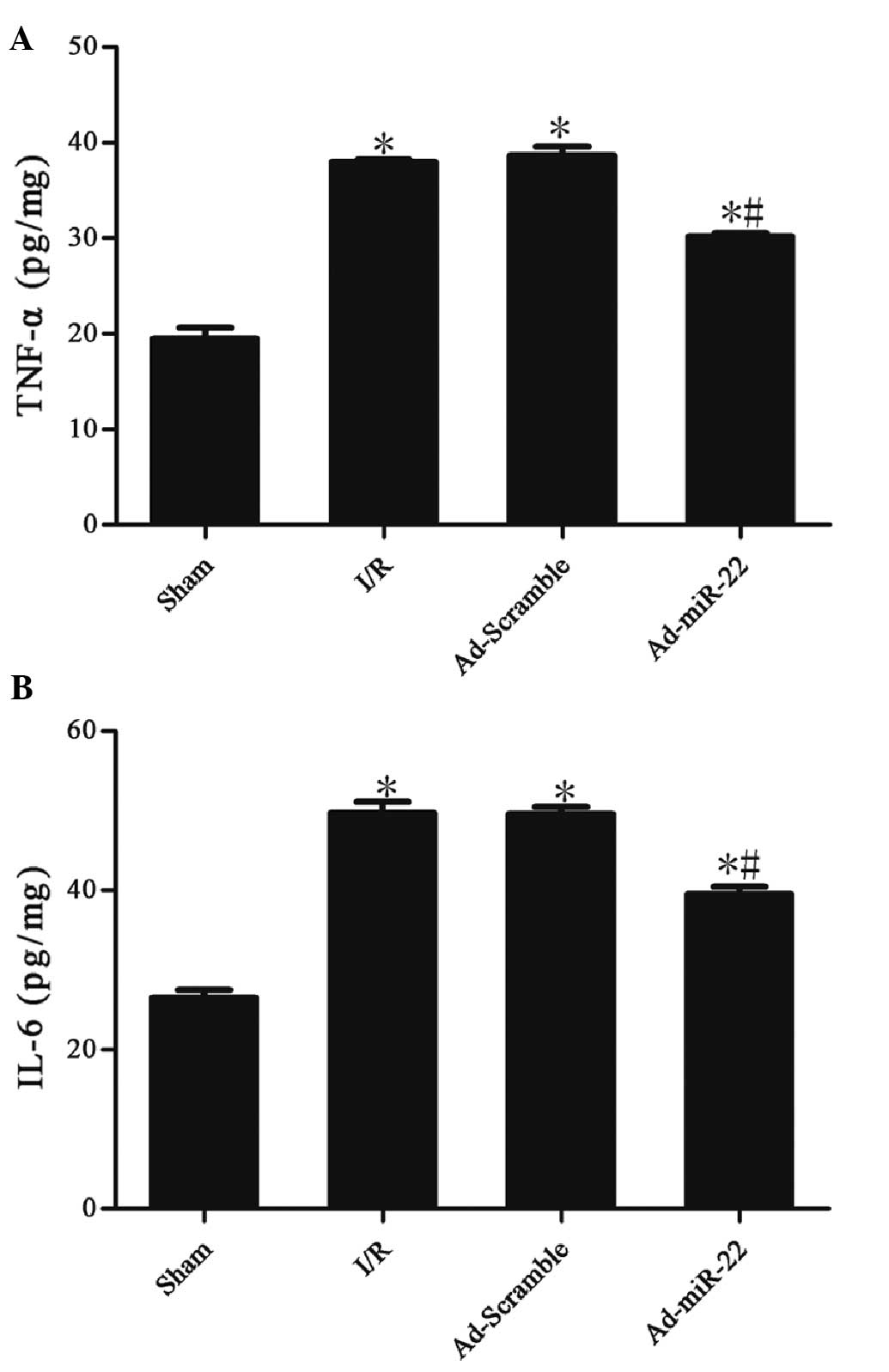
miR-22 reduces the levels of TNF-α and
IL-6
Myocardial I/R induced a significant increase in the
concentrations of TNF-α (37.99±0.13 vs. 19.43±0.69 pg/mg;
P<0.05) and IL-6 (49.66±0.85 vs. 26.45±0.60 pg/mg; P<0.05) in
the I/R group in comparison with the sham group. Myocardial
delivery of miR-22 significantly inhibited TNF-α (30.15±0.21 vs.
37.99±0.13 pg/mg; P<0.05) and IL-6 (39.50±0.55 vs. 49.66±0.85
pg/mg; P<0.05) expression compared with the I/R group. However,
adenoviral transfection of Ad-Scramble did not have a significant
effect on the two aforementioned cytokines in comparison with the
I/R group (Ad-Scramble vs. I/R group; P>0.05). These data
suggest that miR-22 could inhibit the production of inflammation
cytokines (Table II and Fig. 6).
| Table II.Myocardial TNF-α and IL-6 expression
after 4 h reperfusion in the four groups. |
Table II.
Myocardial TNF-α and IL-6 expression
after 4 h reperfusion in the four groups.
| Group | TNF-α (pg/mg) | IL-6 (pg/mg) |
|---|
| Sham | 19.43±0.69 | 26.45±0.60 |
| I/R |
37.99±0.13a |
49.66±0.85a |
| Ad-Scramble |
38.65±0.56a |
49.61±0.51a |
| Ad-miR-22 |
30.15±0.21a,b |
39.50±0.55a,b |
Discussion
It has previously been demonstrated that
inflammation pathways have a significant role in the
pathophysiological process of myocardial I/R injury (19). Experimental and clinical evidence has
indicated that anti-inflammatory actions may attenuate I/R injury
(5,6). Utilizing adenovirus-associated vectors,
the present study demonstrated that selective overexpression of
miR-22 induced promisingly cardioprotective properties as well as
an anti-inflammatory role in ameliorating myocardial I/R injury
in vivo. After increasing the levels of miR-22, the infarct
size and disordered morphology and myocardial enzyme levels were
reduced. Meanwhile, concomitant p38 MAPK, CBP, c-Jun-AP-1,
p-c-Jun-AP-1 suppression and inflammation cytokine (TNF-α and IL-6)
reduction occurred. These major findings demonstrated that the
cardioprotective effect of miR-22 against inflammation is, at least
partly, functionally attributed to its suppression of the p38
MAPK/CBP/c-Jun-AP-1 signaling pathway.
A number of miRNAs have been demonstrated to be
involved in myocardial I/R injury and miR-22 was only one of these
reported to regulate I/R injury (12,20). Our
previous study also demonstrated that adenovirus-mediated miR-22
overexpression protected against myocardial I/R injury through an
anti-apoptosis mechanism in rats by targeting CBP (12). However, the molecular mechanisms
involved in the cardioprotective effect of miR-22 are complicated
and are far from fully understood.
As a cardiac-enriched miRNA, miR-22 has various
targets in cardiomyocytes that were identified using TargetScan
(12). p38 MAPK and CBP are both
targets (Table I). In the present
study, p38 MAPK and CBP were upregulated in the I/R group; however,
both were suppressed following the delivery of miR-22. The
transcription factor, AP-1, had a similar variation tendency with
p38 MAPK and CBP in the four groups. This indicated that AP-1 was
also suppressed after p38 MAPK and CBP was suppressed by
miR-22.
p38 MAPK, which is one of the most functional
members of the MAPK family, has a key role in the progression of
I/R injury and is also involved in the activation of the
transcription factor, AP-1 (21,22). The
CBP gene is widely expressed and is important in the cardiovascular
system as it interacts with a variety of diverse transcriptional
factors, including AP-1 and p53 (12,23–25). Our
previously published data also demonstrated that CBP may be
important in myocardial I/R injury by influencing p53 (12). In summary, p38 MAPK and CBP may both
activate AP-1. Furthermore, p38 MAPK may not only activate AP-1
directly but may also activate CBP, and as a consequence activate
AP-1 (26). AP-1 is a regulator of
cytokine expression and is an important modulator in inflammatory
diseases, including rheumatoid arthritis, psoriasis, psoriatic
arthritis and myocardial I/R injury (16). c-Jun is considered to be a dominant
component of the c-Jun-AP-1 transcription factor complex and the
phosphorylation of this complex is the most important regulator of
c-Jun-AP-1, which suggests transcriptional activity (17,27).
Hence, suppressing c-Jun-AP-1 activity reduces I/R induced
myocardial injury by suppressing the inflammation caused by
p-c-Jun-AP-1. The present study revealed that the production of
inflammatory cytokines (TNF-α and IL-6) decreased with the
suppression of p38 MAPK, CBP, c-Jun-AP-1 and p-c-Jun-AP-1 compared
with the Sham group and after the overexpression of miR-22. These
results indicate that anti-inflammation action may be an additional
cardioprotective effect induced by miR-22 in myocardial I/R injury,
and the mechanism is associated with the p38 MAPK/CBP/c-Jun-AP-1
signaling pathway.
In conclusion, the results of the present study
suggests that miR-22 is capable of diminishing myocardial I/R
injury induced by inflammation in rat models by directly targeting
p38 MAPK and CBP. Overexpression of miR-22 leads to a significant
repression of the p38 MAPK/CBP/c-Jun-AP-1 signaling pathway and
results in the amelioration of inflammation. These findings suggest
that overexpression of miR-22 may be a desirable therapeutic
approach for the treatment of myocardial I/R injury.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81170133, 81200088
and 81470387), the Master's Degree Paper Pew Foundation of China
Three Gorges University (grant no. 2015PY052) and Hubei Province's
Outstanding Medical Academic Leader program of China (grant no.
201304).
References
|
1
|
Ndrepepa G: Improving myocardial injury,
infarct size, and myocardial salvage in the era of primary PCI for
STEMI. Coron Artery Dis. 26:341–355. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kloner RA: Does reperfusion injury exist
in humans? J Am Coll Cardiol. 21:537–545. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion - from mechanism to translation. Nat Med. 17:1391–1401.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu H, Zhai C, Qian G, Gu A, Liu J, Ying F,
Xu W, Jin D, Wang H, Hu H, Zhang Y and Tang G: Protective effects
of tanshinone IIA on myocardial ischemia reperfusion injury by
reducing oxidative stress, HMGB1 expression and inflammatory
reaction. Pharm Biol. 53:1752–1758. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Du X, Hu X and Wei J: Anti-inflammatory
effect of exendin-4 postconditioning during myocardial ischemia and
reperfusion. Mol Biol Rep. 41:3853–3857. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Doddakula KK, Neary PM, Wang JH, Sookhai
S, O'Donnell A, Aherne T, Bouchier-Hayes DJ and Redmond HP: The
antiendotoxin agent taurolidine potentially reduces
ischemia/reperfusion injury through its metabolite taurine.
Surgery. 148:567–572. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fan ZX and Yang J: Microribonucleic acids
and vascular restenosis. Saudi Med J. 35:796–801. 2014.PubMed/NCBI
|
|
8
|
Suzuki HI and Miyazono K: Emerging
complexity of microRNA generation cascades. J Biochem. 149:15–25.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peterson SM, Thompson JA, Ufkin ML,
Sathyanarayana P, Liaw L and Congdon CB: Common features of
microRNA target prediction tools. Front Genet. 5:232014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thum T and Condorelli G: Long noncoding
RNAs and microRNAs in cardiovascular pathophysiology. Circ Res.
116:751–762. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lorenzen JM, Batkai S and Thum T:
Regulation of cardiac and renal ischemia-reperfusion injury by
microRNAs. Free Radic Biol Med. 64:78–84. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang J, Chen L, Yang J, Ding J, Li S, Wu
H, Zhang J, Fan Z, Dong W and Li X: MicroRNA-22 targeting CBP
protects against myocardial ischemia-reperfusion injury through
anti-apoptosis in rats. Mol Biol Rep. 41:555–561. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu X, Shah A, Gangwani MR, Silverstein
PS, Fu M and Kumar A: HIV-1 Nef induces CCL5 production in
astrocytes through p38-MAPK and PI3K/Akt pathway and utilizes
NF-kB, CEBP and AP-1 transcription factors. Sci Rep.
4:44502014.PubMed/NCBI
|
|
14
|
McManus KJ and Hendzel MJ: CBP, a
transcriptional coactivator and acetyltransferase. Biochem Cell
Biol. 79:253–266. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sugden PH and Clerk A: ‘Stress-responsive’
mitogen-activated protein kinases (c-Jun N-terminal kinases and p38
mitogen-activated protein kinases) in the myocardium. Circ Res.
83:345–352. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zenz R, Eferl R, Scheinecker C, Redlich K,
Smolen J, Schonthaler HB, Kenner L, Tschachler E and Wagner EF:
Activator protein 1 (Fos/Jun) functions in inflammatory bone and
skin disease. Arthritis Res Ther. 10:2012008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Karin M: The regulation of AP-1 activity
by mitogen-activated protein kinases. J Biol Chem. 270:16483–16486.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCt method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma L, Liu H, Xie Z, Yang S, Xu W, Hou J
and Yu B: Ginsenoside Rb3 protects cardiomyocytes against
ischemia-reperfusion injury via the inhibition of JNK-mediated
NF-κB pathway: A mouse cardiomyocyte model. PLoS One.
9:e1036282014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang W, Shao J, Bai X and Zhang G:
Expression of plasma microRNA-1/21/208a/499 in myocardial ischemic
reperfusion injury. Cardiology. 130:237–241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kumphune S, Surinkaew S, Chattipakorn SC
and Chattipakorn N: Inhibition of p38 MAPK activation protects
cardiac mitochondria from ischemia/reperfusion injury. Pharm Biol.
53:1831–1841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Surinkaew S, Kumphune S, Chattipakorn S
and Chattipakorn N: Inhibition of p38 MAPK during ischemia, but not
reperfusion, effectively attenuates fatal arrhythmia in
ischemia/reperfusion heart. J Cardiovasc Pharmacol. 61:133–141.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang J, Jiang H, Chen SS, Chen J, Li WQ,
Xu SK and Wang JC: Lentivirus-mediated RNAi targeting CREB binding
protein attenuates neointimal formation and promotes
re-endothelialization in balloon injured rat carotid artery. Cell
Physiol Biochem. 26:441–448. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ait-Si-Ali S, Ramirez S, Barre FX, Dkhissi
F, Magnaghi-Jaulin L, Girault JA, Robin P, Knibiehler M, Pritchard
LL, Ducommun B, Trouche D and Harel-Bellan A: Histone
acetyltransferase activity of CBP is controlled by cycle-dependent
kinases and oncoprotein E1A. Nature. 396:184–186. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Avantaggiati ML, Ogryzko V, Gardner K,
Giordano A, Levine AS and Kelly K: Recruitment of p300/CBP in
p53-dependent signal pathways. Cell. 89:1175–1184. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kappelmann M, Bosserhoff A and Kuphal S:
AP-1/c-Jun transcription factors: Regulation and function in
malignant melanoma. Eur J Cell Biol. 93:76–81. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Karin M and Gallagher E: From JNK to pay
dirt: Jun kinases, their biochemistry, physiology and clinical
importance. IUBMB Life. 57:283–295. 2005. View Article : Google Scholar : PubMed/NCBI
|