Introduction
Inherited metabolic disorders (IMD) result from
mutations of genes associated with metabolism, cofactors, channels,
carriers or receptors in metabolic pathways, which are the basis of
metabolic disorders of anabolic or catabolic pathways (1,2). These
mutations may be responsible for different types of biochemical
abnormalities, including the accumulation of intermediate and/or
bypass metabolites, or a deficiency of terminal metabolites,
leading to metabolic crises, such as lactic acidosis,
hyperammonemia syndrome and ketoacidosis (3,4).
Biochemical abnormalities can be classified as those involving
organic acids, amino acids, fatty acids, carbohydrates, purines and
pyrimidines. Genotype-phenotype correlations based on expression
analysis of structural and functional mutations are gradually being
recognized in different IMDs and subtypes of the disease, and
>500 types of IMD have been identified so far (5). In all regions, it is reported that the
overall incidence of neonatals with IMD is 1 in 1,000–2,500
newborns, while an estimated 16–20 million newborns are confirmed
in China each year, indicating that effective and applicable
measures are warranted due to the high-risk of infant IMD (6).
The majority of the abnormal metabolites associated
with IMD are excreted in the urine; urine samples are easy to
collect noninvasively without intrusive operations, so are
therefore favored by researchers worldwide (7). Currently, the cross-association of
substances with different diseases requires a preferable strategy
to concurrently analyze characteristic metabolites in urine.
However, expensive analytical instruments in combination with the
long established ‘modified urease pretreatment method’ (8) and barriers to the fourth generation
chemical analysis methodology have seriously hampered clinical and
scientific development associated with IMD in China (9–11).
Metabolomics is a new systematic approach for the study of changes
in all components of intermediate molecular weight and terminal
metabolites (<1,000 Mr) (12) for the early screening and monitoring
of IMD in children. Metabolomics is characterized by high
throughput, sensitivity and selectivity. The simultaneous analysis
of metabolites in urine and the study of various types of IMD have
become popular research topics. As an advantageous analytical tool,
gas chromatography/mass spectrometry (GC/MS) is able to analyze a
variety of complex biological materials, including many metabolites
that are volatile and thermally stable (13–15).
Furthermore, GC/MS with retention locking time techniques
(GC-MS-RTL) and associated software have provided a strong
foundation for metabolomics research (16). However, related sample pretreatment
technology has become a limitation that requires investigation. The
GC/MS technique is limited for the analysis of thermally non-stable
amino acids, α-ketoacids and α-aldehyde acids, volatile and
semi-volatile organic acids, and polar metabolites.
Clinical technologies for simultaneous testing have
been reported for different types of materials, diseases and small
molecules in the urine (17,18). However, simultaneous analysis
procedures for the clinical analysis of urine have rarely been
reported for the hundreds of IMDs in China. The substances in urine
comprise five categories of small molecules with differing
compositions, properties and structure. A two-step chemical
derivatization method (19) for
urine pretreatment has achieved the ability to synchronously screen
organic acids, fatty acids, carbohydrates, purines and pyrimidines.
The clinical significance of this two-step chemical derivatization
method merits exploration in children with IMD.
The present study was performed to identify clinical
screening and monitoring values for five categories of IMDs using
the self-developed two-step derivatization method in urine. The
study aimed to explore potential markers for distinguishing
phenylketonuria, ornithine transcarbamylase (OTC) deficiency,
neonatal intrahepatic cholestasis caused by citrin deficiency
(NICCD), β-ureidopropionase deficiency, and mitochondrial metabolic
disorders with 8993T>G mutants from control subjects. The method
enabled the simultaneous examination of urinary organic acids
(including α-ketoacids and α-aldehyde acids), amino acids, fatty
acids, carbohydrates, purines and pyrimidines in 100-µl urine
samples.
Materials and methods
Ethics statement
The protocol of the current study has been approved
by the Ethics Committee of the Hunan Province Technical Institute
of Clinical Preventive and Treatment for Children's IMDs (Changsha,
China). Written informed consent was obtained from each child's
parents or guardians prior to the performance of the following
experimental procedures.
Source of material
From October 2010 to May 2014, children with
IMD-related symptoms treated in the Maternal and Child Health
Hospital of Hunan Province were selected and enrolled in the study,
including children who were stunted, or had mental retardation,
jaundice delay, epilepsy or seizures, cerebral palsy and autism
spectrum disorders. Inclusion criteria for those eligible children
were as follows: i) Children with low birth weight, preterm
children and severely malnourished IMD-risk children; ii) children
with normal or abnormal liver and kidney function; and iii)
children whose ages ranged between 0 and 12 years. The male to
female ratio was ~2.5:1. Morning urine and postprandial urine (2.5
h after a meal) samples (3–10 ml) were separately collected and
stored in a refrigerator at −70°C (Haier Group, Qingdao,
China).
The main focus of the research was the analysis of
pathogenic genes, including those associated with phenylketonuria
(c.782G>A), OTC deficiency (c.912G>T), NICCD (c.851-854del4;
NG_012247.1:g.A92352A/G), thymine-uracil aciduria (c.C216C/T),
β-urea acid deficiency diseases (c.C792C/A; c.G977G/A) and
mitochondrial metabolic disorders (m.8993T>G; m.10191T>
C).
Sample pretreatment
Urine samples (100 µl, containing 2.5 mmol/l
creatinine) were added to 30.0 µl (1.2 U/µl) urease, and then
incubated at 37°C for ~30 min. Following the addition of 50 µl of
the internal standards tropic acid solution and heptadecanoic acid
solution (0.5 mg/ml, respectively; Sigma-Aldrich; Merck Millipore,
Darmstadt, Germany) and mixing, 800 µl ethanol was added, followed
by centrifugation at 12,000 × g for 15 min at room
temperature (37°C). Subsequently, 40 µl hydroxylamine hydrochloride
solution (0.04 mol/l) and 60 µl Ba(OH)2 solution (0.05
mol/l) were supplied, and a solution of oxime compound was thus
generated. The liquid was heated for 60 min at 60°C, and the
supernatant was extracted into a small sample preparation bottle
placed under a nitrogen blowing instrument and carefully dried at
60°C. The residue was then combined with 100 µl double-derivatizing
reagent
N,O-bis(trimethylsilyl)trifluoroacetamide-trimethylchlorosilane
(BSTFA-TMCS, 99:1; Sigma-Aldrich; Merck Millipore), 20 µl pyridine
co-solvent and other self-made silylated complex liquids [including
BSTFA (bis-trimethylsilyl-trifluoroacetamide), MSTFA
(N-methyl-trimethylsilyltrifluoroacetamide), MTBSTFA
(N-(ter-Butyldimethylsilyl)-N-methyltrifluoroacetamide) and TMCS
(trimethyl chlorosilane)], and then placed at 80°C under a drying
heater to initiate the silanization derivatization process, as
previously described (19).
Subsequently, the derivatized products were transferred into a
bottle, diluted to a volume of 500 µl, and 1.0 µl derivatized
products were taken for analysis.
GC/MS conditions
Instrumentation used in this procedure comprised a
GC/MS instrument (7890-5975C Agilent Technologies, Inc., Santa
Clara, CA, USA), with a J&W HP-5 capillary column (column
length, 60 mm; film thickness, 25 mm; inner diameter, 0.25 µm). The
carrier gas was helium with a high purity (99.995%) at a flow rate
of 1.5 ml/min; the inlet temperature was 250°C, and the detailed
temperature program was as follows: An initial column oven
temperature of 60°C for 4 min, followed by a 6–15°C/min rate of
temperature increase up to 320°C for 10 min. The interface
temperature was 300°C, and the electronic ionization (EI) source
energy was 70 eV with an ion-source temperature of 230°C.
Basic parameters of data
processing
Using the SCAN mode, the retention times of the
chromatographic peaks for tropic acid and heptadecanoic acid were
determined. For tropic acid-TMS2, the retention time was 26.0340
min and its characteristic ion mass-to-charge ratios were 103, 115
and 280. Furthermore, for heptadecanoic acid-TMS1, the retention
time was 36.1070 min and characteristic ion mass-to-charge ratios
were 132, 145 and 327. Using the selecting ion scanning (SIM) mode
for qualitative and quantitative analysis, drift in the retention
time was automatically corrected by the software, using retention
time locking technology. Therefore, the retention time of tropic
acid TMS2 was locked at 29.9280±0.0100 min and that of
heptadecanoic acid-TMS1 was locked at 36.0000±0.0100 min; the ratio
of five different peak areas for 80, 90, 100, 110 and 120 µl of
12.9 mmol/l tropate to the peak area for 100 µl of 2.5 mmol/l
creatinine was used to create a calibration curve (metabolomic data
normalization), as an internal standard method of quality control,
as described in our two previously published papers (8,20). The
ratio of the peak areas of the metabolites to the peak area of 100
µl of 2.5 mmol/l creatinine were used as parameters in a
semi-quantitative analysis, in accordance with the establishment of
a principal component analysis (PCA) model.
Pattern recognition results and the Physician's
Guide to the Laboratory Diagnosis of Metabolic Diseases (21) were used to identify the
metabolites.
Statistical analysis
Independent-sample t-tests were used for the
comparison of the metabolite levels to identify significant
differences between the preliminary study group and
gender/age-matched controls. Statistical analysis of some
characteristic metabolites has been described in detail in our
previous papers (8,20). A PCA model was constructed using
differentially expressed compounds, with P<0.05 considered to
indicate a statistically significant difference. Deconvolution
Reporting Software (version A.02.00), an Automated Mass Spectral
Deconvolution and Identification System (5975C-7890A) and Mass
Profiler Professional software (version 2.0; all Agilent
Technologies, Inc.) were used.
Results
Simultaneous analysis using two-step
derivatization in phenylalanine hydroxylase deficiency
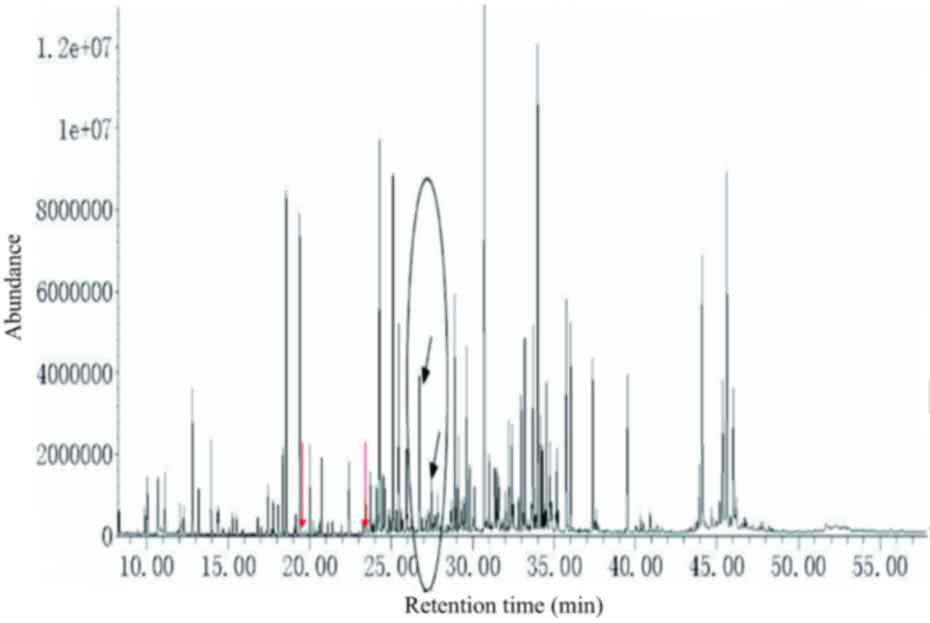
The urine from patients with phenylketonuria
(c.782G>A) presented a wide range of peaks. Test results for
patients with phenylketonuria indicated the presence of not only
organic acids, but also amines, amino acids and fatty acids
(Fig. 1). The urine metabolic
spectrum indicated the presence of N-acetylphenylalanine, tyrosine,
phenylacetate, mandelate, o-hydroxyphenylacetate, phenyllactate,
phenylpyruvate, phenylacetylglutamine and homovanillate. Thus, the
present method was able to detect various metabolites in the urine
simultaneously.
 | Figure 1.Urine metabolic spectrum from a
patient with classic phenylketonuria (c.782G>A). From left to
right, peaks for phenylacetic acid, mandelic acid,
ortho-hydroxyphenylacetic acid, phenyllactic acid, phenylalanine,
phenylpyruvic acid, N-acetylphenylalanine and phenylacetylglutamin
(and its bypass metabolite products, including tyrosine,
tryptophan, vitamins and secondary metabolities) are indicated by
arrows. All the indicated peaks, with the exception of
N-acetylphenylalanine, were markedly increased. |
Simultaneous analysis using two-step
derivatization in urea cycle disorders
The metabolites present in the urine from patients
with OTC deficiency (c.912G>T) were simultaneously presented
under the current experimental conditions. The results identified
the presence of uracil, orotate and uridine, among other
metabolites, in the urine (Fig.
2).
Simultaneous analysis using two-step
derivatization in citrin deficiency
In the urine samples from patients with citrin
deficiency (c.851-854del4; NG_012247.1: g.A92352A/G) galactose,
galactonate and galactitol were detected, which might be due to
abnormal metabolic pathways for galactose, in addition to glucose,
gluconate and glucuronate (glucose disorders), and
4-hydroxyphenylacetate, 4-hydroxyphenyllactate,
4-hydroxyphenylpyruvate, N-acetyltyrosine, phenylalanine and
phenylacetate (tyrosine and phenylalanine disorders). The urine
metabolic spectrum (Fig. 3) showed
peaks for galactose, galactonate, galactitol, glucose, gluconate,
glucuronate, 4-hydroxyphenylacetate, 4-hydroxyphenyllactate,
4-hydroxyphenylpyruvate, tyrosine and N-acetyltyrosine (Fig. 3).
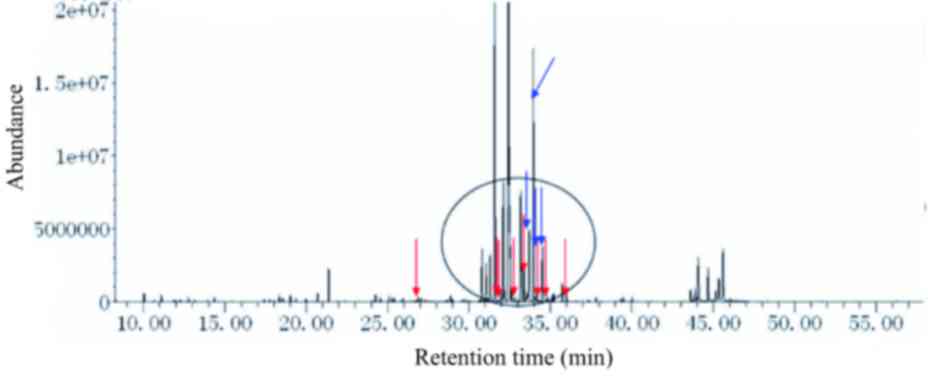 | Figure 3.Urine metabolic spectrum from a
patient with neonatal intrahepatic cholestasis caused by citrin
deficiency (c.851-854del4). From left to right, peaks for
4-hydroxyphenylacetate, galactonate, 4-hydroxyphenyllactate,
4-hydroxyphenylpyruvate, galactitol, galactose-oxime-1,
glucose-oxime-1, and glucose-oxime-2 are indicated by red arrows.
Gluconate, glucuronate, galactose-oxime-2 and N-acetyltyrosine are
indicated by blue arrows. The main biomarkers of the disorders are
circled, excluding phenylalanine, tyrosine, methionine and
ornithine. Urine levels of galactose-oxime-1 and galactose-oxime-2
were markedly increased. |
Simultaneous analysis using two-step
derivatization in purine and pyrimidine deficiencies
The urine from patients with thymine-uracil aciduria
(c.C216C/T), β-ureidopropionase deficiency (c.C792C/A and
c.G977G/A) presented results under the current analytical
conditions. The results indicated the presence of uracil, thymine,
5-hydroxymethyl-uracil, dihydrouracil, dihydrothymine,
β-ureidopropionate, β-aminoisobutyrate and β-alanine, among others
(Fig. 4). These results indicated
that metabolites associated with the metabolic pathways of purines
and pyrimidines can be detected appropriately using this two-step
derivatization method.
Simultaneous analysis using two-step
derivatization in mitochondrial metabolic disorders
The urine from patients with mitochondrial metabolic
disorders (e.g., m..8993T>G, m.10191T> C, m.14487 T>C,
m.14693 A>G) was clearly presented under the current
experimental conditions. Results which are closely correlated with
mitochondrial metabolic disorders include lactate pyruvate,
β-hydroxybutyrate, acetoacetate, ethylmalonate, methylmalonate,
adipate, azelate and sebate were observed. In addition,
α-ketoglutarate (α-KG) and succinate were also detected (Fig. 5). Both α-KG and succinate were
clearly increased to varying degrees in the urine of outpatients
with mitochondrial metabolic disorders.
Methodological evaluation using
two-step derivatization
Repeated injection of the same urine sample
containing standards of α-KG was conducted 9 times and the relative
standard deviations (RSDs) of the retention time and peak area of
α-KG were found to be 3 and 1.3%, respectively, indicating that the
apparatus had a good level of precision. Following the preparation
of 9 urine samples in parallel, the RSDs of the retention time and
peak area of α-KG were found to be 0.8 and 3.9%, respectively,
indicating that the reproducibility of the sample preparation
method was also good. When a blank urine sample was taken, α-KG
added and the recovery measured, the average recoveries were 93.1,
99.8 and 89.6% (n=9), respectively, for 3.3788, 6.7576 and 10.1364
µmol/l α-KG, and the RSD was <7.5% (n=9).
Discussion
Since this two-step derivatization method was
devised, it has been applied to the analysis of >3,320 cases
from the Hunan Province of China, and elsewhere. Our previous
investigations supported the ability of two-step derivatization to
simultaneously screen and monitor biochemical metabolic markers of
five categories of IMDs (8,22). At present, classic phenylketonuria,
OTC deficiency diseases, citrin deficiency, β-ureidopropionase
deficiency, and 8993T>G mitochondrial metabolic disorders have
been detected, and 69 single diseases and 516 cases of children
with IMDs, such as citrin deficiency disease (NG_012247.1
g.A92352A/G) and other genetic analyses have been confirmed with
testing of gene mutations. We have detected the presence of one or
more secondary metabolic diseases in some individuals, which has
enabled a comprehensive understanding of their complications and
effective treatment.
However, it difficult to investigate all aspects of
metabolic diseases due to the wide variety of metabolites, namely
amino acids, organic acids, fats, carbohydrates, purines and
pyrimidines. Therefore, this study investigated classic
phenylketonuria, OTC deficiency diseases, citrin deficiency,
β-ureidopropionase deficiency and 8993T>G mitochondrial
metabolic disorders, focusing on the analysis of relevant
biomarkers using the two-step derivatization of urine samples.
There are two types of defects associated with
phenylketonuria and hyperphenylalaninemia, which are correlated
with the tetrahydrobiopterin (BH4) anabolic pathway or
phenylalanine catabolic pathway (23,24). A
wide range of biomarkers for phenylketonuria (c.782G>A) were
presented in the present study, including phenylalanine,
N-acetylphenylalanine, tyrosine, phenylacetate, mandelate,
o-hydroxyphenylacetate, phenyllactate, phenylpyruvate,
phenylacetylglutamine and homovanillate. These biomarkers of
phenylketonuria have been detected previously through mass
spectrometry with electrospray ionization (15) and high-performance liquid
chromatography with a GC-clamped primer combined with other
analytical methods (25).
The present study focused on classical
phenylketonuria, which has been described in detail in our previous
study (20). Some biomarkers were
identified as being helpful for secondary differential disorders in
certain complications associated with classical phenylketonuria.
These are increased 4-hydroxyphenylacetate, 4-hydroxyphenylpyruvate
and 4-hydroxyphenyllactate (secondary tyrosinemia type III, or
vitamin C deficiency), 5-hydroxyindoleacetate,
5-hydroxyindolepyruvate and 5-hydroxyindolepropionate (secondary
tryptophan deficiency), lactate, pyruvate and alanine (vitamin B1
deficiency), glutarate, succinate and α-KG (vitamin B2 deficiency),
quinolinate, 4-hydroxyquinolinate and 4,8-dihydroxyquinolinate
(vitamin B6 deficiency), and methylmalonate and 2-methylcitrate
(vitamin B12 deficiency), respectively. Furthermore, various
biomarkers were used to discern classical phenylketonuria
(increased phenylacetate), hyperphenylalaninemia (increased
phenylalanine) and BH4-defects (increased 5-hydroxyindoleacetate,
5-hydroxyindolepyruvate and 5-hydroxyindolepropionate). Thus, the
analysis of urine samples using two-step derivatization
pretreatment under optimal conditions enabled the screening of
numerous potential biomarkers in phenylketonuria and
hyperphenylalaninemia.
There are >10 types of defect in the urea cycle,
which have been shown to be associated with various metabolic
pathways (26–30). The activities of enzymes including
N-acetylglutamate synthetase (NAGs), carbamoyl phosphate synthetase
I (CPS-I), argininosuccinate synthetase, argininosuccinate lyase,
OTC and argininase are severely lacking in the urea cycle, leading
to the accumulation of ammonia and other abnormal metabolites. When
screening for the urea cycle and related disorders, there are two
key challenges, which are the low solubility of the metabolites,
and their sensitivity to oxidation and reduction. Most importantly,
orotate is characterized by thermal non-stability, high sensitivity
to oxidation and reduction and poor solubility in organic solvents,
and so has not been precisely tested using conventional methods
(31).
In the present study, at least six biomarkers were
found to be helpful for severe OTC deficiency and its secondary
β-ureidopropionase deficiency, including uracil, orotate, uridine,
β-ureidopropionate, β-aminoisobutyrate and β-alanine. Reports of
these OTC biomarkers in the literature are rare (32). In the present study, a PCA model was
constructed using uracil, orotate and uridine, respectively.
In addition, CPS-I deficiency and NAG deficiency may
cause hyperammonemia (33), Even
though we did not find carbamyl phosphate accumulation in these two
disorders, the present method was able to detect elevated
glutamine, alanine and asparagine, combined with hyperammonemia, so
as to enable a differential diagnosis.
Citrin deficiency is a defect of mitochondrial
carrier protein glutamate-aspartate carrier that was discovered in
1999 (34). Citrin has several
functions, which include participation in the biosynthesis of urea,
protein and nucleic acids (35), and
the provision of adequate NADH for mitochondrial membranes
(36). Therefore, citrin dysfunction
affects synthetic or catabolic pathways, leading to a series of
metabolic dysfunctions and varying degrees of clinical
manifestations (37). The most
common mutants associated with citrin deficiency have been
identified (c.1177+1G>A and 851-854del) (38).
In general, three critical metabolic pathways were
identified as potential biomarkers for citrin deficiency in the
present study: Galactose, galactonate and galactitol; glucose,
gluconate and glucuronate; and 4-hydroxyphenylacetate,
4-hydroxyphenyllactate and 4-hydroxyphenylpyruvate. Differences in
their expression was detected between classical patients and normal
controls, with a PCA model constructed using these biomarkers.
Notably, 4-hydroxyphenyllactate and 4-hydroxyphenylpyruvate are
readily oxidized and reduced metabolites.
Other biomarkers, such as methionine, lysine,
threonine, citrulline, arginine and ornithine, for non-classical or
classical citrin deficiency have been reported in previous studies
(39,40). However, lysine, citrulline and
ornithine are characterized by a lack of thermal stability, and
ease of oxidation and reduction and, to the best of our knowledge,
have not been precisely tested using conventional derivatization.
With respect to derivatization, in the present study the urine
samples were pretreated at room temperature, and minute pH-value
variation was required to obtain optimal conditions. Furthermore,
the present study newly identified certain metabolites of
phenylacetate, N-acetyltyrosine, adenine and adenosine, which have
not been reported in previous studies.
Metabolic disorders of purines and pyrimidines are
autosomal recessive inheritance disorders (41). There are three defects associated
with pyrimidines, namely dihydropyrimidine dehydrogenase,
dihydropyrimidinase and 3-ureidopropionase deficiencies (42,43).
There are numerous purine-associated disorders, including orotic
aciduria (44,45). When considering biomarkers for these
disorders, it is notable that thymine, 5-hydroxymethyl-uracil,
dihydrouracil, dihydrothymine and uracil are characterized by poor
solubility in organic solvents and so they have not been accurately
tested using conventional methods. Furthermore, orotate is oxidized
and reduced readily in pretreated urine to form, for example,
β-ureidopropionate, β-ureidoisobutyrate, β-aminoisobutyrate, and
β-alanine. Some of these changes could be classified as secondary
purine and pyrimidine metabolic disorders, as the previously
described OTC deficiency increases orotate levels.
Using the two-step derivatization method of the
present study, concentrations of thymine, 5-hydroxymethyl-uracil,
β-ureidopropionate, β-ureidoisobutyrate and orotate in patients
could be detected at concentration ranges up to a hundred-fold
those of normal controls, with a PCA model constructed using
thymine, β-ureidopropionate and uracil, respectively.
Thus, the present method was found to be helpful for
the analysis of purine and pyrimidine deficiencies. So far, we have
examined only 3 cases with thymine-uracil aciduria combined with
β-ureidopropionase deficiency (c.C216C/T and c.G977G/A), two cases
with thymine-uracil aciduria (c.C216C/T), two cases with
β-ureidopropionase deficiency (c.C792C/A combined c.G977G/A), and
two cases with orotic aciduria to be examined. However, the
prevalence of these metabolic disorders in Europe is reportedly
only 26/3.2 million (46–48), suggesting that conventional methods
by urine could have been affected in the course of investigation
and thus lead to a false negative or incorrect result, which
requires further investigation. Therefore, urine samples were
pretreated at room temperature, and fine pH-value variation was
required to obtain the optimal conditions.
Mitochondrial metabolic disorders belong to the
maternal or Mendelian inherited class of diseases (49,50).
Approximately 50% of patients succumb to these disorders by the age
of 3 years. Mutations associated with Leigh syndrome are located in
genes including MT-ATP6, MT-ND2 and MT-CO3 by Mendelian inheritance
(51,52). It has been reported that 10–20%
patients with Leigh syndrome have a MT-ATP6 m.8993T>G mutation
(53). When there is a severe defect
of the complex IV respiratory chain enzyme of the mitochondrial
oxidative phosphorylation system, the enzyme cannot convert ADP
into ATP successfully as that of a healthy individual would. Thus,
a chronic progressive disorder of mitochondrial energy metabolism
occurs, with a series of clinical manifestations of mitochondrial
energy (54,55). Notably, estimable biomarkers for
mitochondrial metabolic disorders remain to be verified;
metabolites such as lactate, pyruvate and the lactate:pyruvate
ratio; β-hydroxybutyrate, acetoacetate and the
β-hydroxybutyrate:acetoacetate ratio, fumarate, malate, citrate,
aconitate, ethylmalonate, methylmalonate, adipate, azelate,
sebacate and 4-hydroxyphenyllactate have been reported in urine
(21). In particular, many of the
aforementioned changes might be attributable to secondary
mitochondrial metabolic disorders (for example, vitamin B2 or
vitamin B1 deficiency, valproate hyperlipidemia or propionic
acidemia).
Despite the lack of specificity in qualitative
parameters, findings for quantitative parameters identified
markedly increased concentrations of α-KG and succinate using the
present two-step derivatization method. These two biomarkers have
also been reported in previous studies (56,57);
however, they cannot be verified as biomarkers for mitochondrial
metabolic disorders in the present study, due to the limited number
of cases (n=31). Therefore, they require further investigation. In
the 31 cases with mitochondrial metabolic disorders (including
m..8993T>G, m.10191T> C, m.14487 T>C, m.14693 A>G and
CoQ10 deficiency), the two-step derivatization process of the
present study revealed that concentrations of α-KG alone or
combined with succinate in untreated patients could be >5 to
130-fold higher than those in normal controls, based on the
construction of a PCA model using α-KG (<5-fold) and combined
succinate with α-KG (>5 to 130-fold). It should be noted that
their levels were not notably decreased following the
administration of conventional vitamins.
In the quantitation procedure, the oximation
processing time of α-KG was clearly reduced. Notably, α-KG has not
previously been precisely examined using conventional MS-based
methods with oximation in urine as it is thermally unstable, and
readily undergoes oxidation and reduction. The present study
resolved this technical problem using our own oximation reagents;
thus, α-KG required only <3 min for oximation in the urine
samples, whereas, oximation in previous studies has been reported
to take from 4 to 48 h (19). Also,
it is widely reported that α-KG is converted at least partially to
α-hydroxyglutarate during the conventional pretreatment used for
urinary organic acids (58), as its
oxidative and reductive reactions are increased under heating at
higher pH-values. Under the optimal conditions in the present
study, oximation was conducted at room temperature for <3 min,
as described in detail in our previous two studies (8,20).
Biochemical substances that contain α-KG, glyoxylate
and other α-keto acids and aldehydes have extremely unstable
structures (59). Ion exchange,
organic extraction, urease pretreatment and modified urease
pretreatment methods in the analysis of urine have not been adapted
to traditional oximation, and it is difficult to obtain stable
GC/MS data for these categories of substances in the laboratory.
However, the traditional method of oximation requires high
temperatures and a relatively long reaction time, which greatly
affects the combined analysis of semi-volatile, readily
decomposable substances. Therefore, only α-KG was used as an
example to evaluate the method.
In conclusion, urine analysis with two-step
derivatization has demonstrated an improved ability for the
analysis of thermally unstable amino acids, α-ketoacids, α-aldehyde
acids, and volatile and semi-volatile organic acids, which expands
the range of compounds able to be simultaneously analyzed by GC/MS.
In China, the novel technology described in this study has been
applied effectively for the clinical screening and monitoring of
five types of small molecule in children with IMDs. It has also
provided a more comprehensive and reliable biochemical analysis
tool than any previously used in China. Furthermore, it has
provided an effective and reliable monitoring platform for use
metabolomic studies of mitochondrial and nutritional metabolic
disorders.
Acknowledgements
This work was supported by Hunan Province Technical
Institute of Clinical Preventive and Treatment for Children's
Inherited Metabolic Disorders. This work was financially supported
by Hunan Province Department of Science and Technology (grant nos.
2013XC83-11, 2014SK4044 and 2015SK2032).
References
|
1
|
Pampols T: Inherited metabolic rare
disease. Adv Exp Med Biol. 686:397–431. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pérez B, Rodriguez-Pascau L, Vilageliu L,
Grinberg D, Ugarte M and Desviat LR: Present and future of
antisense therapy for splicing modulation in inherited metabolic
disease. J Inherit Metab Dis. 33:397–403. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rodrigues JV, Henriques BJ, Lucas TG and
Gomes CM: Cofactors and metabolites as protein folding helpers in
metabolic diseases. Curr Top Med Chem. 12:2546–2559. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cassiman D: Gene transfer for inborn
errors of metabolism of the liver: The clinical perspective. Curr
Pharm Des. 17:2550–2557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Blau N, Duran M, Gibson KM and
Dionisi-Vici C: Physician's Guide to the Diagnosis, Treatment, and
Follow-Up of Inherited Metabolic Diseases. Springer; Heidelberg:
2014, View Article : Google Scholar
|
|
6
|
Song SM, Yoon HR, Lee A and Lee KR:
Seven-year experience with inherited metabolic disorders screening
by tandem mass spectrometry. J Genet Med. 5:21–25. 2008.
|
|
7
|
Janeckova H, Kalivodova A, Najdekr L,
Friedecky D, Hron K, Bruheim P and Adam T: Untargeted metabolomic
analysis of urine samples in the diagnosis of some inherited
metabolic disorders. Biomed Pap Med Fac Univ Palacky Olomouc Czech
Repub. 159:582–585. 2015.PubMed/NCBI
|
|
8
|
Xiong X, Sheng X, Liu D, Zeng T, Peng Y
and Wang Y: A GC/MS-based metabolomic approach for reliable
diagnosis of phenylketonuria. Anal Bioanal Chem. 407:8825–8833.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kuhara T: Diagnosis of inborn errors of
metabolism using filter paper urine, urease treatment, isotope
dilution and gas chromatography-mass spectrometry. J Chromatogr B
Biomed Sci Appl. 758:3–25. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kuhara T: Diagnosis and monitoring of
inborn errors of metabolism using urease-pretreatment of urine,
isotope dilution, and gas chromatography-mass spectrometry. J
Chromatogr B Analyt Technol Biomed Life Sci. 781:497–517. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kuhara T, Ohse M, Inoue Y, Yorifuji T,
Sakura N, Mitsubuchi H, Endo F and Ishimatu J: Gas
chromatographic-mass spectrometric newborn screening for propionic
acidaemia by targeting methylcitrate in dried filter-paper urine
samples. J Inherit Metab Dis. 25:98–106. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fryčák P, Lemr K, Adam T and Hušková R:
Diagnostics of some inherited metabolic disorders by mass
spectrometry using modern ionisation techniques. Chemicke Listy.
97:93–100. 2003.
|
|
13
|
Bruheim P, Kvitvang HF and Villas-Boas SG:
Stable isotope coded derivatizing reagents as internal standards in
metabolite profiling. J Chromatogr A. 1296:196–203. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bouatra S, Aziat F, Mandal R, Guo AC,
Wilson MR, Knox C, Bjorndahl TC, Krishnamurthy R, Saleem F, Liu P,
et al: The human urine metabolome. PLoS One. 8:e730762013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Janečková H, Hron K, Wojtowicz P, Hlídková
E, Barešová A, Friedecký D, Zídková L, Hornik P, Behúlová D,
Procházková D, et al: Targeted metabolomic analysis of plasma
samples for the diagnosis of inherited metabolic disorders. J
Chromatogr A. 1226:11–17. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qun G: Evaluation of automated sample
preparation, retention time locked GC-MS and automated data
analysis for the metabolomic study of Arabidopsis species. J
Chromatogr A. 1218:3247–3254. 2010.
|
|
17
|
Scher HI, Nasso SF, Rubin EH and Simon R:
Adaptive clinical trial designs for simultaneous testing of matched
diagnostics and therapeutics. Clin Cancer Res. 17:6634–6640. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rosser CJ, Dai Y, Miyake M, Zhang G and
Goodison S: Simultaneous multi-analyte urinary protein assay for
bladder cancer detection. BMC Biotechnol. 14:81–89. 2013.
|
|
19
|
Wang YC: A two-step derivatization on
simultaneous analysis for organic acids, fatty acids, amino acid,
carbohydrates, pyrimidines, and purines in complicated organic
compounds. CN patent ZL201210114246.2. Filed April 18, 2012; issued
December 18, 2013.
|
|
20
|
Xiong X, Liu D, Wang Y, Zeng T and Peng Y:
Urinary 3-(3-hydroxyphenyl)-3-hydroxypropionic acid,
3-hydroxyphenylacetic acid, and 3-hydroxyhippuric acid are elevated
in children with autism spectrum disorders. Biomed Research
International. 2016:94854122016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Blau N, Duran M, Blaskovics ME and Gibson
MK: Physician's Guide to the Laboratory Diagnosis of Metabolic
Diseases. 2nd. Chapman and Hall Medical; London: 1996
|
|
22
|
Xiong X, Liu D, Wang Y, Zeng T and Peng Y:
Urinary 3-(3-hydroxyphenyl)-3-hydroxypropionic acid,
3-hydroxyphenylacetic acid, and 3-hydroxyhippuric acid are elevated
in children with autism spectrum disorders. BioMed Res Int.
2016:1–8. 2016. View Article : Google Scholar
|
|
23
|
Trujillano D, Perez B, González J,
Tornador C, Navarrete R, Escaramis E, Ossowski S, Armengol L,
Cornejo V, Desviat LR, et al: Accurate molecular diagnosis of
phenylketonuria and tetrahydrobiopterin-deficient
hyperphenylalaninemias using high-throughput targeted sequencing.
Eur J Hum Genet. 22:528–534. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Setoodeh A, Yarali B, Rabbani A, Khatami S
and Shams S: Tetrahydrobiopterin responsiveness in a series of 53
cases of phenylketonuria and hyperphenylalaninemia in Iran. Mol
Genet Metab Rep. 2:77–79. 2015. View Article : Google Scholar
|
|
25
|
Okano Y, Kudo S, Nishi Y, Sakaguchi T and
Aso K: Molecular characterization of phenylketonuria and
tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency
in Japan. J Hum Genet. 56:306–312. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yaplito-Lee J, Chow CW and Boneh A:
Histopathological findings in livers of patients with urea cycle
disorders. Mol Genet Metab. 108:161–165. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nagasaka H, Yorifuji T, Egawa H, Inui A,
Fujisawa T, Komatsu H, Tsukahara H, Uemoto S and Inomata Y:
Characteristics of NO cycle coupling with urea cycle in
non-hyperammonemic carriers of ornithine transcarbamylase
deficiency. Mol Genet Metab. 109:251–254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mukhtar A, Dabbous H, El Sayed R,
Aboulfetouh F, Bahaa M, Abdelaal A, Fathy M and El-Meteini M: A
novel mutation of the ornithine transcarbamylase gene leading to
fatal hyperammonemia in a liver transplant recipient. Am J
Transplant. 13:1084–1087. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Paine SM, Grünewald S and Jacques TS:
Antenatal neurodevelopmental defects in ornithine transcarbamylase
deficiency. Neuropathol Appl Neurobiol. 38:509–512. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mhanni AA, Prasad C and Rockman-Greenberg
C: Ornithine transcarbamylase deficiency presenting as recurrent
abdominal pain in childhood. Pediatr Emerg Care. 27:850–853. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Swarts L, Leisegang F, Owen EP and
Henderson HE: An OTC deficiency ‘phenocopy’ in association with
Klinefelter syndrome. J Inherit Metab Dis. 30:1012007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang LL, Morizono H, Lin JP, Bell P, Jones
D, McMenamin D, Yu HW, Batshaw ML and Wilson JM: Preclinical
evaluation of a clinical candidate AAV8 vector for Ornithine
Transcarbamylase (OTC) deficiency reveals functional enzyme from
each persisting vector genome. Mol Genet Metab. 105:203–211. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vaidyanathan K: Molecular diagnosis of
urea cycle disorders: Current global scenario. Indian J Biochem
Biophys. 50:357–362. 2013.PubMed/NCBI
|
|
34
|
Kobayashi K, Sinasac DS, Iijima M, Boright
AP, Begum L, Lee JR, Yasuda T, Ikeda S, Hirano R, Terazono H, et
al: The gene mutated in adult-onset type II citrullinaemia encodes
a putative mitochondrial carrier protein. Nat Genet. 22:159–163.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Contreras L, Gomez-Puertas P, Iijima M,
Kobayashi K, Saheki T and Satrustegui J: Ca2+ Activation
kinetics of the two aspartate-glutamate mitochondrial carriers,
aralar and citrin: Role in the heart malate-aspartate NADH shuttle.
J Biol Chem. 282:7098–7106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Saheki T, Iijima M, Li MX, Kobayashi K,
Horiuchi M, Ushikai M, Okumura F, Meng XJ, Inoue I, Tajima A, et
al: Citrin/mitochondrial glycerol-3-phosphate dehydrogenase double
knock-out mice recapitulate features of human citrin deficiency. J
Biol Chem. 282:25041–25052. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Song YZ, Li BX, Chen FP, Liu SR, Sheng JS,
Ushikai M, Zhang CH, Zhang T, Wang ZN, Kobayashi K, et al: Neonatal
intrahepatic cholestasis caused by citrin deficiency: Clinical and
laboratory investigation of 13 subjects in mainland of China. Dig
Liver Dis. 41:683–689. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yamaguchi N, Kobayashi K, Yasuda T, Nishi
I, Iijima M, Nakagawa M, Osame M, Kondo I and Saheki T: Screening
of SLC25A13 mutations in early and late onset patients with citrin
deficiency and in the Japanese population: Identification of two
novel mutations and establishment of multiple DNA diagnosis methods
for nine mutations. Hum Mutat. 19:122–130. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang MH, Gong JY and Wang JS: Citrin
deficiency presenting as acute liver failure in an eight-month-old
infant. World J Gastroenterol. 21:7331–7334. 2015.PubMed/NCBI
|
|
40
|
Wang LY, Chen NI, Chen PW, Chiang SC, Hwu
WL, Lee NC and Chien YH: Newborn screening for citrin deficiency
and carnitine uptake defect using second-tier molecular tests. BMC
Med Genet. 14:1–6. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wert SE, Whitsett JA and Nogee LM: Genetic
disorders of surfactant dysfunction. Pediatr Dev Pathol.
12:253–274. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen BC, Rawi Mohd R, Meinsma R, Meijer J,
Hennekam RC and van Kuilenburg AB: Dihydropyrimidine dehydrogenase
deficiency in two malaysian siblings with abnormal MRI findings.
Mol Syndromol. 5:299–303. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chang HS, Shibata T, Arai S, Zhang CH,
Yabuki A, Mitani S, Higo T, Sunagawa K, Mizukami K and Yamato O:
Dihydropyrimidinase deficiency: The first feline case of
dihydropyrimidinuria with clinical and molecular findings. JIMD
Rep. 6:21–26. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Micheli V, Camici M, Tozzi MG, Ipata PL,
Sestini S, Bertelli M and Pompucci G: Neurological disorders of
purine and pyrimidine metabolism. Curr Top Med Chem. 11:923–947.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Porcu S, Corda M, Lilliu F, Contini L, Era
B, Traldi P and Fais A: Increase in urinary purines and pyrimidines
in patients with methylmalonic aciduria combined with
homocystinuria. Clin Chim Acta. 411:853–858. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nakajima Y, Meijer J, Dobritzsch D, Ito T,
Meinsma R, Abeling NGG, Roelofsen J, Zoetekouw L, Watanabe Y,
Tashiro K, et al: Clinical, biochemical and molecular analysis of
13 Japanese patients with β-ureidopropionase deficiency
demonstrates high prevalence of the c.977G>A (p.R326Q) mutation.
J Inherit Metab Dis. 37:801–812. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jurecka A: Inborn errors of purine and
pyrimidine metabolism. J Inherit Metab Dis. 32:247–263. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Balasubramaniam S, Duley JA and
Christodoulou J: Inborn errors of pyrimidine metabolism: clinical
update and therapy. J Inherit Metab Dis. 37:687–698. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ruhoy IS and Saneto RP: The genetics of
Leigh syndrome and its implications for clinical practice and risk
management. Appl Clin Genet. 7:221–234. 2014.PubMed/NCBI
|
|
50
|
Honzik T, Tesarova M, Vinsova K, Hansikova
H, Magner M, Kratochvilova H, Zamecnik J, Zeman J and Jesina P:
Different laboratory and muscle biopsy findings in a family with an
m.8851T>C mutation in the mitochondrial MTATP6 gene. Mol Genet
Metab. 108:102–105. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hejzlarová K, Kaplanová V, Nůsková H,
Kovářová N, Ješina P, Drahota Z, Mráček T, Seneca S and Houštěk J:
Alteration of structure and function of ATP synthase and cytochrome
c oxidase by lack of Fo-a and Cox3 subunits caused by mitochondrial
DNA 9205delTA mutation. Biochem J. 466:601–611. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chol M, Lebon S, Bénit P, Chretien D, de
Lonlay P, Goldenberg A, Odent S, Hertz-Pannier L, Vincent-Delorme
C, Cormier-Daire V, et al: The mitochondrial DNA G13513A MELAS
mutation in the NADH dehydrogenase 5 gene is a frequent cause of
Leigh-like syndrome with isolated complex I deficiency. J Med
Genet. 40:188–191. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kara B, Arikan M, Maraş H, Abacı N,
Cakıris A and Ustek D: Whole mitochondrial genome analysis of a
family with NARP/MILS caused by m.8993T>C mutation in the
MT-ATP6 gene. Mol Genet Metab. 107:389–393. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Anglin RE, Garside SL, Tarnopolsky MA,
Mazurek MF and Rosebush PI: The psychiatric manifestations of
mitochondrial disorders: A case and review of the literature. J
Clin Psychiatry. 73:506–512. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hulgan T, Haubrich R, Riddler SA, Tebas P,
Ritchie MD, McComsey GA, Haas DW and Canter JA: European
mitochondrial DNA haplogroups and metabolic changes during
antiretroviral therapy in AIDS Clinical Trials Group Study A5142.
AIDS. 25:37–47. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Xiao MT, Yang H, Xu W, Ma SH, Lin HP, Zhu
HG, Liu LX, Liu Y, Yang C, Xu YH, et al: Inhibition of
α-KG-dependent histone and DNA demethylases by fumarate and
succinate that are accumulated in mutations of FH and SDH tumor
suppressors. Genes Dev. 29:1326–1338. 2012. View Article : Google Scholar
|
|
57
|
MacKenzie ED, Selak MA, Tennant DA, Payne
LJ, Crosby S, Frederiksen CM, Watson DG and Gottlieb E:
Cell-permeating α-ketoglutarate derivatives alleviate pseudohypoxia
in succinate dehydrogenase-deficient cells. Mol Cell Biol.
27:3282–3289. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Leonardi R, Subramanian C, Jackowski S and
Rock CO: Cancer-associated isocitrate dehydrogenase mutations
inactivate NADPH-dependent reductive carboxylation. J Biol Chem.
287:14615–14620. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Baughn AD, Garforth SJ, Vilchèze C and
Jacobs WR Jr: An anaerobic-type alpha-ketoglutarate ferredoxin
oxidoreductase completes the oxidative tricarboxylic acid cycle of
Mycobacterium tuberculosis. PLoS Pathog. 5:e10006622009.
View Article : Google Scholar : PubMed/NCBI
|