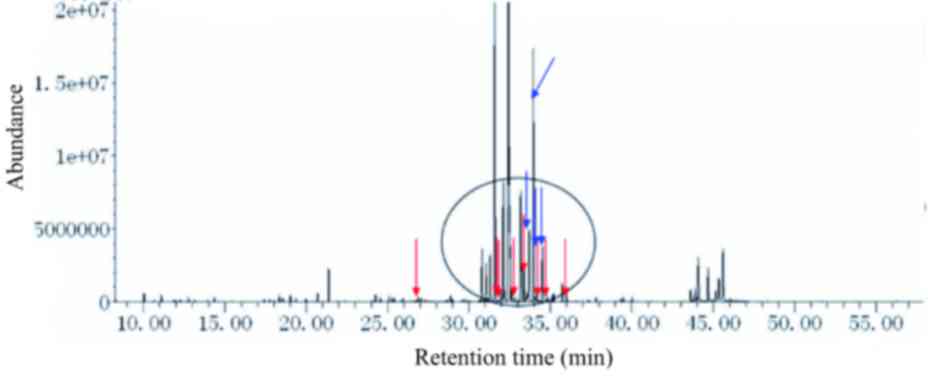
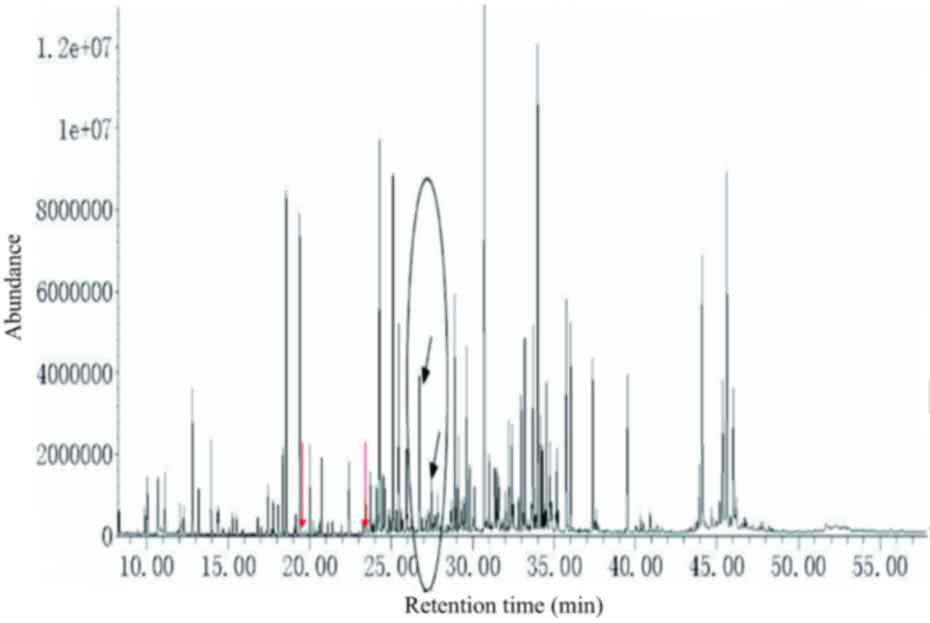
|
1
|
Pampols T: Inherited metabolic rare
disease. Adv Exp Med Biol. 686:397–431. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pérez B, Rodriguez-Pascau L, Vilageliu L,
Grinberg D, Ugarte M and Desviat LR: Present and future of
antisense therapy for splicing modulation in inherited metabolic
disease. J Inherit Metab Dis. 33:397–403. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rodrigues JV, Henriques BJ, Lucas TG and
Gomes CM: Cofactors and metabolites as protein folding helpers in
metabolic diseases. Curr Top Med Chem. 12:2546–2559. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cassiman D: Gene transfer for inborn
errors of metabolism of the liver: The clinical perspective. Curr
Pharm Des. 17:2550–2557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Blau N, Duran M, Gibson KM and
Dionisi-Vici C: Physician's Guide to the Diagnosis, Treatment, and
Follow-Up of Inherited Metabolic Diseases. Springer; Heidelberg:
2014, View Article : Google Scholar
|
|
6
|
Song SM, Yoon HR, Lee A and Lee KR:
Seven-year experience with inherited metabolic disorders screening
by tandem mass spectrometry. J Genet Med. 5:21–25. 2008.
|
|
7
|
Janeckova H, Kalivodova A, Najdekr L,
Friedecky D, Hron K, Bruheim P and Adam T: Untargeted metabolomic
analysis of urine samples in the diagnosis of some inherited
metabolic disorders. Biomed Pap Med Fac Univ Palacky Olomouc Czech
Repub. 159:582–585. 2015.PubMed/NCBI
|
|
8
|
Xiong X, Sheng X, Liu D, Zeng T, Peng Y
and Wang Y: A GC/MS-based metabolomic approach for reliable
diagnosis of phenylketonuria. Anal Bioanal Chem. 407:8825–8833.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kuhara T: Diagnosis of inborn errors of
metabolism using filter paper urine, urease treatment, isotope
dilution and gas chromatography-mass spectrometry. J Chromatogr B
Biomed Sci Appl. 758:3–25. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kuhara T: Diagnosis and monitoring of
inborn errors of metabolism using urease-pretreatment of urine,
isotope dilution, and gas chromatography-mass spectrometry. J
Chromatogr B Analyt Technol Biomed Life Sci. 781:497–517. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kuhara T, Ohse M, Inoue Y, Yorifuji T,
Sakura N, Mitsubuchi H, Endo F and Ishimatu J: Gas
chromatographic-mass spectrometric newborn screening for propionic
acidaemia by targeting methylcitrate in dried filter-paper urine
samples. J Inherit Metab Dis. 25:98–106. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fryčák P, Lemr K, Adam T and Hušková R:
Diagnostics of some inherited metabolic disorders by mass
spectrometry using modern ionisation techniques. Chemicke Listy.
97:93–100. 2003.
|
|
13
|
Bruheim P, Kvitvang HF and Villas-Boas SG:
Stable isotope coded derivatizing reagents as internal standards in
metabolite profiling. J Chromatogr A. 1296:196–203. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bouatra S, Aziat F, Mandal R, Guo AC,
Wilson MR, Knox C, Bjorndahl TC, Krishnamurthy R, Saleem F, Liu P,
et al: The human urine metabolome. PLoS One. 8:e730762013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Janečková H, Hron K, Wojtowicz P, Hlídková
E, Barešová A, Friedecký D, Zídková L, Hornik P, Behúlová D,
Procházková D, et al: Targeted metabolomic analysis of plasma
samples for the diagnosis of inherited metabolic disorders. J
Chromatogr A. 1226:11–17. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qun G: Evaluation of automated sample
preparation, retention time locked GC-MS and automated data
analysis for the metabolomic study of Arabidopsis species. J
Chromatogr A. 1218:3247–3254. 2010.
|
|
17
|
Scher HI, Nasso SF, Rubin EH and Simon R:
Adaptive clinical trial designs for simultaneous testing of matched
diagnostics and therapeutics. Clin Cancer Res. 17:6634–6640. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rosser CJ, Dai Y, Miyake M, Zhang G and
Goodison S: Simultaneous multi-analyte urinary protein assay for
bladder cancer detection. BMC Biotechnol. 14:81–89. 2013.
|
|
19
|
Wang YC: A two-step derivatization on
simultaneous analysis for organic acids, fatty acids, amino acid,
carbohydrates, pyrimidines, and purines in complicated organic
compounds. CN patent ZL201210114246.2. Filed April 18, 2012; issued
December 18, 2013.
|
|
20
|
Xiong X, Liu D, Wang Y, Zeng T and Peng Y:
Urinary 3-(3-hydroxyphenyl)-3-hydroxypropionic acid,
3-hydroxyphenylacetic acid, and 3-hydroxyhippuric acid are elevated
in children with autism spectrum disorders. Biomed Research
International. 2016:94854122016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Blau N, Duran M, Blaskovics ME and Gibson
MK: Physician's Guide to the Laboratory Diagnosis of Metabolic
Diseases. 2nd. Chapman and Hall Medical; London: 1996
|
|
22
|
Xiong X, Liu D, Wang Y, Zeng T and Peng Y:
Urinary 3-(3-hydroxyphenyl)-3-hydroxypropionic acid,
3-hydroxyphenylacetic acid, and 3-hydroxyhippuric acid are elevated
in children with autism spectrum disorders. BioMed Res Int.
2016:1–8. 2016. View Article : Google Scholar
|
|
23
|
Trujillano D, Perez B, González J,
Tornador C, Navarrete R, Escaramis E, Ossowski S, Armengol L,
Cornejo V, Desviat LR, et al: Accurate molecular diagnosis of
phenylketonuria and tetrahydrobiopterin-deficient
hyperphenylalaninemias using high-throughput targeted sequencing.
Eur J Hum Genet. 22:528–534. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Setoodeh A, Yarali B, Rabbani A, Khatami S
and Shams S: Tetrahydrobiopterin responsiveness in a series of 53
cases of phenylketonuria and hyperphenylalaninemia in Iran. Mol
Genet Metab Rep. 2:77–79. 2015. View Article : Google Scholar
|
|
25
|
Okano Y, Kudo S, Nishi Y, Sakaguchi T and
Aso K: Molecular characterization of phenylketonuria and
tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency
in Japan. J Hum Genet. 56:306–312. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yaplito-Lee J, Chow CW and Boneh A:
Histopathological findings in livers of patients with urea cycle
disorders. Mol Genet Metab. 108:161–165. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nagasaka H, Yorifuji T, Egawa H, Inui A,
Fujisawa T, Komatsu H, Tsukahara H, Uemoto S and Inomata Y:
Characteristics of NO cycle coupling with urea cycle in
non-hyperammonemic carriers of ornithine transcarbamylase
deficiency. Mol Genet Metab. 109:251–254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mukhtar A, Dabbous H, El Sayed R,
Aboulfetouh F, Bahaa M, Abdelaal A, Fathy M and El-Meteini M: A
novel mutation of the ornithine transcarbamylase gene leading to
fatal hyperammonemia in a liver transplant recipient. Am J
Transplant. 13:1084–1087. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Paine SM, Grünewald S and Jacques TS:
Antenatal neurodevelopmental defects in ornithine transcarbamylase
deficiency. Neuropathol Appl Neurobiol. 38:509–512. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mhanni AA, Prasad C and Rockman-Greenberg
C: Ornithine transcarbamylase deficiency presenting as recurrent
abdominal pain in childhood. Pediatr Emerg Care. 27:850–853. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Swarts L, Leisegang F, Owen EP and
Henderson HE: An OTC deficiency ‘phenocopy’ in association with
Klinefelter syndrome. J Inherit Metab Dis. 30:1012007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang LL, Morizono H, Lin JP, Bell P, Jones
D, McMenamin D, Yu HW, Batshaw ML and Wilson JM: Preclinical
evaluation of a clinical candidate AAV8 vector for Ornithine
Transcarbamylase (OTC) deficiency reveals functional enzyme from
each persisting vector genome. Mol Genet Metab. 105:203–211. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vaidyanathan K: Molecular diagnosis of
urea cycle disorders: Current global scenario. Indian J Biochem
Biophys. 50:357–362. 2013.PubMed/NCBI
|
|
34
|
Kobayashi K, Sinasac DS, Iijima M, Boright
AP, Begum L, Lee JR, Yasuda T, Ikeda S, Hirano R, Terazono H, et
al: The gene mutated in adult-onset type II citrullinaemia encodes
a putative mitochondrial carrier protein. Nat Genet. 22:159–163.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Contreras L, Gomez-Puertas P, Iijima M,
Kobayashi K, Saheki T and Satrustegui J: Ca2+ Activation
kinetics of the two aspartate-glutamate mitochondrial carriers,
aralar and citrin: Role in the heart malate-aspartate NADH shuttle.
J Biol Chem. 282:7098–7106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Saheki T, Iijima M, Li MX, Kobayashi K,
Horiuchi M, Ushikai M, Okumura F, Meng XJ, Inoue I, Tajima A, et
al: Citrin/mitochondrial glycerol-3-phosphate dehydrogenase double
knock-out mice recapitulate features of human citrin deficiency. J
Biol Chem. 282:25041–25052. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Song YZ, Li BX, Chen FP, Liu SR, Sheng JS,
Ushikai M, Zhang CH, Zhang T, Wang ZN, Kobayashi K, et al: Neonatal
intrahepatic cholestasis caused by citrin deficiency: Clinical and
laboratory investigation of 13 subjects in mainland of China. Dig
Liver Dis. 41:683–689. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yamaguchi N, Kobayashi K, Yasuda T, Nishi
I, Iijima M, Nakagawa M, Osame M, Kondo I and Saheki T: Screening
of SLC25A13 mutations in early and late onset patients with citrin
deficiency and in the Japanese population: Identification of two
novel mutations and establishment of multiple DNA diagnosis methods
for nine mutations. Hum Mutat. 19:122–130. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang MH, Gong JY and Wang JS: Citrin
deficiency presenting as acute liver failure in an eight-month-old
infant. World J Gastroenterol. 21:7331–7334. 2015.PubMed/NCBI
|
|
40
|
Wang LY, Chen NI, Chen PW, Chiang SC, Hwu
WL, Lee NC and Chien YH: Newborn screening for citrin deficiency
and carnitine uptake defect using second-tier molecular tests. BMC
Med Genet. 14:1–6. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wert SE, Whitsett JA and Nogee LM: Genetic
disorders of surfactant dysfunction. Pediatr Dev Pathol.
12:253–274. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen BC, Rawi Mohd R, Meinsma R, Meijer J,
Hennekam RC and van Kuilenburg AB: Dihydropyrimidine dehydrogenase
deficiency in two malaysian siblings with abnormal MRI findings.
Mol Syndromol. 5:299–303. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chang HS, Shibata T, Arai S, Zhang CH,
Yabuki A, Mitani S, Higo T, Sunagawa K, Mizukami K and Yamato O:
Dihydropyrimidinase deficiency: The first feline case of
dihydropyrimidinuria with clinical and molecular findings. JIMD
Rep. 6:21–26. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Micheli V, Camici M, Tozzi MG, Ipata PL,
Sestini S, Bertelli M and Pompucci G: Neurological disorders of
purine and pyrimidine metabolism. Curr Top Med Chem. 11:923–947.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Porcu S, Corda M, Lilliu F, Contini L, Era
B, Traldi P and Fais A: Increase in urinary purines and pyrimidines
in patients with methylmalonic aciduria combined with
homocystinuria. Clin Chim Acta. 411:853–858. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nakajima Y, Meijer J, Dobritzsch D, Ito T,
Meinsma R, Abeling NGG, Roelofsen J, Zoetekouw L, Watanabe Y,
Tashiro K, et al: Clinical, biochemical and molecular analysis of
13 Japanese patients with β-ureidopropionase deficiency
demonstrates high prevalence of the c.977G>A (p.R326Q) mutation.
J Inherit Metab Dis. 37:801–812. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jurecka A: Inborn errors of purine and
pyrimidine metabolism. J Inherit Metab Dis. 32:247–263. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Balasubramaniam S, Duley JA and
Christodoulou J: Inborn errors of pyrimidine metabolism: clinical
update and therapy. J Inherit Metab Dis. 37:687–698. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ruhoy IS and Saneto RP: The genetics of
Leigh syndrome and its implications for clinical practice and risk
management. Appl Clin Genet. 7:221–234. 2014.PubMed/NCBI
|
|
50
|
Honzik T, Tesarova M, Vinsova K, Hansikova
H, Magner M, Kratochvilova H, Zamecnik J, Zeman J and Jesina P:
Different laboratory and muscle biopsy findings in a family with an
m.8851T>C mutation in the mitochondrial MTATP6 gene. Mol Genet
Metab. 108:102–105. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hejzlarová K, Kaplanová V, Nůsková H,
Kovářová N, Ješina P, Drahota Z, Mráček T, Seneca S and Houštěk J:
Alteration of structure and function of ATP synthase and cytochrome
c oxidase by lack of Fo-a and Cox3 subunits caused by mitochondrial
DNA 9205delTA mutation. Biochem J. 466:601–611. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chol M, Lebon S, Bénit P, Chretien D, de
Lonlay P, Goldenberg A, Odent S, Hertz-Pannier L, Vincent-Delorme
C, Cormier-Daire V, et al: The mitochondrial DNA G13513A MELAS
mutation in the NADH dehydrogenase 5 gene is a frequent cause of
Leigh-like syndrome with isolated complex I deficiency. J Med
Genet. 40:188–191. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kara B, Arikan M, Maraş H, Abacı N,
Cakıris A and Ustek D: Whole mitochondrial genome analysis of a
family with NARP/MILS caused by m.8993T>C mutation in the
MT-ATP6 gene. Mol Genet Metab. 107:389–393. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Anglin RE, Garside SL, Tarnopolsky MA,
Mazurek MF and Rosebush PI: The psychiatric manifestations of
mitochondrial disorders: A case and review of the literature. J
Clin Psychiatry. 73:506–512. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hulgan T, Haubrich R, Riddler SA, Tebas P,
Ritchie MD, McComsey GA, Haas DW and Canter JA: European
mitochondrial DNA haplogroups and metabolic changes during
antiretroviral therapy in AIDS Clinical Trials Group Study A5142.
AIDS. 25:37–47. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Xiao MT, Yang H, Xu W, Ma SH, Lin HP, Zhu
HG, Liu LX, Liu Y, Yang C, Xu YH, et al: Inhibition of
α-KG-dependent histone and DNA demethylases by fumarate and
succinate that are accumulated in mutations of FH and SDH tumor
suppressors. Genes Dev. 29:1326–1338. 2012. View Article : Google Scholar
|
|
57
|
MacKenzie ED, Selak MA, Tennant DA, Payne
LJ, Crosby S, Frederiksen CM, Watson DG and Gottlieb E:
Cell-permeating α-ketoglutarate derivatives alleviate pseudohypoxia
in succinate dehydrogenase-deficient cells. Mol Cell Biol.
27:3282–3289. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Leonardi R, Subramanian C, Jackowski S and
Rock CO: Cancer-associated isocitrate dehydrogenase mutations
inactivate NADPH-dependent reductive carboxylation. J Biol Chem.
287:14615–14620. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Baughn AD, Garforth SJ, Vilchèze C and
Jacobs WR Jr: An anaerobic-type alpha-ketoglutarate ferredoxin
oxidoreductase completes the oxidative tricarboxylic acid cycle of
Mycobacterium tuberculosis. PLoS Pathog. 5:e10006622009.
View Article : Google Scholar : PubMed/NCBI
|