Introduction
Chronic wounds, such as diabetic foot ulcers and
venous and arterial leg ulcers, pose a major health risk to
patients and place marked financial, resource and time burdens on
the healthcare system. In industrialised countries, ~1% of the
population experiences a non-healing wound, which accounts for a
significant part of the healthcare budget (1). In 1998, it was estimated that chronic
wounds would cost the National Health Service in the United Kingdom
(UK) a total of £1 billion per year (2). Between 2005 and 2006, there were
200,000 individuals in the UK with chronic wounds, accounting for
£2.3–3.1 billion per year, or 3% of the total estimated out-turn
expenditure on health for the same period (£89.4 billion) (2,3). The
cost may become much higher as the number of people suffering from
chronic wounds in the UK increases; for example, in 2011, this
figure was estimated to be >600,000 (4).
Advances in molecular biology have helped to
elucidate the complexities of wound biology. Menke et al
(5) describe the nature of wound
healing biology as ‘complex, multiscale, multitemporal and
hierarchical’. Past failures in wound diagnosis and monotherapies
may be due to the under appreciation of this complexity. The
signalling mechanisms of growth factors have been well studied
(6,7), and reduced levels in the wound
environment may be partially responsible for the failure of certain
wounds to heal. It has been demonstrated that chronic ulcers
exhibit reduced levels of platelet derived growth factor, epidermal
growth factor, basic fibroblast growth factor, and transforming
growth factor β compared with acute wounds, typically as a result
of trapping or degradation (6,7).
The imbalance between proteinases and their
inhibitors with excessive proteinase activity in chronic wounds,
which is potentially due to the overexpression of matrix
metalloproteinase, results in abnormal degradation of the
extracellular matrix (8–10). Dermal fibroblasts exhibit an
age-related decrease in proliferation potential, or cell
senescence, and it has previously been demonstrated that
fibroblasts isolated from chronic wounds have a decreased or
non-existent replicative ability (11). In addition, cell senescence, in the
form of epidermal arrest, has been demonstrated in keratinocytes
(12). Further studies have shown
that gene arrays of wound edges are able to guide specific cell
subpopulations, which aid in targeting therapy and debridement
(13,14). Charles et al (15) compared the gene expression patterns
of five non-healing venous leg ulcers with five healing venous leg
ulcers, and 15 genes were identified that were differentially
expressed in the keratinocytes at the non-healing wound edge. Among
these was the S100 calcium-binding protein A7 gene (Psoriasin),
which expresses protein products that have been proposed to be
associated with keratinocyte differentiation.
Psoriasin, which is also known as S100A7, belongs to
the S100 protein family of calcium-responsive signalling proteins.
It was originally discovered in unfractionated non-cultured
keratinocytes as a novel protein with a low molecular weight (11.4
kDa) (16,17). It was found to be overexpressed in
psoriatic keratinocytes and expressed at low levels in normal
proliferating keratinocytes and foetal skin (16). Although Psoriasin was initially
studied as a secreted protein in psoriatic skin (17), it was later detected in the cytoplasm
and nucleus of keratinocytes and breast epithelial cells (18), suggesting that this protein has
multiple functions. Psoriasin is homologous with the S100 genes as
it encodes small cytoplasmic and secreted proteins that share
EF-hand-helix-loop-helix domains, which are necessary for calcium
binding (19). A total of 13 of the
S100 proteins, including Psoriasin, are encoded within the
epidermal differentiation complex of human chromosome 1q21.2-q22
(18,20,21).
Many of these genes products, including Psoriasin, serve important
roles in exerting the effects of calcium on cell growth and
differentiation (22).
Although Psoriasin protein overexpression was
initially discovered in patients with psoriasis (16), it has subsequently been discovered to
be associated with other inflammatory skin conditions (23). Secreted Psoriasin has been
demonstrated to have a chemotactic influence on inflammatory cells,
suggesting a link with inflammatory skin diseases (24). The levels of Psoriasin in
pre-cancerous lesions of the breast and skin, as demonstrated in
multiple studies (25–28), indicate that Psoriasin expression is
low in normal epithelium and increased in pre-invasive carcinoma,
specifically act in keratosis and breast carcinoma in situ,
and that high Psoriasin levels are associated with unfavourable
histological features and a worse clinical outcome in patients with
breast cancer (29–33). This suggests that Psoriasin may serve
an important role in cancer progression.
Expression of Psoriasin in normal keratinocytes is
known to be upregulated in response to calcium and retinoic acid
stimuli, and in abnormal pathways of differentiation in culture
(18,25,34).
Among normal tissues, Psoriasin has restricted expression in the
epithelial component of tissues, such as skin, breast and bladder
(23,25,29,35).
Furthermore, expression is elevated in the differentiating layers
compared with the basal cells, where Psoriasin is absent, thereby
indicating that it may have a specific association with
differentiation (25,36). Expression of Psoriasin in the context
of wound healing was initially observed based on the regenerative
similarities among wounds and psoriatic keratinocyte (37,38). The
effects of Psoriasin in chronic venous ulcers were previously
studied by Dressel et al (39) via immunohistochemical (IHC) staining,
demonstrating a significant induction of Psoriasin among seven
chronic venous wound margin biopsies compared with normal skin
biopsies.
Psoriasin is important in many basic cellular
functions and keratinocytes are associated with the
re-epithelisation of wound edges. Psoriasin expression in the
epidermis and its association with the regulation of survival,
adhesion and motility of various types of cells, suggests that it
may have a function in aiding wound healing. The aim of the present
study was to investigate the specific role of Psoriasin in
keratinocyte cellular functions and its implication in chronic
wounds.
Materials and methods
Materials and cell line
A universal IHC kit (Elite ABC Kit) was purchased
from Vector Laboratories, Ltd. (Peterborough, UK). Total RNA
isolation reagent (TRIzol) was purchased from Sigma-Aldrich (Merck
Millipore, Darmstadt, Germany), and reverse transcription kits
(iScript) were obtained from Bio-Rad Laboratories, Inc. (Hercules,
CA, USA). Small inhibitors for neural Wiskott-Aldrich syndrome
protein (N-WASP), focal adhesion kinase (FAK) and rho-associated
protein kinase (ROCK) were purchased from Merck Millipore. The
HaCaT human keratinocyte cell line was purchased from The German
Cancer Research Center (Heidelberg, Germany). Cells were maintained
in Dubecco's modified Eagle medium (DMEM), which was supplemented
with penicillin, streptomycin and 10% fetal calf serum (PAA
Laboratories, Ltd.; GE Healthcare Life Sciences, Chalfont, UK).
Cells were incubated at 37°C and 95% humidity in an atmosphere
containing 5% CO2. Primer sequences are provided in
Table I.
 | Table I.Primer sequences used for PCR. |
Table I.
Primer sequences used for PCR.
| Primer | Forward | Reverse |
|---|
| Psoriasin |
5′-GAGGTCCATAATAGGCATGA-3′ |
5′-AGCAAGGACAGAAACTCAGA-3′ |
| Psoriasin
(qPCR) |
5′-TGTGACAAAAAGGGCACAAA-3′ |
5′-ACTGAACCTGACCGTACACCCAGCAAGGACAGAAACTC-3′ |
| GAPDH |
5′-ATGATATCGCCGCGCTCGTC-3′ |
5′-GCTCGGTCAGGATCTTCA-3′ |
| GAPDH (qPCR) |
5′-CTGAGTACGTCGTGGAGTC-3′ |
5′-ACTGAACCTGACCGTACAGAGATGATGACCCTTTTG-3′ |
| Psoriasin
ribozyme |
5′-CTGCAGTCACAGGCACTAAGG |
5′-ACTAGTGGCTGGTGTTTGAC |
|
|
AAGTTGGGCTGATGAGTCCGTGAGGA-3′ |
ATTTCGTCCTCACGGACT-3′ |
Chronic wound tissues and skin
biopsies
Skin biopsies were obtained from patients attending
the University Hospital of Wales (Cardiff, UK) wound healing clinic
as described previously (40,41).
Ethical approval was granted by the Local Research Ethics Committee
and written informed consent was obtained from each patient.
Biopsies were taken from 14 patients with chronic leg ulcers and
used during the present study. All wounds were present for ≥6
months, displaying no evidence of healing occurring six weeks prior
to biopsy and had a minimum area of 4 cm2 prior to
biopsy with no clinical indications of infection. Venous disease
was diagnosed by duplex ultrasonography using a Viamo system
(Toshiba Medical Systems, Ltd., Crawley, UK). Following the
administration of local anaesthetic (1% lidocaine; Hameln
Pharmaceuticals, Ltd., Gloucester, UK), 6 mm punch biopsies,
incorporating epidermis and dermis at the wound edge with adjacent
granulation tissue, were harvested from the wound margin under
aseptic conditions. Single wedge biopsies were obtained from 10
patients with acute surgical wounds following excision of pilonidal
disease. These wounds were determined to be clinically
non-infected. Biopsies were harvested from the edge of the healing
wound within six weeks of excision surgery. Normal, unwounded skin
was also obtained and examined as a control to provide a comparison
with wound tissue. Under local anaesthetic (1% lidocaine), 3 mm
punch biopsies were taken from the inner aspect of the upper arm of
10 healthy volunteers working within the Wound Healing Research
Unit at the University Hospital of Wales.
IHC staining
Frozen sections from wound tissues were fixed in an
acetone/methanol solution and rehydrated in wash buffer (Mena Path
Autowash buffer; A. Menarini Diagnostics, Ltd., Winnersh, UK) and
placed in a wash buffer solution containing 10% horse serum (Vector
Laboratories, Ltd.) to block non-specific antigen binding. An
avidin/biotin complex (ABC) IHC kit (PK-6200, Vector Laboratories,
Ltd.) was used. An anti-Psoriasin polyclonal antibody, was diluted
in a buffer that contained 1% horse serum and 0.1% Tween 20
(Sigma-Aldrich; Merck Millipore) at 1:40 dilution. Following 1 h of
incubation at room temperature with the primary antibodies, the
slides were washed four times in wash buffer and incubated at room
temperature with a universal biotinylated secondary antibody
provided in the IHC kit (1:500; Vector Laboratories, Ltd.) for 30
min. Avidin and biotin were added through the addition of the ABC
complex following washing. A 3,3′-diaminobenzidinecolour developing
system was used to indirectly detect protein staining. A series of
graded alcohols were used to dehydrate sections, which were
subsequently cleared in xylene, mounted and examined using a light
microscope equipped with a digital camera (Olympus Corporation,
Tokyo, Japan). Semi-quantification was performed using ImageJ
software (version 1.5; National Institutes of Health, Bethesda, MD,
USA), with normalisation of data to the background.
Psoriasin knockdown in HaCaT
cells
Anti-Psoriasin hammerhead ribozymes were designed
based on the secondary structure of Psoriasin mRNA. The ribozymes
were synthesised using a touchdown PCR procedure and subsequently
cloned into a mammalian expression vector (pEF6/His TOPO vector;
Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
constructed anti-Psoriasin transgenes and empty vectors were
transfected into HaCaT cells according to a previously reported
procedure (42). Following a period
of blasticidin selection (5 µg/ml; Sigma-Aldrich; Merck Millipore),
the cells were maintained in DMEM containing 0.5 µg/ml blasticidin.
The selected transfectants were verified for the knockdown of
Psoriasin (HaCaTPSOkd). HaCaTPSOkd, together
with the control cells transfected with empty vectors
(HaCaTpEF) and wild type cells (HaCaTWT),
were used in the following experiments. Three independent
transfections were performed to verify the knockdown of Psoriasin
using the anti-Psoriasin ribozymes.
RNA extraction, reverse transcription
(RT), conventional polymerase chain reaction (PCR) and quantitative
(q)PCR)
RNA isolation from cells or tissues was performed
using an ABgene Total RNA Isolation Reagent (TRIR) Kit (ABgene;
Thermo Fisher Scientific, Inc.) in accordance with the
manufacturer's protocol. Briefly, cells were cultured in 25
cm2 flasks until 85–90% confluent. The growth medium was
subsequently removed and 1 ml of TRIR reagent was added to the
monolayer to lyse the cells. Following RNA isolation, RNA was
quantified using a UV1101 photometer (WPA Biochrom; Biochrom, Ltd.,
Cambridge, UK) at 260 nm. cDNA was synthesised using an iScript
cDNA synthesis kit (Bio-Rad Laboratories) for a standardised 0.5 µg
RNA in a 20 µl-reaction. Conventional PCR was subsequently used to
verify Psoriasin expression in transfected cells using REDTaq
ReadyMix PCR reaction mixture comprising 20 mM Tris-HCl (pH 8.3)
with 100 mM KCl, 3 mM MgCl2, 0.002% gelatin, 0.4 mM dNTP
mix (dATP, dCTP, dGTP, TTP), stabilizers, and 0.06 U/ml Taq DNA
Polymerase (R2523; Sigma-Aldrich; Merck Millipore) and a T-Cy
thermocycler (Creacon Technologies, B.V., Emmen, the Netherlands).
Conditions for conventional PCR to amplify transcripts of Psoriasin
were: 36 cycles at 94°C for 30 sec, 55°C for 20 sec, 72°C for 30
sec and a final extension phase of 7 min at 72°C. GAPDH was used as
a housekeeping gene. PCR products were separated on a 1.5% agarose
gel and stained with ethidium bromide prior to examination under UV
light.
qPCR was used to determine Psoriasin
transcripts in wound tissues following a previously reported method
(43)
An iCycler IQ system (Bio-Rad Laboratories, Inc.)
was used to determine Psorisain transcript levels. Each reaction
contained 5 µl of the 2x concentrated HotstarTaq-master mix
(ABgene; Thermo Fisher Scientific, Inc.), 1 µl of forward primer
(10 pmol/µl), 1 µl reverse primer (1 pmol/µl), 1 µl of a FAM-tagged
universal probe (10 pmol/µl; Intergen Co., Purchase, NY, USA), and
cDNA samples. Conditions for qPCR were as follows: An initial 10
min 95°C denature followed by 80 cycles of 95°C for 15 sec, 55°C
for 35 sec and 72°C for 20 sec. Psoriasin transcript levels were
normalised against corresponding GAPDH quantity. Primer sequences
are provided in Table I.
SDS-PAGE and western blot
analysis
Cellular protein was extracted and lysed in
Ca2+ and Mg2+ free HEPES buffer containing
0.5% SDS, 1% Triton X-100, 2 mM CaCl2, 100 µg/ml
phenylmethylsulfonyl fluoride, 1 mg/ml leupeptin, 1 mg/ml aprotinin
and, 10 mM sodium orthovanadate (Sigma-Aldrich; Merck Millipore) on
a rotor wheel for 1 h. Insoluble proteins were removed via
centrifugation at 13,000 × g for 4°C for 15 min and quantified
using a DC Protein Assay kit (Bio-Rad Laboratories, Inc.). Samples
were standardised and diluted in Laemmli 2x concentrate sample
buffer (Sigma-Aldrich; Merck Millipore) and boiled for 5 min.
Following this, 20 µg total protein of each sample were separated
by 12% SDS-PAGE. The separated proteins were transferred to a
nitrocellulose membrane via western blotting. Gels were removed
from the electrophoretic tank and unclipped from the loading
cassette, and the stacking gel was removed. The resulting gel was
then carefully laid on top of a pre-cut Hybond nitrocellulose
membrane (GE Healthcare Life Sciences) in an SD20 Maxi System
blotting unit (SemiDRY; Wolf Laboratories, Ltd., York, UK). Blots
were probed with anti-Psoriasin (1:250; ab13680; Abcam, Cambridge,
UK) and anti-GAPDH (1:500, SC-47724, Insight Biotechnology, Ltd.,
Wembley, UK) mouse monoclonal antibodies following an overnight
incubation with a blocking buffer which was 10% skimmed milk in
tris-buffered saline (TBS) at 4°C. Peroxidase-conjugated anti-mouse
antibody (1:1,000; A9044; Sigma-Aldrich; Merck Millipore) was then
added prior to visualising protein bands using the Supersignal West
Dura system (Thermo Fisher Scientific, Inc.) and a gel imaging
system (UVItec, Ltd., Cambridge, UK). The experiment was repeated
three times at least for each subline.
In vitro growth assay
An in vitro growth assay was used to
determine HaCaT cell growth. A total of 3,000 cells per well were
seeded into 96 well plates. Triplicate plates were set up and
incubated at 37°C for three and five-day periods prior to analyses.
Following incubation, the plates were fixed in 4% formaldehyde
(v/v), stained with 0.5% (w/v) crystal violet and treated with 10%
acetic acid (v/v). Absorbance at 540 nm was determined using an
ELx800 multi-plate reader (BioTek Instruments Inc., Winnoski, VT,
USA).
Adhesion and migration tests using
electric cell-substrate impedance sensing (ECIS) analysis
The ECIS 9600 system (Applied Biophysics Inc., Troy,
NY, USA) was used to monitorthe adhesion and migration of HaCaT
cells, as previously described (44,45).
Briefly, HaCaT cells were seeded onto ECIS 96W1E arrays and
adhesion of cells to the culture surface and electrodes was
monitored via measuring electrical resistance. Once a confluent
monolayer had been formed, the cells were damaged by applying
electric current (1,400 µA, 60 kHz) for 20 sec to create a break in
the cell monolayer. The rate of change in impedance as cells
migrated back onto the electrode was subsequently monitored and
analysed.
Statistical analysis
The Minitab 14 statistical package (Minitab, Inc.,
State College, PA, USA) was used to identify statistically
significant differences between the test groups using a two-sample,
two-tailed, Student's t-test. In vitro functional assays
were repeated a minimum of three times. P<0.05 was considered to
indicate a statistically significant difference.
Results
Expression of Psoriasin in chronic
wounds
Frozen sections of chronic wound tissue samples were
stained to detect Psoriasin, as described previously, and tissue
sections from the wound edge and distal edge were compared. IHC
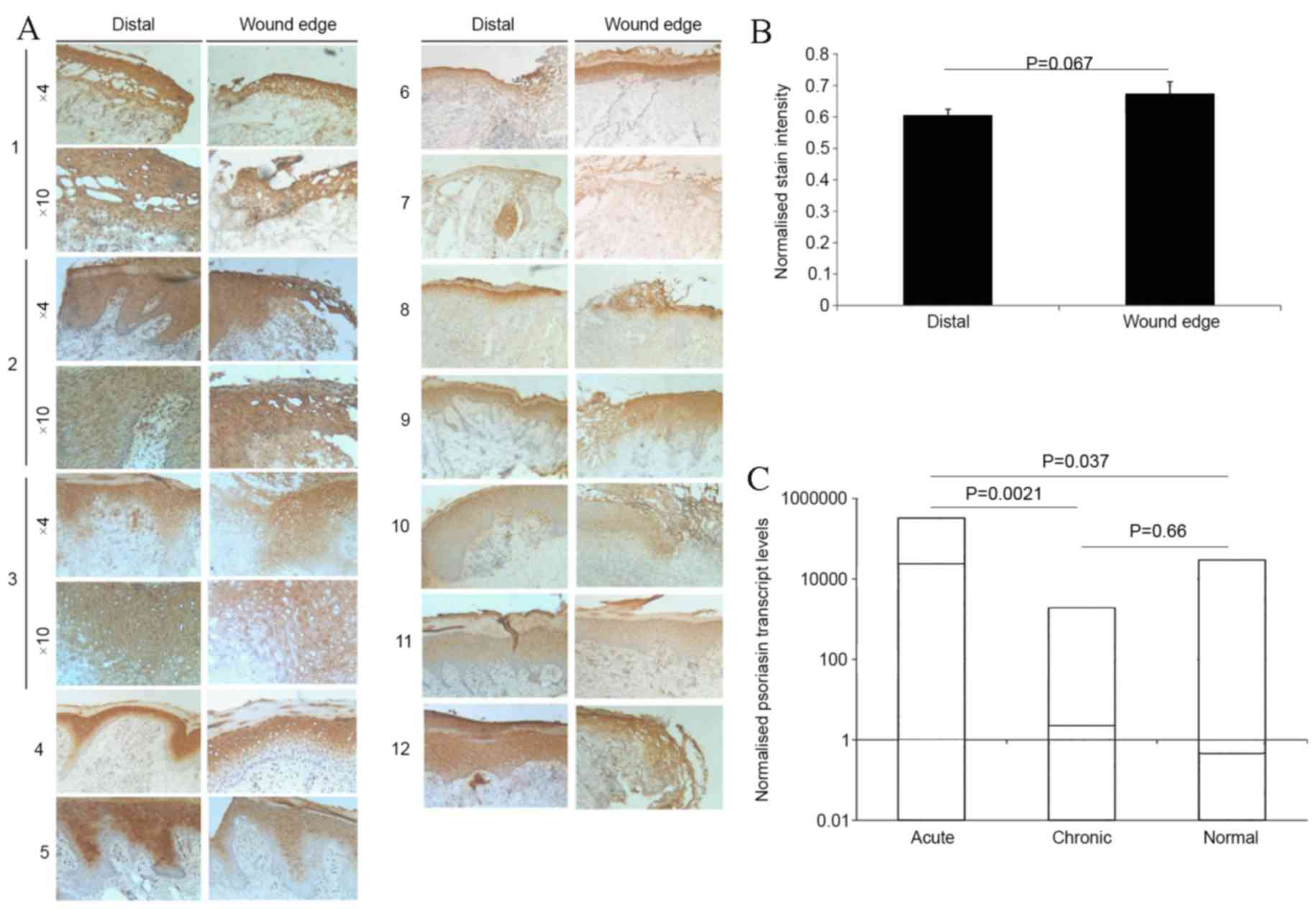
stained images from the 12 wound samples are presented in Fig. 1A. Staining of Psoriasin revealed
elevated protein levels throughout all epidermal layers in wounds,
with the exception of the stratum basale. Furthermore,
keratinocytes also demonstrated an increase in Psoriasin staining,
in accordance with the secretory nature of the protein. Initial
comparisons between samples revealed a less intense immunostaining
of Psoriasin at the distal edges of the wounds compared with the
wound edges. Semi-quantification using ImageJ software revealed an
increase ≤19.8% in the intensity of Psoriasin staining within the
epidermal layers at the wound edge, in comparison with the
epidermal staining at the distal wound (Fig. 1B). This indicates an upregulation of
Psoriasin in the wound edges. Psoriasin mRNA transcript in tissues
taken from the edge of acute and chronic wounds and normal skin was
analysed (Fig. 1C). In acute wounds,
there was a significant increase in Psoriasin mRNA expression
(P=0.0021) compared with the chronic wound phenotype. A significant
increase was also observed in Psoriasin expression between the
acute wounds and the normal skin controls (P=0.037). Furthermore,
Psoriasin expression was markedly lower in chronic wounds compared
with the normal controls.
Knockdown of Psoriasin in HaCaT
cells
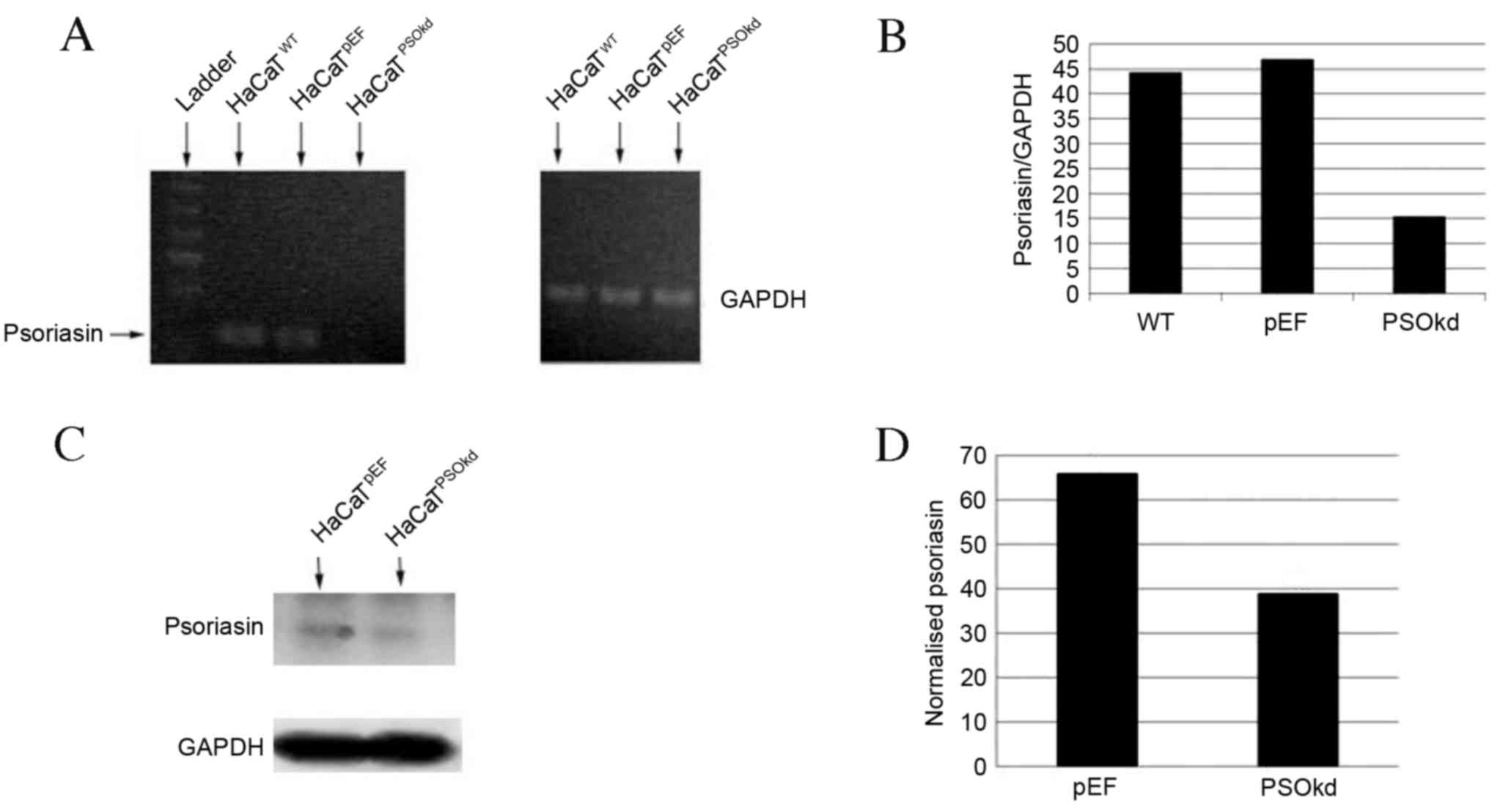
The presence of Psoriasin in HaCaT keratinocytes was
confirmed using RT-qPCR. To examine the functional role of
Psoriasin in keratinocytes, HaCaT cells were transfected with
anti-Psoriasin ribozyme transgenes. HaCaT Psoriasin knockdown in
the HaCaTPSOkd cells was seen and its mRNA expression
compared with the HaCaTWT and HaCaTpEF
control cells using RT-qPCR (Fig. 2A and
B). Resulting protein levels of Psoriasin in
HaCaTpEF and HaCaTPSOkd cells were evaluated
using western blot analysis. As illustrated in Fig. 2C, Psoriasin protein was detected in
the HaCaTpEF control cells with a corresponding
reduction of Psoriasin protein levels in the HaCaTPSOkd
cells. Semi-quantifications showed a 41% reduction of Psoriasin
protein expression in HaCaTPSOkd cells compared with the
control cells (Fig. 2D).
Influence of Psoriasin knockdown on in
vitro growth, adhesion and migration of HaCaT cells
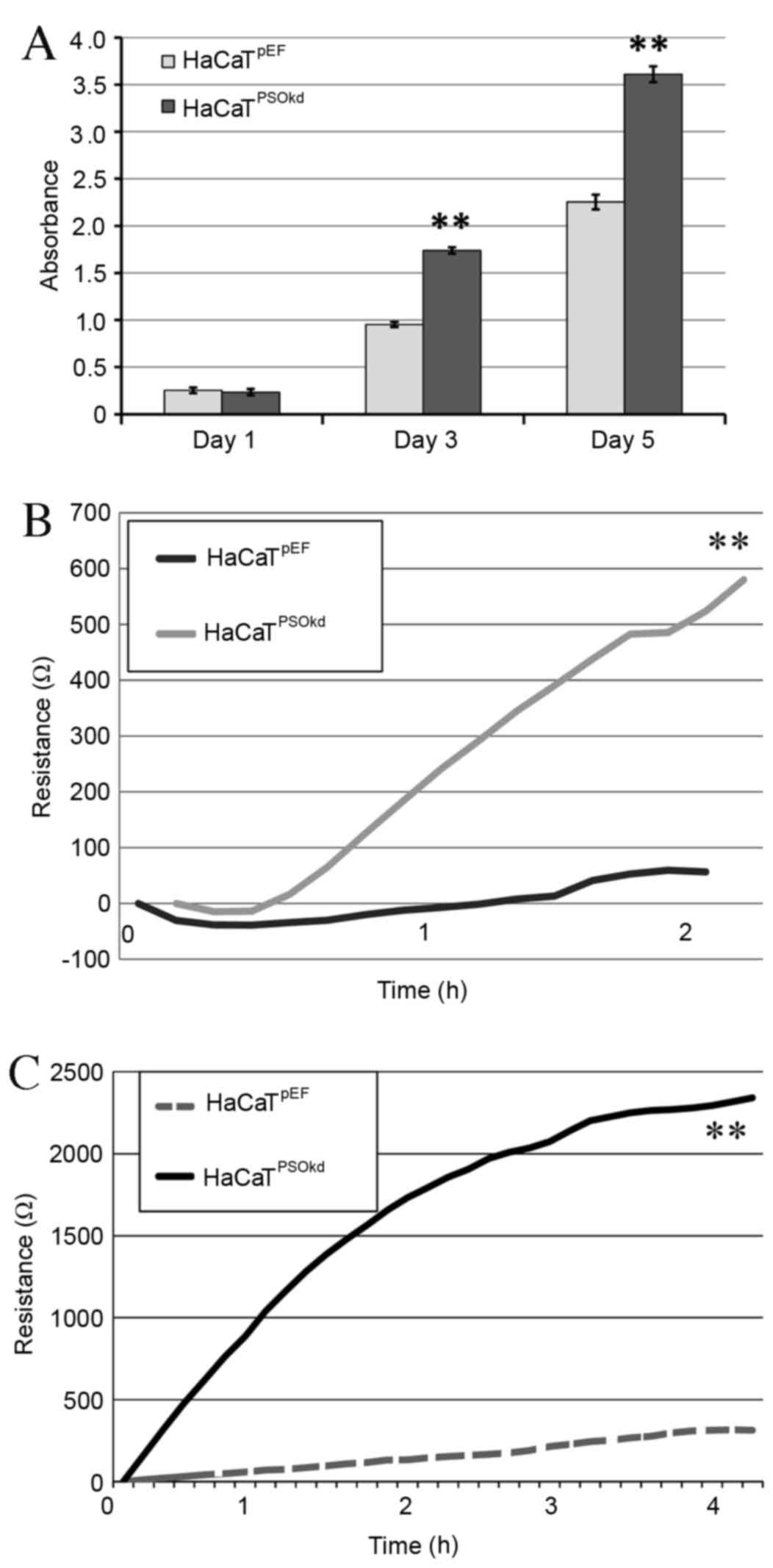
Using the colorimetric growth assay method described
previously, the effects of Psoriasin knockdown on HaCaT cells were
evaluated and compared with the HaCaTpEF controls.
Psoriasin knockdown significantly enhanced the growth rate of
HaCaTPSOkd cells at three and five days compared with
control HaCaTpEF cells (both P<0.01; Fig. 3A).
ECIS assay was performed to evaluate cell adhesion
and migration. Cell adhesion was calculated by assessing the
increase in the mean resistance at 2 h following cell seeding, and
increased adhesion was demonstrated in the Psoriasin knockdown
cells compared with the control cells (Fig. 3B). At 4–5 h, when adhesion was
complete, as determined by the plateauing of resistance readings,
the confluent monolayer of cells was damaged by passing a high
voltage current through it via an electrode. Migration was then
calculated from the subsequent mean resistances recorded at 0, 1,
2, 3 and 4 h (Fig. 3C). It was shown
that the migration of HaCaTPSOkd cells increased
significantly (P<0.01) when compared with HaCaTpEF
cells.
Association of N-WASP, ROCK and FAK
with Psoriasin-regulated cell adhesion and migration
To elucidate the underlying mechanisms that alter
the adhesion and migration of HaCaT cells following knockdown of
Psoriasin, small molecule inhibitors were used to evaluate the
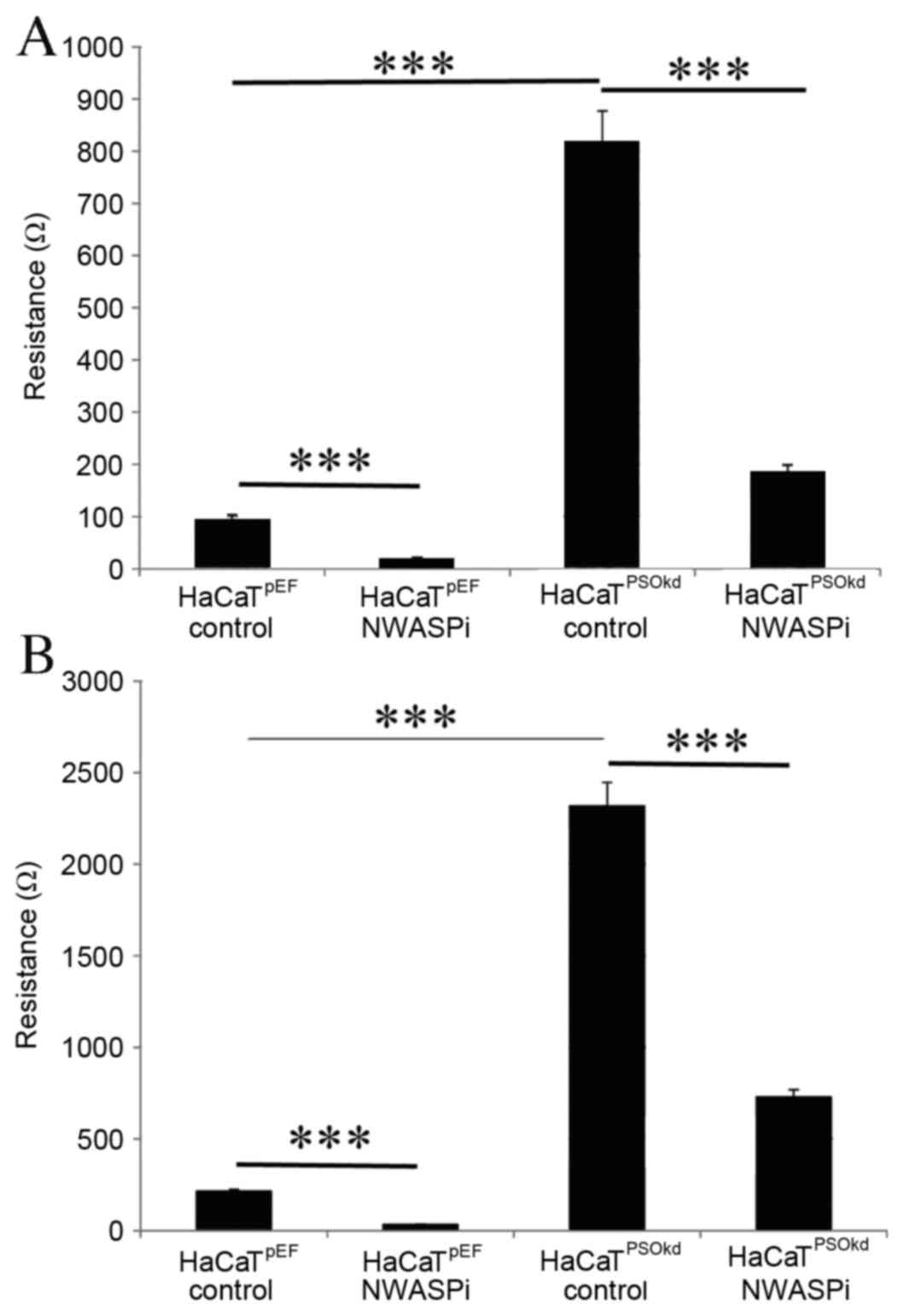
roles of some associated molecules. N-WASP inhibitor wiskostatin
was used at a concentration of 200 nM. In the presence of the
N-WASP inhibitor, HaCaTPSOkd demonstrated a significant
reduction in adhesion rate (P<0.001; Fig. 4A). The relative percentage of
inhibition compared with the corresponding untreated cell line was
79% for HaCaTpEF and 77.4% for HaCaTPSOkd,
which is significant. N-WASP inhibition induced a reduction in the
rate of keratinocyte migration across a wound similar to the effect
on adhesion (Fig. 4B). The effect of
inhibition in both cell lines was significant (P<0.001), and
comparing the relative percentile of reduction
(HaCaTpEF, 85%; HaCaTPSOkd, 68.6%), it
appears that N-WASP inhibition is less effective at reducing cell
migration in the Psoriasin knockdown cells.
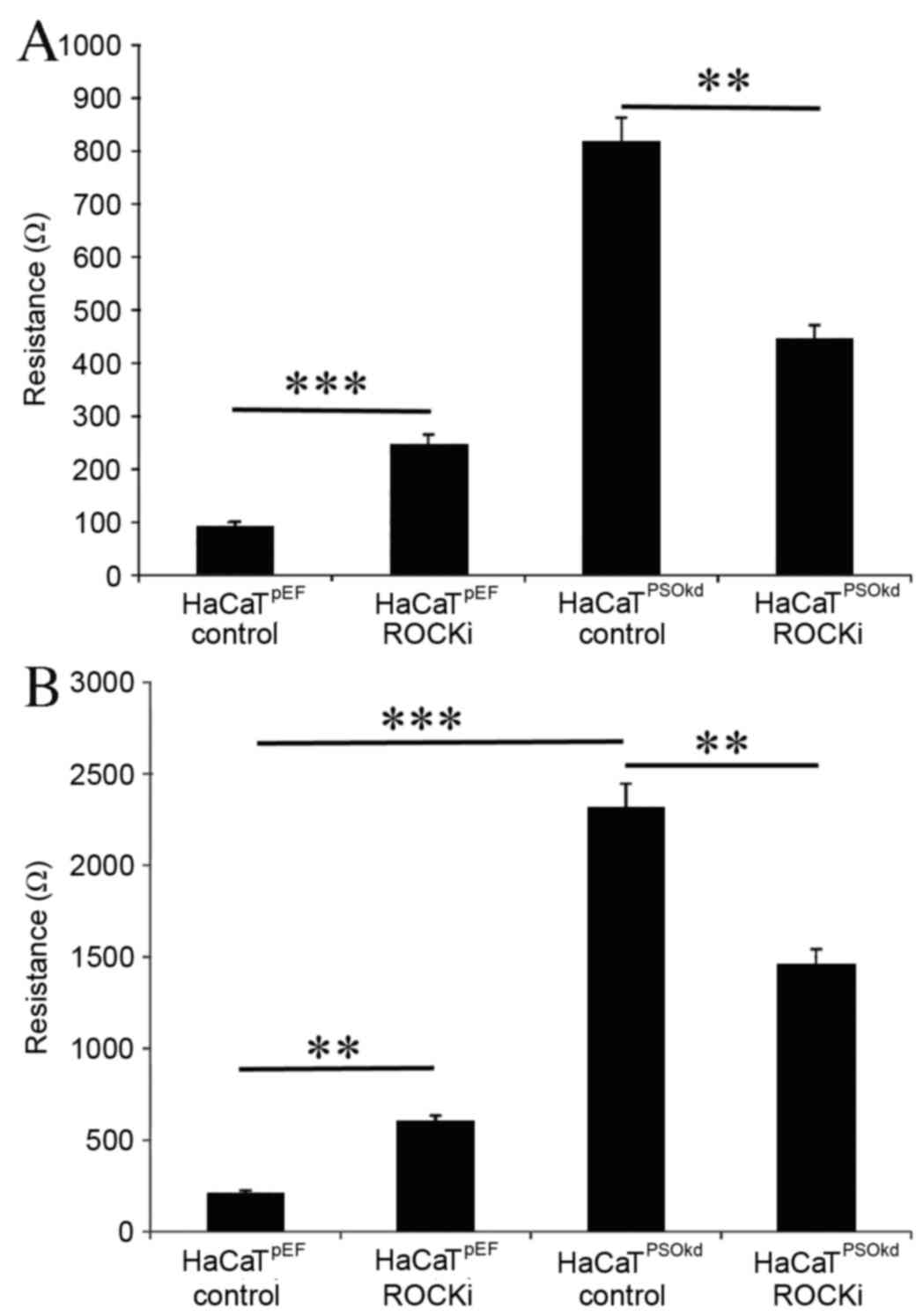
ROCK inhibitor significantly increased (P<0.001)
the rate of cell adhesion in the HaCaTpEF cell line by a
factor of 163%, as demonstrated in Fig.
5A. However, in the HaCaTPSOkd cells ROCK inhibitor
induced a significant reduction (P<0.01) in the rate of cell
adhesion, with a percentile difference of 45%. The HaCaT cell
adhesion rate in the HaCaTPSOkd group remained higher
compared with that of the HaCaTpEF control group. The
pattern of influence on migration was similar to that seen for
adhesion (Fig. 5B).
HaCaTpEF cells continued to demonstrate a significant
increase (P<0.01) in cell migration in the presence of ROCK
inhibitor, whereas the HaCaTPSOkd cells exhibited a
reduction in cell migration in the presence of ROCK inhibitor
(P<0.01).
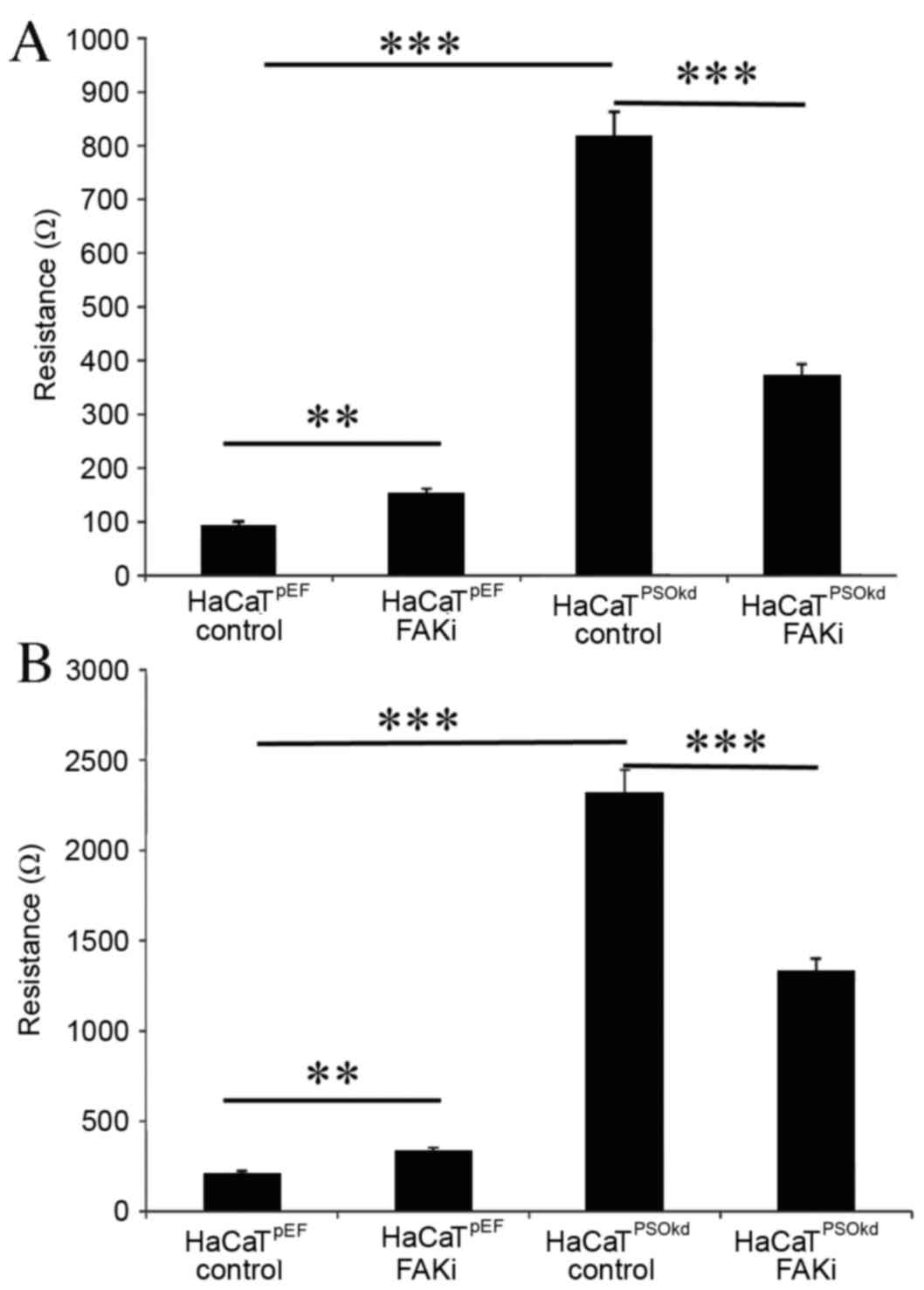
HaCaTpEF cell adhesion was increased with
FAK inhibition, as illustrated in Fig.
6A. However, FAK inhibition induced a reduction in cell
adhesion rate in HaCaTPSOkd cells. Both changes were
significant (P<0.001) compared with untreated equivalents,
ranging from ≤38% in the HaCaTpEF cell group to ≤54% in
the HaCaTPSOkd group. A similar pattern to that of
adhesion was observed in cell migration (Fig. 6B) and HaCaTpEF cell
migration was significantly increased by FAK inhibition
(P<0.001).
Discussion
The expression of the S100 family calcium responsive
signalling protein Psoriasin was studied in normal and chronic
wound tissue. IHC staining for Psoriasin was visualized, and
results were in accordance with the known expression patterns of
Psoriasin in wound tissue (39). The
present study demonstrated a 19.8% increase in Psoriasin levels at
the leading wound edge of the chronic wound compared with the
distal end. This is consistent with previous reports of increased
Psoriasin expression within wounds (39). However, within the chronic wound
phenotype, Psoriasin mRNA levels are not significantly increased
when compared with normal skin. This suggests that chronic wounds
may be influenced by reduced Psoriasin expression; however, further
study is required to clarify its value as a chronic wound
biomarker. The findings of the present study correlate with the
available data that Psoriasin expression is significantly increased
in acute wounds (39). This suggests
that Psoriasin has a notable influence at the leading wound edge in
chronic wounds, and in acute wounds. Furthermore, Psoriasin may
have an application as a biomarker for wound phenotyping and may
also be used therapeutically to alter cell behaviour and increase
wound healing rates. In conjunction with other cells types, the
role of keratinocytes is critical in the healing of chronic
cutaneous wounds. Their main function is to re-epithelialize the
wound surface, thereby re-establishing epidermal cover, integrity
and barrier function. This process encompasses a series of steps
including proliferation, migration and differentiation (46,47). To
study some of these effects in vitro, the naturally
immortalised HaCaT cell line was used.
In order to examine the effects of Psoriasin in
keratinocytes, an in vitro model downregulating Psoriasin in
HaCaT was used. Psoriasin knockdown was chosen as Psoriasinis
already expressed in normal keratinocytes. The proliferation
crystal violet assay identified a significant increase in the
growth rate of HaCaTPSOkd cells compared with
HaCaTpEF controls. Together with the expression pattern
seen in the IHC of Psoriasin in wounds, this suggests that
Psoriasin may serve differential roles in different locations
within wound tissues, such as cells proximal and distal to wound
edges. Upregulation of Psoriasin production and secretion was
initially noted among psoriatic skin lesions (16) and was subsequently attributed to
inflammation (23,24). However, further evidence of elevated
Psoriasin expression in bladder and breast cancer was observed
(17,29). Furthermore, an increase in Psoriasin
expression levels was demonstrated among precancerous skin lesions
and malignant epithelial tumours (squamous and basal cell
carcinomas) (28), independent of
differentiation and inflammation. It has therefore been suggested
that this overexpression may be an important factor in tumour
proliferation and progression.
Similarly, when compared with the corresponding
HaCaT pEF6 control, in vitro ECIS migration assays show that
downregulation of Psoriasin in HaCaT cells results in a significant
increase in the rate of cell migration across a wounded area. At
present, this increased migratory effect has only been noted in
association with Psoriasin overexpression, similar to cell
proliferation, which has also led to attributing Psoriasin to
cancer progression. In addition, adhesion was found to be
significantly increased, which is in accordance with previous
findings where loss of adhesion, attributed to cancer cell
invasiveness and progression, was associated with the upregulation
of Psoriasin. The in vitro findings of the present study
therefore suggest that increased cellular adhesion associated with
downregulation of Psoriasin may explain the phenomenon of cancer
progression from reduced adhesion, as has been noted with Psoriasin
overexpression.
The results of the present study conflict somewhat,
in that significantly lower levels of Psoriasin are present in
chronic wound tissue compared with acute wound tissue and ribozyme
suppression of Psoriasin expression in HaCaT cells enhanced cell
migration and growth, which are traits typically found in healing
tissues. The mechanisms underlying this are currently unknown,
however it may be hypothesized that these findings will likely be
due to the inherent differences between in vitro models and
complex tissue, comprising multiple cell types, cytokine and growth
factors. It is possible that Psoriasin may have a prominent role in
HaCaT biology, although this may only be obvious or significant
when various other complex factors and interactions are also
considered.
The high expression of Psoriasin in abnormal and
proliferative lesions of squamous epithelia (18,26,48)
suggest that it serves a role in the regulation of cell growth and
survival. The contrasting evidence of increased migration and cell
proliferation with Psoriasin knockdown may suggest that an
alternative pathway, which is blocked by Psoriasin, stimulates cell
growth and migration. However, the increased adhesion with
knockdown suggests that Psoriasin downregulation is unlikely to be
associated with cancer proliferation. Overall, Psoriasin function
appears to be important in maintaining cellular homeostasis,
particularly with respect to cell proliferation, migration,
adhesion and differentiation.
N-WASP is a member of the Wiskott-Aldrich syndrome
family of proteins, which are widely involved in signal
transduction from receptors on the cell surface to the actin
cytoskeleton. The actin cytoskeleton is a dynamic filament network
that is essential for cell movement, polarisation, morphogenesis
and cell division, which was first described as a 65 kDa protein
from the brain that bound to SH3 domains of Ash/Grb2 (49). Having a sequence that was 50%
homologous with Wiskott- Aldrich syndrome protein (WASP), this
novel protein was termed N-WASP (50,51).
N-WASP has been implicated in various
actin-dependent processes, such as filopodium formation and the
motility of Shigella (52).
N-WASP and complexes with other proteins, such as Arp2/3, comprise
a core mechanism for the stimulation of actin polymerisation and
actin assembly (53,54). A reduction of N-WASP has been shown
to be associated with a greater malignant potential in breast
cancer via its role in cell migration and invasion, and interaction
with FAK (55,56). In keratinocytes, knockout of N-WASP
has previously been shown to reduce cellular proliferation
(57). However, in-house studies
(Jiang et al, unpublished data), have revealed increased
keratinocyte migration in vitro with N-WASP inhibition by
using the inhibitor wiskostatin. Similarly, inhibiting N-WASP by
applying inhibitors topically and via the intraperitoneal route to
mice enhanced wound closure rates (Jiang et al, unpublished
data). As a result of these findings, a patent has been put in
place to develop this product into a licensed treatment for
hard-to-heal ulcers (Cardiff University patent, March 2009, ID 090
4886.9) (58).
ROCK1 and 2 occur in mammals (59) and are part of a group of kinases
belonging to the AGC family of serine-threonine kinases. They are
primarily involved in cell shape regulation, actin organisation on
the cytoskeleton and hence cell migration. ROCK1 has a wide range
of cellular functions including cellular contractility, migration,
cytokinesis and cell-cell adhesion, via its downstream effector
function of small GTPase Rho, which is a major cytoskeleton
regulator (60,61). ROCK 2 is primarily located in the
brain and heart (62,63). These kinasesalso serve an important
role in smooth muscle contractility, neuronal development and nerve
generation (64). Elevated ROCK
protein levels in human breast, hepatocellular, bowel and bladder
cancers have been demonstrated to correlate with increased tumour
grade and poor overall survival rates (64,65). In
keratinocytes, differentiation is prevented and proliferation
increased by ROCK inhibition, and a two-fold upregulation in
Psoriasin following activation of ROCK2 has also been reported
(66). ROCK-signalling pathways
serve important roles in various human diseases and have been
considered as potential targets for the treatment of these
diseases, including cancer, leading to increased interest in the
pharmacological potential of ROCK inhibitors (67). In terms of wound healing, ROCK
inhibitors have previously been demonstrated to enhance corneal
endothelial healing (68).
FAK, also known as protein tyrosine kinase 2, is a
focal adhesion-associated protein kinase encoded in humans by the
PTK2 gene. This cytoplasmic protein tyrosine kinase is found
concentrated in the focal adhesions that form among cells attaching
to extracellular matrix (ECM) constituents (69). Most cells express FAK, and activation
of the FAK tyrosine kinase promote cell contacts with ECM and
promotes cell migration (69). The
most well-characterised mechanism promoting FAK activation is
integrin receptor clustering upon the binding of cells to ECM
proteins, which leads to FAK dimerisation, autophosphorylation,
SRC-family kinase binding and activated complex formation (69–71).
FAK is a multifunctional regulator of cell
signalling within a tumour microenvironment, and is overexpressed
and activated in several advanced stage-solid cancers (72). An increase in FAK mRNA levels has
been demonstrated in serous ovarian tumours, invasive breast
cancers, head and neck squamous cell carcinoma, colorectal
malignancy and various other human malignancies (72). At the cellular level, FAK is thought
to be associated with various signalling pathways that promote
cancer growth and metastasis. It increases cell motility via
ARP2/3, affects survival via p53 and MDM2 and induces cell cycle
progression via cyclin D1 SRC-ERK or JUN (72). Small molecule FAK inhibitors have
chemotherapeutic potential, as indicated in mouse models where FAK
inhibition has been demonstrated to prevent tumour growth,
metastasis, vascular permeability and angiogenesis (73–75).
Among keratinocytes FAK has previously been revealed to be
necessary for cell survival in vitro due to massive
apoptosis, although this is not true in vivo. The same study
group established FAK expression in mouse epidermis, thinner
epidermis/hair cycle irregularities with no effect on wound healing
rates in FAK knockdown mice (76).
To our knowledge, no reports currently exist studying interactions
between FAK and Psoriasin.
In conclusion, Psoriasin is expressed in
keratinocytes and is a fundamental regulator of keratinocyte
migration. Significant increases in the rate of keratinocyte
adhesion, migration and growth have been observed in
Psoriasin-deficient cells. N-WASP, FAK, and ROCK proteins serve
certain roles in the Psoriasin-regulated cell adhesion and
motility, implicating that Psoriasin may be associated with wound
healing, thereby endorsing Psoriasin as a molecule of interest and
a potential wound biomarkers.
Acknowledgements
The authors wish to thank Cancer Research Wales,
Welsh Government A4B Scheme and Welsh Life Science Network-Ser
Cymru for supporting the present study.
References
|
1
|
Gottrup F and Apelqvist J: The challenge
of using randomized trials in wound healing. Br J Surg. 97:303–304.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Leaper DJ, Harding KJ and Harding KG: The
future of wound healingWounds: Biology and Management. Leaper DJ
and Harding KJ: Oxford University Press; Oxford: pp. 1911998
|
|
3
|
Posnett J and Franks PJ: The costs of skin
breakdown and ulceration in the UKSkin Breakdown: The Silent
Epidemic. Pownall M: Hull: Smith & Nephew Foundation; pp. 6–12.
2007
|
|
4
|
Abstract of Statistics-Quarter.
3:2011.
|
|
5
|
Menke NB, Ward KR, Witten TM, Bonchev DG
and Diegelmann RF: Impaired wound healing. Clin Dermatol. 25:19–25.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bucalo B, Eaglstein WH and Falanga V:
Inhibition of cell proliferation by chronic wound fluid. Wound
Repair Regen. 1:181–186. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harding KG, Morris HL and Patel GK:
Science, medicine and the future: Healing chronic wounds. BMJ.
324:160–163. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vaalamo M, Leivo T and Saarialho-Kere U:
Differential expression of tissue inhibitors of metalloproteinases
(TIMP-1, −2, −3 and −4) in normal and aberrant wound healing. Hum
Pathol. 30:795–802. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ravanti L and Kähäri VM: Matrix
metalloproteinases in wound repair (review). Int J Mol Med.
6:391–407. 2000.PubMed/NCBI
|
|
10
|
Brandner JM, Zacheja S, Houdek P, Moll I
and Lobmann R: Expression of matrix metalloproteinases, cytokines,
and connexins in diabetic and nondiabetic human keratinocytes
before and after transplantation into an ex vivo wound-healing
model. Diabetes Care. 31:114–120. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Telgenhoff D and Shroot B: Cellular
senescence mechanisms in chronic wound healing. Cell Death Differ.
12:695–698. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Herrick SE, Sloan P, McGurk M, Freak L,
McCollum CN and Ferguson MW: Sequential changes in histologic
pattern and extracellular matrix deposition during the healing of
chronic venous ulcers. Am J Pathol. 141:1085–1095. 1992.PubMed/NCBI
|
|
13
|
Brem H, Stojadinovic O, Diegelmann RF,
Entero H, Lee B, Pastar I, Golinko M, Rosenberg H and Tomic-Canic
M: Molecular markers in patients with chronic wounds to guide
surgical debridement. Mol Med. 13:30–39. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tomic-Canic M, Ayello EA, Stojadinovic O,
Golinko MS and Brem H: Using gene transcription patterns (bar
coding scans) to guide wound debridement and healing. Adv Skin
Wound Care. 21:487–494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Charles CA, Tomic-Canic M, Vincek V,
Nassiri M, Stojadinovic O, Eaglstein WH and Kirsner RS: A gene
signature of nonhealing venous ulcers: Potential diagnostic
markers. J Am Acad Dermatol. 59:758–771. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Madsen P, Rasmussen HH, Leffers H, Honoré
B, Dejgaard K, Olsen E, Kiil J, Walbum E, Andersen AH, Basse B, et
al: Molecular cloning, occurrence, and expression of a novel
partially secreted protein ‘psoriasin’ that is highly up-regulated
in psoriatic skin. J Invest Dermatol. 97:701–712. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Celis JE, Cruger D, Kiil J, Lauridsen JB,
Ratz G, Basse B and Celis A: Identification of a group of proteins
that are strongly up-regulated in total epidermal keratinocytes
from psoriatic skin. FEBS Lett. 262:159–164. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Watson PH, Leygue ER and Murphy LC:
Psoriasin (S100A7). Int J Biochem Cell Biol. 30:567–571. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schäfer BW and Heizmann CW: The S100
family of EF-hand calcium-binding proteins: Functions and
pathology. Trends Biochem Sci. 21:134–140. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wicki R, Schafer BW, Erne P and Heizmann
CW: Characterization of the human and mouse cDNAs coding for
S100A13, a new member of the S100 protein family. Biochem Biophys
Res Commun. 227:594–599. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Børglum AD, Flint T, Madsen P, Celis JE
and Kruse TA: Refined mapping of the psoriasin gene S100A7 to
chromosome 1cen-q21. Hum Genet. 96:592–596. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Heizmann CW and Hunziker W: Intracellular
calcium-binding proteins: More sites than insights. Trends Biochem
Sci. 16:98–103. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Algermissen B, Sitzmann J, LeMotte P and
Czarnetzki B: Differential expression of CRABP II, psoriasin and
cytokeratin 1 mRNA in human skin diseases. Arch Dermatol Res.
288:426–430. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jinquan T, Vorum H, Larsen CG, Madsen P,
Rasmussen HH, Gesser B, Etzerodt M, Honoré B, Celis JE and
Thestrup-Pedersen K: Psoriasin: A novel chemotactic protein. J
Invest Dermatol. 107:5–10. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Alowami S, Qing G, Emberley E, Snell L and
Watson PH: Psoriasin (S100A7) expression is altered during skin
tumorigenesis. BMC Dermatol. 3:12003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Enerback C, Porter DA, Seth P, Sgroi D,
Gaudet J, Weremowicz S, Morton CC, Schnitt S, Pitts RL, Stampl J,
et al: Psoriasin expression in mammary epithelial cells in vitro
and in vivo. Cancer Res. 62:43–47. 2002.PubMed/NCBI
|
|
27
|
Leygue E, Snell L, Hiller T, Dotzlaw H,
Hole K, Murphy LC and Watson PH: Differential expression of
psoriasin messenger RNA between in situ and invasive human breast
carcinoma. Cancer Res. 56:4606–4609. 1996.PubMed/NCBI
|
|
28
|
Moubayed N, Weichenthal M, Harder J,
Wandel E, Sticherling M and Gläser R: Psoriasin (S100A7) is
significantly up-regulated in human epithelial skin tumours. J
Cancer Res Clin Oncol. 133:253–261. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Al-Haddad S, Zhang Z, Leygue E, Snell L,
Huang A, Niu Y, Hiller-Hitchcock T, Hole K, Murphy LC and Watson
PH: Psoriasin (S100A7) expression and invasive breast cancer. Am J
Pathol. 155:2057–2066. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Carlsson H, Yhr M, Petersson S, Collins N,
Polyak K and Enerbäck C: Psoriasin (S100A7) and calgranulin-B
(S100A9) induction is dependent on reactive oxygen species and is
downregulated by Bcl-2 and antioxidants. Cancer Biol Ther.
4:998–1005. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Emberley ED, Alowami S, Snell L, Murphy LC
and Watson PH: S100A7 (psoriasin) expression is associated with
aggressive features and alteration of Jab1 in ductal carcinoma in
situ of the breast. Breast Cancer Res. 6:R308–R315. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Emberley ED, Niu Y, Leygue E, Tomes L,
Gietz RD, Murphy LC and Watson PH: Psoriasin interacts with Jab1
and influences breast cancer progression. Cancer Res. 63:1954–1961.
2003.PubMed/NCBI
|
|
33
|
Jiang WG, Watkins G, Douglas-Jones A and
Mansel RE: Psoriasin is aberrantly expressed in human breast cancer
and is related to clinical outcomes. Int J Oncol. 25:81–85.
2004.PubMed/NCBI
|
|
34
|
Hoffmann HJ, Olsen E, Etzerodt M, Madsen
P, Thøgersen HC, Kruse T and Celis JE: Psoriasin binds calcium and
is upregulated by calcium to levels that resemble those observed in
normal skin. J Invest Dermatol. 103:370–375. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rasmussen HH, Orntoft TF, Wolf H and Celis
JE: Towards a comprehensive database of proteins from the urine of
patients with bladder cancer. J Urol. 155:2113–2119. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Eckert RL, Broome AM, Ruse M, Robinson N,
Ryan D and Lee K: S100 proteins in the epidermis. J Invest
Dermatol. 123:23–33. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mansbridge JN and Knapp AM: Changes in
keratinocyte maturation during wound healing. J Invest Dermatol.
89:253–263. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
McKay IA and Leigh IM: Altered
keratinocyte growth and differentiation in psoriasis. Clin
Dermatol. 13:105–114. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dressel S, Harder J, Cordes J, Wittersheim
M, Meyer-Hoffert U, Sunderkötter C and Gläser R: Differential
expression of antimicrobial peptides in margins of chronic wounds.
Exp Dermatol. 19:628–632. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Conway K, Ruge F, Price P, Harding KG and
Jiang WG: Hepatocyte growth factor regulation: An integral part of
why wounds become chronic. Wound Repair Regen. 15:683–692. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Conway KP, Price P, Harding KG and Jiang
WG: The role of vascular endothelial growth inhibitor in wound
healing. Int Wound J. 4:55–64. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ye L, Sun PH, Martin TA, Sanders AJ, Mason
MD and Jiang WG: Psoriasin (S100A7) is a positive regulator of
survival and invasion of prostate cancer cells. Urol Oncol.
31:1576–1583. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiang WG, Douglas-Jones A and Mansel RE:
Expression of peroxisome-proliferator activated receptor-gamma
(PPARγ) and the PPARgamma co-activator, PGC-1, in human breast
cancer correlates with clinical outcomes. Int J Cancer.
106:752–757. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jiang WG, Martin TA, Lewis-Russell JM,
Douglas-Jones A, Ye L and Mansel RE: Eplin-alpha expression in
human breast cancer, the impact on cellular migration and clinical
outcome. Mol Cancer. 7:712008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Keese CR, Wegener J, Walker SR and Giaever
I: Electrical wound-healing assay for cells in vitro. Proc Natl
Acad Sci USA. 101:1554–1559. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bereiter-Hahn J, MatolsyA G and Richards
Sylvia K: Epidermal cell migration and wound repairBiology of the
Integument 2 Vertebrates. Berlin: Springer-Verlag; pp. 444–447.
1986
|
|
47
|
Patel GK, Wilson CH, Harding KG, Finlay AY
and Bowden PE: Numerous keratinocyte subtypes involved in wound
re-epithelialization. J Invest Dermatol. 126:497–502. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ostergaard M, Wolf H, Orntoft TF and Celis
JE: Psoriasin (S100A7): A putative urinary marker for the follow-up
of patients with bladder squamous cell carcinomas. Electrophoresis.
20:349–354. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Miki H, Miura K and Takenawa T: N-WASP, a
novel actin-depolymerizing protein, regulates the cortical
cytoskeletal rearrangement in a PIP2-dependent manner downstream of
tyrosine kinases. EMBO J. 15:5326–5335. 1996.PubMed/NCBI
|
|
50
|
Fukuoka M, Miki H and Takenawa T:
Identification of N-WASP homologs in human and rat brain. Gene.
196:43–48. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fukuoka M, Suetsugu S, Miki H, Fukami K,
Endo T and Takenawa T: A novel neural Wiskott-Aldrich syndrome
protein (N-WASP) binding protein, WISH, induces Arp2/3 complex
activation independent of Cdc42. J Cell Biol. 152:471–482. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Uruno T, Liu J, Li Y, Smith N and Zhan X:
Sequential interaction of actin-related proteins 2 and 3 (Arp2/3)
complex with neural Wiscott-Aldrich syndrome protein (N-WASP) and
cortactin during branched actin filament network formation. J Biol
Chem. 278:26086–26093. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Weaver AM, Heuser JE, Karginov AV, Lee WL,
Parsons JT and Cooper JA: Interaction of cortactin and N-WASp with
Arp2/3 complex. Current Biol. 12:1270–1278. 2002. View Article : Google Scholar
|
|
54
|
Rohatgi R, Ma L, Miki H, Lopez M,
Kirchhausen T, Takenawa T and Kirschner MW: The interaction between
N-WASP and the Arp2/3 complex links Cdc42-dependent signals to
actin assembly. Cell. 97:221–231. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Martin TA, Pereira G, Watkins G, Mansel RE
and Jiang WG: N-WASP is a putative tumour suppressor in breast
cancer cells, in vitro and in vivo and is associated with clinical
outcome in patients with breast cancer. Clin Exp Metastasis.
25:97–108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sanchez AM, Flamini MI, Baldacci C, Goglia
L, Genazzani AR and Simoncini T: Estrogen receptor-alpha promotes
breast cancer cell motility and invasion via focal adhesion kinase
and N-WASP. Mol Endocrinol. 24:2114–2125. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lefever T, Pedersen E, Basse A, Paus R,
Quondamatteo F, Stanley AC, Langbein L, Wu X, Wehland J, Lommel S
and Brakebusch C: N-WASP is a novel regulator of hair-follicle
cycling that controls antiproliferative TGF{beta} pathways. J Cell
Sci. 123:128–140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jiang W and Harding K: Method and kit for
the classification and prognosis of wounds. Journal. 2010.
|
|
59
|
Reese KA, Reddy S and Rock JA:
Endometriosis in an adolescent population: The Emory experience. J
Pediatr Adolesc Gynecol. 9:125–128. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Riento K and Ridley AJ: Rocks:
Multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol.
4:446–456. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yasui Y, Amano M, Nagata K, Inagaki N,
Nakamura H, Saya H, Kaibuchi K and Inagaki M: Roles of
Rho-associated kinase in cytokinesis; mutations in Rho-associated
kinase phosphorylation sites impair cytokinetic segregation of
glial filaments. J Cell Biol. 143:1249–1258. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhou Z, Meng Y, Asrar S, Todorovski Z and
Jia Z: A critical role of Rho-kinase ROCK2 in the regulation of
spine and synaptic function. Neuropharmacology. 56:81–89. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhao Z and Rivkees SA: Rho-associated
kinases play a role in endocardial cell differentiation and
migration. Dev Biol. 275:183–191. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Morgan-Fisher M, Wewer UM and Yoneda A:
Regulation of ROCK activity in cancer. J Histochem Cytochem.
61:185–198. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Lane J, Martin TA, Watkins G, Mansel RE
and Jiang WG: The expression and prognostic value of ROCK I and
ROCK II and their role in human breast cancer. Int J Oncol.
33:585–593. 2008.PubMed/NCBI
|
|
66
|
McMullan R, Lax S, Robertson VH, Radford
DJ, Broad S, Watt FM, Rowles A, Croft DR, Olson MF and Hotchin NA:
Keratinocyte differentiation is regulated by the Rho and ROCK
signaling pathway. Curr Biol. 13:2185–2189. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hahmann C and Schroeter T: Rho-kinase
inhibitors as therapeutics: From pan inhibition to isoform
selectivity. Cell Mol Life Sci. 67:171–177. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Okumura N, Koizumi N, Ueno M, Sakamoto Y,
Takahashi H, Hirata K, Torii R, Hamuro J and Kinoshita S:
Enhancement of corneal endothelium wound healing by Rho-associated
kinase (ROCK) inhibitor eye drops. Br J Ophthalmol. 95:1006–1009.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Brami-Cherrier K, Gervasi N, Arsenieva D,
Walkiewicz K, Boutterin MC, Ortega A, Leonard PG, Seantier B, Gasmi
L, Bouceba T, et al: FAK dimerization controls its kinase-dependent
functions at focal adhesions. EMBO J. 33:356–370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Parsons JT: Focal adhesion kinase: The
first ten years. J Cell Sci. 116:1409–1416. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Schaller MD: Cellular functions of FAK
kinases: Insight into molecular mechanisms and novel functions. J
Cell Sci. 123:1007–1013. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Sulzmaier FJ, Jean C and Schlaepfer DD:
FAK in cancer: Mechanistic findings and clinical applications. Nat
Rev Cancer. 14:598–610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Parsons JT, Slack-Davis J, Tilghman R and
Roberts WG: Focal adhesion kinase: Targeting adhesion signaling
pathways for therapeutic intervention. Clin Cancer Res. 14:627–632.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Shi Q, Hjelmeland AB, Keir ST, Song L,
Wickman S, Jackson D, Ohmori O, Bigner DD, Friedman HS and Rich JN:
A novel low-molecular weight inhibitor of focal adhesion kinase,
TAE226, inhibits glioma growth. Mol Carcinog. 46:488–496. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Roberts WG, Ung E, Whalen P, Cooper B,
Hulford C, Autry C, Richter D, Emerson E, Lin J, Kath J, et al:
Antitumor activity and pharmacology of a selective focal adhesion
kinase inhibitor, PF-562,271. Cancer Res. 68:1935–1944. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Essayem S, Kovacic-Milivojevic B,
Baumbusch C, McDonagh S, Dolganov G, Howerton K, Larocque N, Mauro
T, Ramirez A, Ramos DM, et al: Hair cycle and wound healing in mice
with a keratinocyte-restricted deletion of FAK. Oncogene.
25:1081–1089. 2006. View Article : Google Scholar : PubMed/NCBI
|