Introduction
Hyperglycemic conditions evoked by diabetes mellitus
disrupt the role of endothelial cells in the protection of blood
vessels and is therefore associated with cardiovascular disease.
Diabetic macrovascular complications, such as myocardial infarction
and stroke, are a major cause of morbidity and mortality (1). Once atherosclerosis has developed, it
is irreversible and the progression of endothelial damage
continues. Thus, even if blood sugar levels are well controlled
thereafter, the natural course of macrovascular complications
cannot be altered (2). In early
stage diabetes mellitus, nitric oxide (NO), an endothelium-derived
relaxing factor, is important in relaxing and protecting the blood
vessels. NO is produced as a result of an enzymatic reaction of the
L-arginine amino acid, facilitated by nitric oxide synthase (NOS).
Endothelial cells possess two forms of NOS: Constitutive or
endothelial NOS (cNOS or eNOS; type III) and inducible NOS (iNOS;
type II) (3). Under normal
conditions, NO has anti-inflammatory and anti-proliferative
effects, which protect blood vessels by inhibiting smooth muscle
hyperplasia and scavenging superoxide anion. NO also inhibits
leukocyte adhesion to vascular endothelium, exerting an
anti-thrombotic effect (3). However,
abnormal conditions, such as inflammation and hyperglycemia, lead
to a decrease in the synthesis of NO. This may lead to the
development of vascular diseases including inflammation,
thrombosis, vascular hypertrophy, stenosis and vasoconstriction
(4).
Diabetic vascular complications are caused by
endothelial cell dysfunction. Initially, the damage caused by
lesions is reversible (4), thus
early treatment to correct endothelial dysfunction is important to
prevent diabetic macrovascular complications, and reduce the
morbidity and mortality caused by cardiovascular diseases (5).
Incretin hormones, such as glucose-dependent
insulinotropic polypeptide (GIP) and glucagon-like peptide-1
(GLP-1), have become the subjects of attention. GIP and GLP-1 are
intestinal hormones that control blood sugar by stimulating insulin
release from the pancreas (6).
Incretin hormones have receptors in a number of organs and have
been assessed with regards to control of blood sugar levels
(7). GIP and GLP-1 receptors are
present in vascular endothelial cells. It has been demonstrated
that GIP is directly involved in the physiology of blood vessels;
controlling the blood flow rate of the hepatic portal veins and
increasing nutrient absorption (8).
GLP-1 has a protective effect on blood vessels by acting on the
endothelial cells (9). However, to
the best of our knowledge, the current data do not sufficiently
clarify the effects that GIP and GLP-1 have on endothelial cells in
patients with hyperglycemia.
Therefore, the present study aimed to investigate
whether the incretin hormones GLP-1 and GIP improve endothelial
cell dysfunction caused by hyperglycemia.
Materials and methods
Cell culture and treatments
Hamster-derived insulin-secreting HIT-T15 cells
(Calbiochem; EMD Millipore, Billerica, MA, USA) were maintained in
RPMI 1,640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) containing 11.1 mM glucose supplemented with 10%
heat-inactivated fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), penicillin (100 U/ml), and streptomycin (100
µg/ml), in a humidified atmosphere containing 5% CO2 at
37°C. These cells were used as a comparison when determining
whether GIP and GLP-1 were expressed in HUVECs.
Passage 6–10 cells of human umbilical vein
endothelial cells (HUVECs) supplied by Lonza (Walkersville, MD,
USA) were cultured with medium from the EGM-2™ Bullet kit™ (Lonza).
The kit contained 10% fetal bovine serum, vascular endothelial
growth factor, human fibroblastic growth factor B, hydrocortisone,
R3-insulin-like growth factor-I, ascorbic acid, GA-1,000, human
epidermal growth factor and heparin. The conditions for culture
were maintained at 37°C and 95% humidity in 5% CO2. When
the confluence reached 80–90%, subcultures were prepared following
washing with phosphate-buffered saline (PBS) and processing with
trypsin-EDTA. The collected cells were underwent centrifugation at
room temperature, 500 × g for 10 min in order to produce pellets.
The pellets were gently resuspended in EGM-2™ Bullet kit™ medium
once the supernatant was discarded. This medium was replaced every
2 days until confluence was reached (3–5 days). Following treatment
with GIP (Sigma-Aldrich, Merck kGaA, Darmstadt, Germany), GLP-1
(Bachem Americas, Inc., Torrance, CA, USA), or Exendin 9–39, a
specific GLP-1 receptor antagonist (Bachem Americas) in 5.5 or 30
mM glucose, analysis was completed. Exendin 9–39 GLP-1 receptor
antagonist (Saxon Biochemicals, Hannover, Germany) was used to
determine if the change in iNOS and eNOS was due to the GLP-1
receptor agonist. HUVECs were pretreated for 1 h with 1 nM GIP or 3
nM GLP-1, and 50 µM DPPIV inhibitor (Sigma-Aldrich; Merck kGaA,
Darmstadt, Germany). HUVECs were then cultured in medium containing
5.5 or 30 mM glucose for 48 h.
RNA extraction and reverse
transcription-polymerase chain reaction (RT-PCR)
RT-PCR was used to determine the presence of GIP or
GLP-1 receptors in HUVECs. Total RNA was extracted using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) following the manufacturer's protocol. The reverse
transcription reaction was completed at 42°C for 60 min using
ImProm-II™ reverse transcription system (20 µl; Promega
Corporation, Madison, WI, USA), containing AMV reverse
transcriptase, MgCl2 25 mM, reverse transcription 10X
buffer, dNTP mixture 10 mM, recombinant RNasin ribonuclease
inhibitor and oligo (dT)15 primer, before heating them
to 70°C for 5 min followed immediately by cooling on ice so that
reverse transcription enzymes were inactivated and cDNA remained in
a linear strand state. cDNA (1 µg) was amplified in final volume 20
µl with iTaq™ DNA polymerase (Bio-Rad Laboratories Inc., Hercules,
CA, USA) using the following primer sets: GIP receptor: Forward,
5′-AACGAAGTCAAGGCCATTTG-3′ and reverse, 5′-GTCCTCAGCTTGGACAGGAG-3′;
GLP-1 receptor: Forward 5′-GTTCCCCTGCTGTTTGTTGT-3′ and reverse,
5′-TGGCCTTCAGTTTGGATACC-3′; GAPDH forward,
5′-AAGGGTCATCATCTCTGCCC-3′ and reverse, 5′-GTGATGGCATGGACTGTGGT-3′.
PCR reaction procedure began with heating at 95°C for 5 min prior
to cycle starts. Cycles consisted of denaturation at 94°C for 30
sec, annealing at 55°C for 30 sec and elongation at 72°C for 30
sec, repeated 30 times, followed by the extension of generated
strands at 72°C for 5 min. Following the PCR reaction process, the
extended DNA was subjected to electrophoresis at 100V for 20 min
using 1.2% agarose gel.
Reverse transcription-quantitative PCR
(RT-qPCR)
Extraction of total RNA and cDNA synthesis were
performed using the aforementioned method in the previous section.
RT-qPCR primers were synthesized, ~100 bps based on the base
sequence of GenBank (https://www.ncbi.nlm.nih.gov/genbank). Sequences of
the synthesized primers were as follows: iNOS, forward,
5′-ACAAGCCTACCCCTCCAGAT-3′ and reverse, 5′-TCCCGTCAGTTGGTAGGTTC-3′;
eNOS, forward, 5′-CCCTTCAGTGGCTGGTACAT-3′ and reverse
5′-TATCCAGGTCCATGCAGACA-3′; GAPDH, forward
5′-AAGGGTCATCATCTCTGCCC-3′ and reverse 5′-GTGATGGCATGGACTGTGGT-3′.
qPCR was performed with a 20 µl reaction mixture containing 1 µg
cDNA, 10 pmol forward primer, 10 pmol reverse primer and 10 µl Fast
SYBR Green Master Mix (Invitrogen; Thermo Fisher Scientific, Inc.)
using the iQ™5 Optical system (Bio-Rad Laboratories, Inc.). The
reaction procedure was completed as follows: Heating at 95°C for 5
min prior to denaturation at 95°C for 30 sec, annealing at 60°C for
30 sec and elongation at 72°C for 30 sec for 40 cycles. This was
followed by the extension of generated strands at 72°C for 5 min.
Melting curve analysis was performed at a temperature between
65–95°C and a Cq value for each was used to normalize the data to
GAPDH mRNA value following compensation (10).
Western blot analysis
Cells were harvested and then pelleted by
centrifugation at 13,000 rpm for 10–20 sec. Cells were resuspended
in 400 µl PRO-PREP™ solution (Intron Biotechnology, Inc., Seongnam,
Korea), and mix well. Cell lysis was induced by incubation for
10–20 min on freezer at −20°C. Centrifugation was then performed at
2,000 × g at 4°C for 5 min, and supernatant was transferred to a
fresh 1.5 ml tube. The extracted protein was quantified using a
Bradford protein assay kit (Bio-Rad Laboratories, Inc.) and equal
amounts of protein (20 µg/lane) were separated by 20% SDS-PAGE and
transferred to a nitrocellulose membrane (Invitrogen; Thermo Fisher
Scientific, Inc.). The membrane was incubated in 5% non-fat dry
milk-PBS Tween-20 (PBST; 0.01% Tween 20 in PBS) for 1 h at room
temperature to block the non-specific bonding between antibody and
protein. Primary antibodies; anti-iNOS (sc-49055; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), anti-eNOS (9572; Cell
Signaling Technology, Inc., Danvers, MA, USA), anti-phospho-eNOS
(Ser1177) (9517; Cell Signaling Technology, Inc.), anti-GIP
receptor (ab30679; Abcam, Cambridge, UK), anti-GLP-1 receptor
(ab36598; Abcam) and anti-β-actin (internal control; a5441;
Sigma-Aldrich; Merck kGaA) were incubated with the protein at room
temperature for 2 h (anti-β-actin was diluted to 1:5,000 and other
antibodies to 1:500 with blocking buffer). Following washing with
PBST twice for 10 min, the secondary antibodies, anti-mouse
immunoglobulin (Ig) G (sc-51993) and anti-rabbit IgG (sc-358919;
Santa Cruz Biotechnology, Inc.) conjugated with horseradish
peroxidase (HRP) were diluted to 1:500 with blocking buffer and
incubated at room temperature for 1 h. The protein obtained
following the reaction was examined for specific bands following
exposure to light on X-ray film using the Chemiluminescent Reagent
kit (ab79907; Abcam).
MTT assay
An MTT assay (Vybrant® MTT Cell
Proliferation Assay kit, Invitrogen; Thermo Fisher Scientific,
Inc.) was completed.
Assessment of NO production
The activity of NO was determined by measuring the
nitrite (NO2-) concentration in the culture media using
the Griess reagent system (Promega Corporation). Each medium
supernatant (100 ml) was mixed with 50 ml 1% sulfanilamide (in 5%
phosphoric acid, Sigma-Aldrich; Merck kGaA) and 50 ml 0.1%
N-(1-Naphthyl) ethylenediamine dihydrochloride (Santa Cruz
Biotechnology, Inc.) and incubated in the dark at room temperature
for 10 min. Absorbance was measured using a SpectraMax L Microplate
Reader (Molecular Devices, LLC, Sunnyvale, CA, USA) at 540 nm.
NO2 concentration was determined based on the nitrogen
standard curve.
Measurement of cyclic adenosine
monophosphate (cAMP)
Cultured cells were washed with cold PBS twice and
lysed with 0.1 M hydrochloric acid at room temperature for 20 min.
Cells were then collected using a scraper and subjected to
centrifugation at 1,000 × g for 10 min at room temperature. The
supernatant was transferred to new tubes and used for
experimentation in a 96-well plate. cAMP levels in the cells were
measured at a wavelength of 405 nm using an ELISA microplate reader
(Molecular Devices LLC.) using a cyclic AMP ELISA kit (581001;
Cayman Chemical Company, Ann Arbor, MI, USA) according to the
manufacturers protocol.
Statistical analysis
Each experiment was repeated four times and data
were indicated as mean ± standard deviation. All statistical
analysis was performed using the Statistical Package for the Social
software (SPSS; Korean version 20.0; IBM SPSS, Armonk, NY, USA).
Statistical differences were analyzed using one-way analysis of
variance followed by Tukey's test. P<0.05 was determined to
indicate a statistically significant difference.
Results
Expression of GIP and GLP-1 receptors
in HUVEC
Prior to investigating the action of GIP or GLP-1 in
endothelial cells, RT-PCR and western blot analysis were used to
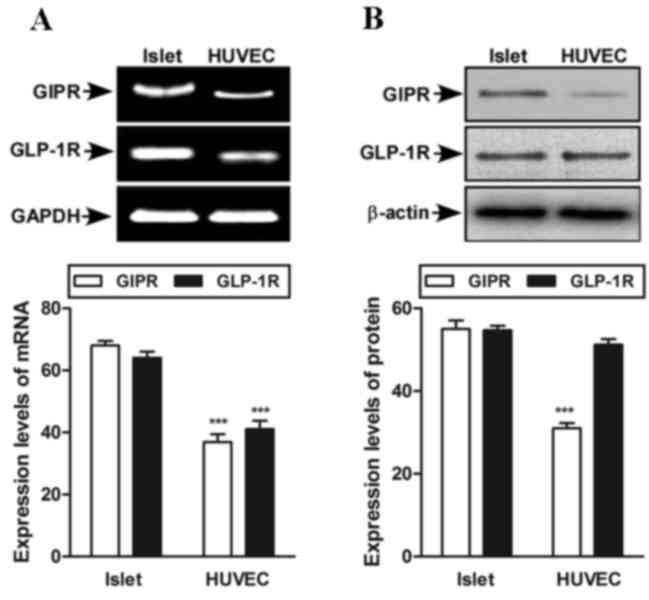
assess whether GIP and GLP-1 receptors were expressed in HUVEC. The
expression of GIP and GLP-1 receptors in HUVECs is lower than in
islet cells (Fig. 1A). Furthermore,
in HUVEC cells, the GLP-1 receptor is more highly expressed than
the GIP receptor (Fig. 1B). Islet
cells were used as positive control. An MTT assay was completed to
eliminate cell death from high glucose. During the low and high
glucose exposure for up to 48 h, the HUVECs survived and the
maintenance of cell morphology was confirmed by MTT and light
microscopy (data not shown).
Effect of high glucose on iNOS and
eNOS mRNA expression and NO production
To compare NOS expression against high glucose
levels, expression of iNOS mRNA and eNOS mRNA were measured by
RT-qPCR. The expression of iNOS and eNOS mRNA was not affected in
normal conditions with 5.5 mM glucose (Fig. 2A). However, the expression of eNOS
mRNA significantly decreased by >50% 12 h following treatment
with 30 mM glucose, indicating a fast reaction (P<0.001;
Fig. 2B). Following treatment with
30 mM glucose, the expression of iNOS mRNA did not increase during
the initial 0–36 h but significantly increased compared with the
baseline value following 48 h (P<0.01; Fig. 2B). The production of nitric oxide,
which is associated with vascular endothelial cell function, was
not affected in normal conditions with 5.5 mM glucose (Fig. 2C) but decreased significantly from 24
h onwards under the high glucose (30 mM) condition (P<0.01;
Fig. 2D).
Effects of GIP or GLP-1 on the
expression of iNOS and eNOS and the production of NO in high
glucose concentrations
Similar to the results described in Fig. 2, a high glucose concentration (30 mM)
had different effects on eNOS and iNOS in HUVEC and led to a
decrease in NO production. In order to determine whether incretin
hormones have a protective effect on the reaction of endothelial
cells to hyperglycemia, the experiment was performed following
treatment with GIP or GLP-1. HUVECs pre-treated with GLP-1
exhibited decreased expression of iNOS mRNA (P<0.01) and an
increase in eNOS mRNA expression under hyperglycemic conditions
(both P<0.001; Fig. 3A). However,
HUVECs pre-treated with GIP did not exhibit any change in iNOS and
eNOS mRNA expressions (Fig. 3A).
Similar results were obtained regarding the expression of iNOS and
eNOS protein (Fig. 3B). The
production of NO also differed between these two treatment groups,
demonstrating a significant increase of NO expression in the GLP-1
treatment group compared with control group (P<0.001; Fig. 3C). However, there was no significant
difference between the NO expression in cells treated with GIP and
control cells in the hyperglycemic condition. To evaluate whether
the GLP-1 acts through GLP-1 receptor or not, Exendin (9–39) was
used. This determined that the effect of GLP-1 on iNOS and eNOS in
HUVEC was reduced significantly compared with treatment with GLP-1
alone. (Fig. 3D).
 | Figure 3.Effect of GIP or GLP-1 on iNOS, eNOS
and NO in normal glucose (5.5 mM) or high-glucose (30 mM). (A)
Expression of iNOS and eNOS mRNA was determined by reverse
transcription-quantitative polymerase chain reaction. (B)
Expression of iNOS, p-eNOS and eNOS proteins was determined by
western blot analysis. β-actin was used as a loading control. (C)
Activity of NO was examined using a Griess reagent system kit. (D)
Following co-treatments with Exendin (9–39), a GLP-1 receptor
antagonist, and GLP-1, the effect of GLP-1 on iNOS and eNOS
expression in HUVEC was observed to decrease in high-glucose cells
(30 mM), compared with treatment with GLP-1 alone. *P<0.05,
**P<0.01, ***P<0.001 vs. control cells of normal glucose (5.5
mM). ###P<0.001 vs. control cells of high-glucose (30
mM). Data are presented as the mean ± standard deviation of four
independent experiments. GIP, glucose-dependent insulinotropic
polypeptide; GLP-1, glucagon-like peptide 1; iNOS, inducible nitric
oxide synthase; eNOS, endothelial nitric oxide synthase; NO, nitric
oxide; HUVEC, human umbilical vein endothelial cells; p-eNOS,
phosphorylated eNOS; DPPIV, dipeptidyl peptidase-4. |
Effect of GIP or GLP-1 on cAMP
concentration in HUVEC
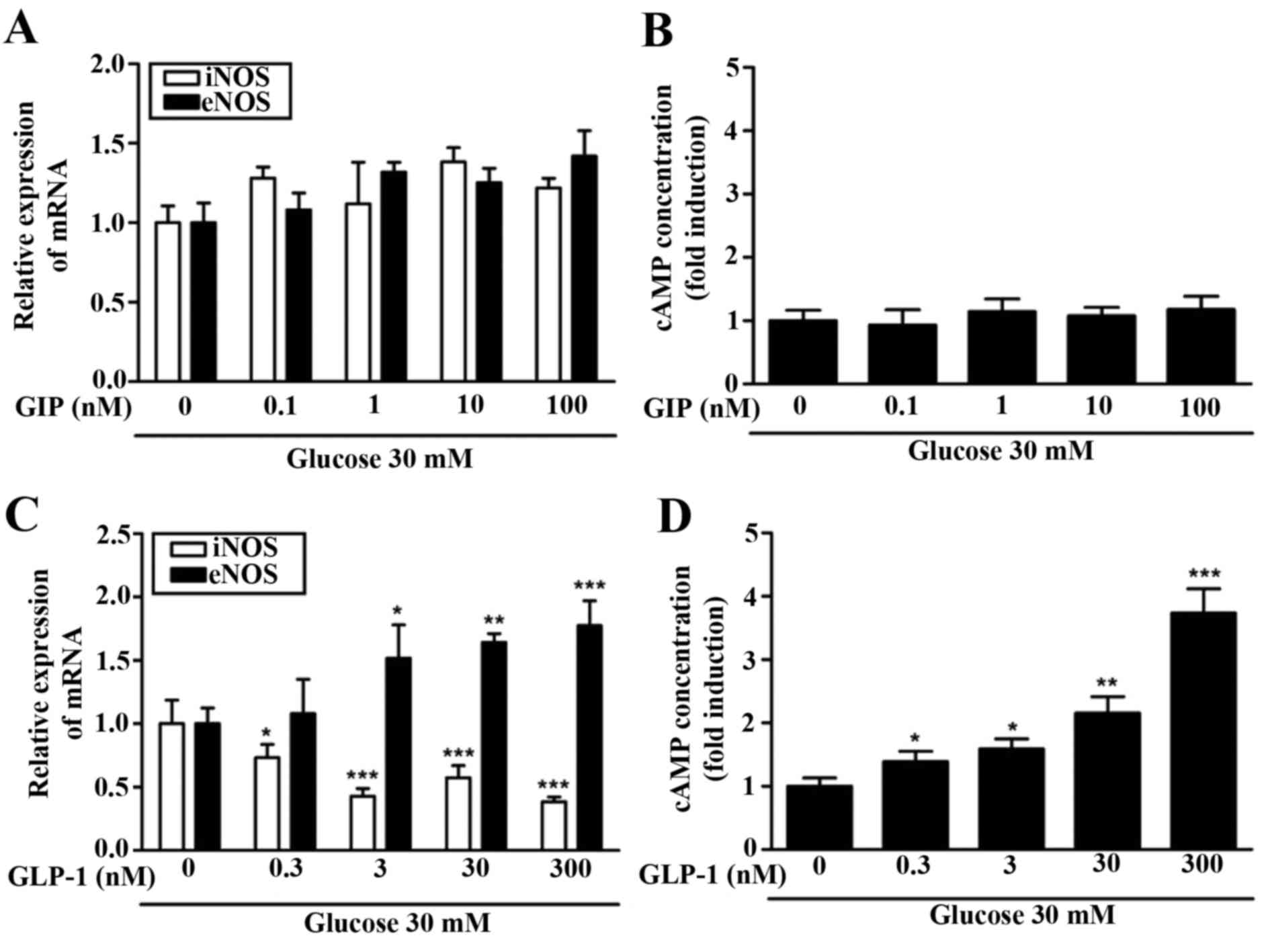
Due to the differing effects of GIP and GLP-1
observed regarding the expression of NO, eNOS and iNOS, the change
in the concentration of cAMP was assessed. Increasing GIP
concentration in the hyperglycemic condition did not alter the
expression of iNOS and eNOS mRNA (Fig.
4A). An increased GIP concentration did not result in any
change in cAMP concentration (Fig.
4B). By contrast, it was observed that the expression of iNOS
and eNOS mRNA differed significantly depending on the concentration
of GLP-1. At higher concentrations of GLP-1 (>0.3 nM), iNOS mRNA
expression decreased significantly (P<0.05) and at
concentrations of GLP-1 >3 nM, eNOS mRNA increased significantly
(P<0.05; Fig. 4C). Furthermore,
cAMP concentration increased in a GLP-1-dependent manner (Fig. 4D). cAMP concentration increased ~x3
in hyperglycemic condition (30 mM glucose) compared with a normal
glucose condition (5.5 mM). The increase in cAMP concentrations
following pre-treatment with GLP-1, but not with GIP treatment,
indicates that there is an association between the two.
Discussion
The current study demonstrates that GLP-1 induced
cAMP activity serves a pivotal role in GLP-1 mediated vascular
protection by increasing NO levels. However, GIP has no effect in
GIP associated with increasing cAMP activity in high glucose
condition. Thus, the results of the present study indicate that
cAMP signaling is required in the protective effect of GLP-1 in
endothelial cells.
It has been demonstrated that vascular endothelial
cells express GIP and GLP-1 receptors and this has been interpreted
to mean that incretin hormones may be useful in the treatment of
vascular diseases (11,12). Therefore, the current study aimed to
examine how the incretin hormones GIP and GLP-1 act to protect
endothelial cells under high glucose conditions and to identify
their mechanism of action using a HUVEC cell line.
GLP-1 receptors are widely expressed in pancreatic
islets, brain, heart, kidney, endothelial cells and the
gastrointestinal tract (13). Some
of the functions of GLP-1 in certain organs have been reported but
its precise functions have not yet been established (14). Since one of the functions of GLP-1 is
to inhibit glucagon and stimulate glucose-induced insulin
secretion, it is used as a fasting or postprandial hypoglycemic
agent. A protective effect of GLP-1 on vascular endothelial cells
has been identified, but its mechanism in endothelial cells remains
unknown (13). A previous clinical
study demonstrated that GLP-1 may lead to the improvement of aorta
pulse wave velocity (PWV) in obese diabetic patients during
short-term treatment (15). Aorta
PWV is the velocity at which the arterial pulse propagates through
the circulatory system and is used clinically as a measure of
arterial stiffness (16). It was
demonstrated that aorta PWV is directly associated with
cardiovascular disease and the early endothelial cell dysfunction
marker in patients with diabetes mellitus (17). In the current study, it was suggested
that GLP-1 protects against hyperglycemia and vascular endothelial
cell dysfunction in hyperglycemic conditions by increasing eNOS
expression and thus increasing levels of NO in endothelial cells.
This protects the endothelial cells from being damaged by the
effects of hyperglycemia. The increase of NO expression in
endothelial cells is associated with an increase in eNOS activation
by GLP-1, which is dependent on the GLP-1 receptor (18). The results of the present study also
demonstrated a GLP-1 dose-dependent increase of eNOS; this effect
was significant at low (5.5 mM) and high (30 nM) glucose
concentrations. By contrast, the effect of GIP was significant at a
low glucose concentration (5.5 mM; P<0.05) but did not cause a
significant change in the amount of iNOS and eNOS at higher glucose
levels (Fig. 3).
Since the two incretin hormones demonstrated
different endothelial cell reactions to a high glucose
concentration, the expression of cAMP was measured. The difference
in cAMP expression between the two hormones at a high concentration
of glucose indicates that GLP-1 is superior to GIP in protecting
endothelial cells. Treatment with GLP-1 also resulted in a
dose-dependent increase of cAMP occurring. Although the effect of
GIP on endothelial vasoconstriction or dilatation may be explained
in connection with the types of vascular endothelial cells or the
expression of receptors, it exhibits the same result that occurs
when blood sugar is controlled normally. As such reactions may be
inhibited at high glucose levels due to unresponsive cAMP, GLP-1 is
effective even in hyperglycemic conditions.
GIP and GLP-1 stimulate insulin secretion through
Ca2+ and cAMP pathways in beta cells (19). In endothelial cells, GIP increases
the level of calcium in a dose-dependent manner, however the
magnitude of this response differs depending on the endothelial
cell type (20). cAMP also increases
insulin secretion in beta cells but it inhibits the mitogenic
effect and bovine fibroblast growth in endothelial cells (21). By contrast, with GIP, cAMP levels did
not increase in endothelial cell lines therefore; the inhibition of
cell proliferation caused by cAMP does not occur (22). The results of the present study
demonstrated no increase of cAMP upon increasing the GIP
concentration at high glucose levels, but GLP-1 increased the cAMP
concentration proportional to its concentration. A previous study
indicated that there was a significant decrease in proliferation
and an increase in the apoptosis of smooth muscle cells in
vitro following treatment with exendin-4, this effect appears
to be mediated through cAMP signaling (23). Ge et al (24) demonstrated the protective effect of
GLP-1 using microvascular endothelial cells whereas the present
study used HUVEC as a macrovascular cell line. GLP-1 also protects
cardiac microvessels against apoptosis and oxidative stress
(24). The protective effects of
GLP-1 are dependent on the downstream inhibition of Rho in a
cAMP/Protein kinase A-dependent manner (25).
Vascular endothelial cells serve an important
function in maintaining vascular homeostasis by controlling
vasomotor, blood coagulation and decomposition, and the
proliferation and migration of inflammatory cells or vascular
smooth muscle (26). In patients
with diabetes however, hyperglycemia causes endothelial dysfunction
and it is difficult to maintain vascular homeostasis. An early
complication of diabetes is lesions of dysfunctional endothelial
cells, which are initially reversible, however early detection and
treatment is required (27). NO is
an important factor for endothelial cell dysfunction in the form of
lesions that appear in the early stage of vascular disease in
patients with diabetes (28).
Controlling blood sugar and NO levels in diabetes patients may help
prevent and treat vascular complications by normalizing the
endothelial function at the early stage of vascular disease
(29).
In conclusion, to the best of our knowledge, the
present study is the first to identify a dose-dependent association
between GIP or GLP-1 and hyperglycemia in HUVEC endothelial cells.
This is associated with the generation of eNOS, iNOS and NO at
different cAMP concentrations.
Acknowledgements
The present study was supported by a grant (L.D.M.,
2011) from the Korean Diabetes Association.
References
|
1
|
Rask-Madsen C and King GL: Mechanisms of
disease: Endothelial dysfunction in insulin resistance and
diabetes. Nat Clin Pract Endocrinol Metab. 3:46–56. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
ACCORD Study Group, ; Gerstein HC, Miller
ME, Genuth S, Ismail-Beigi F, Buse JB, Goff DC Jr..Probstfield JL,
Cushman WC, Ginsberg HN, et al: Long-term effects of intensive
glucose lowering on cardiovascular outcomes. N Engl J Med.
364:818–828. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Förstermann U, Closs EI, Pollock JS,
Nakane M, Schwarz P, Gath I and Kleinert H: Nitric oxide synthase
isozymes: Characterization, purification, molecular cloning and
functions. Hypertension. 23:1121–1131. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wright E Jr..Scism-Bacon JL and Glass LC:
Oxidative stress in type 2 diabetes: The role of fasting and
postprandial glycaemia. Int J Clin Pract. 60:308–314. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schäfer A and Bauersachs J: Endothelial
dysfunction, impaired endogenous platelet inhibition and platelet
activation in diabetes and atherosclerosis. Curr Vasc Pharmacol.
6:52–60. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Davignon J and Ganz P: Role of endothelial
dysfunction in atherosclerosis. Circulation. 109(23 Suppl 1):
III27–III32. 2004.PubMed/NCBI
|
|
7
|
Vilsbøll T, Krarup T, Madsbad S and Holst
JJ: Both GLP-1 and GIP are insulinotropic at basal and postprandial
glucose levels and contribute nearly equally to the incretin effect
of a meal in healthy subjects. Regul Pept. 114:115–121. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim W and Egan JM: The role of incretins
in glucose homeostasis and diabetes treatment. Pharmacol Rev.
60:470–512. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kogire M, Inoue K, Sumi S, Doi R, Yun M,
Kaji H and Tobe T: Effects of gastric inhibitory polypeptide and
glucagon on portal venous and hepatic arterial flow in conscious
dogs. Dig Dis Sci. 37:1666–1670. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ojima A, Matsui T, Maeda S, Takeuchi M and
Yamagishi S: Glucose-dependent insulinotropic polypeptide (GIp)
inhibits signaling pathways of advanced glycation end products
(AGEs) in endothelial cells via its antioxidative properties. Horm
Metab Res. 44:501–505. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Forst T, Weber MM and Pfützner A:
Cardiovascular benefits of GLP-1-based therapies in patients with
diabetes mellitus type 2: Effects on endothelial and vascular
dysfunction beyond glycemic control. Exp Diabetes Res.
2012:6354722012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Holst JJ: The physiology of glucagon-like
peptide 1. Physiol Rev. 87:1409–1439. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hansotia T, Maida A, Flock G, Yamada Y,
Tsukiyama K, Seino Y and Drucker DJ: Extrapancreatic incretin
receptors modulate glucose homeostasis, body weight and energy
expenditure. J Clin Invest. 117:143–152. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hong JY, Park KY, Kim BJ, Hwang WM, Kim DH
and Lim DM: Effects of short-term exenatide treatment on regional
fat distribution, glycated hemoglobin levels and aortic pulse wave
velocity of obese type 2 diabetes mellitus patients. Endocrinol
Metab (Seoul). 31:80–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nichols WW: Clinical measurement of
arterial stiffness obtained from noninvasive pressure waveforms. Am
J Hypertens. 18:3S–10S. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Anderson TJ: Arterial stiffness or
endothelial dysfunction as a surrogate marker of vascular risk. Can
J Cardiol. 22 Suppl B:72B–80B. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding L and Zhang J: Glucagon-like
peptide-1 activates endothelial nitric oxide synthase in human
umbilical vein endothelial cells. Acta Pharmacol Sin. 33:75–81.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
MacDonald PE, El-Kholy W, Riedel MJ,
Salapatek AM, Light PE and Wheeler MB: The multiple actions of
GLP-1 on the process of glucose-stimulated insulin secretion.
Diabetes. 51 Suppl 3:S434–S442. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhong Q, Bollag RJ, Dransfield DT,
Gasalla-Herraiz J, Ding KH, Min L and Isales CM: Glucose-dependent
insulinotropic peptide signaling pathways in endothelial cells.
Peptides. 21:1427–1432. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
D'Angelo G, Lee H and Weiner RI:
CAMP-dependent protein kinase inhibits the mitogenic action of
vascular endothelial growth factor and fibroblast growth factor in
capillary endothelial cells by blocking raf activation. J Cell
Biochem. 67:353–366. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ding KH, Zhong Q and Isales CM:
Glucose-dependent insulinotropic peptide stimulates thymidine
incorporation in endothelial cells: Role of endothelin-1. Am J
Physiol Endocrinol Metab. 285:E390–E396. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Eriksson L, Saxelin R, Röhl S, Roy J,
Caidahl K, Nyström T, Hedin U and Razuvaev A: Glucagon like
peptide-1 receptor activation does not affect re-endothelization
but reduces intimal hyperplasia via direct effects on smooth muscle
cells in a nondiabetic model of arterial injury. J Vasc Res.
52:41–52. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ge GH, Dou HJ, Yang SS, Ma JW, Cheng WB,
Qiao ZY, Hou YM and Fang WY: Glucagon-like peptide-1 protects
against cardiac microvascular endothelial cells injured by high
glucose. Asian Pac J Trop Med. 8:73–78. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang D, Luo P, Wang Y, Li W, Wang C, Sun
D, Zhang R, Su T, Ma X, Zeng C, et al: Glucagon-like peptide-1
protects against cardiac microvascular injury in diabetes via a
cAMP/PKA/Rho-dependent mechanism. Diabetes. 62:1697–1708. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lüscher TF and Barton M: Biology of the
endothelium. Clin Cardiol. 20(11 Suppl 2): II3–II10. 1997.
|
|
27
|
Forbes JM and Cooper ME: Mechanisms of
diabetic complications. Physiol Rev. 93:137–188. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Deanfield JE, Halcox JP and Rabelink TJ:
Endothelial function and dysfunction: Testing and clinical
relevance. Circulation. 115:1285–1295. 2007.PubMed/NCBI
|
|
29
|
Belz GG and Mohr-Kahaly S: Cacoa and dark
chocolate in cardiovascular prevention? Dtsch Med Wochenschr.
136:2657–2663. 2011.(In German). PubMed/NCBI
|