Introduction
Lung ischemia-reperfusion injury (LIRI), a form of
acute sterile lung injury, remains a frequent complication that may
result in morbidity and mortality during lung transplantation
(1), cardiopulmonary bypass
(2), trauma (3), pulmonary embolism (4) and resuscitation in hemorrhagic shock
(5). Previous studies have
demonstrated that dexmedetomidine may protect against lung injury
by promoting the expression of heme oxygenase-1 (6). Pre-administration of dexmedetomidine
prior to ischemia-reperfusion (IR) has been identified to reduce
pulmonary damage and decrease myeloperoxidase activation in lung
tissue, in addition to levels of proinflammatory cytokines in the
bronchoalveolar lavage fluid (7).
The results of this previous study indicated that dexmedetomidine
exerted these effects through the toll-like receptor 4/myeloid
differentiation primary response 88/mitogen-activated protein
kinase signaling pathway (7).
Autophagy is a cell protective mechanism that is
activated in response to stress signals from the endoplasmic
reticulum (8). Autophagy occurs from
the embryonic period (9) and is
fundamental for the growth and differentiation of tissues.
Autophagy is involved in the growth and differentiation of alveolar
cells (10,11). Depletion of energy and an oxygen
deficient environment are triggers for autophagy. It is well known
that autophagic cellular damage during ischemia and reperfusion is
the basis of ischemia-reperfusion injury (IRI). This is an attempt
to survive the severely limiting conditions during ischemia.
Previous studies have investigated the role of autophagy in IRI.
However, it remains controversial whether high levels of autophagy
lessen or aggravate IRI (12,13).
Autophagy occurs at low levels basally to mediate homeostatic
functions, including organelle and protein turnover. Furthermore,
autophagy increases when intracellular nutrition and energy are
deficient; for example, in hypoxia or starvation. Autophagy can
also trigger apoptosis (14).
A previous study demonstrated that ischemia and
immediate reperfusion can trigger autophagy, and that autophagy
participates in renal IRI (12).
Furthermore, autophagy serves a protective role in LIRI (15). However, in a previous study indicated
that autophagy was activated and served a role in the
pathophysiological process of LIRI (16). Whether autophagy is associated with
the protective effect of dexmedetomidine in LIRI remains unclear.
The present study aimed to investigate the underlying molecular
mechanism of the protective effect of dexmedetomidine in LIRI.
Materials and methods
Animals
The present study was approved by the Animal Care
and Use Committee of Henan Provincial People's Hospital (Zhengzhou,
China). Adult (aged 8–10 weeks) male Sprague-Dawley rats (n=48;
weight, 250–300 g) were provided by Zhengzhou University Laboratory
Animal Center (Zhengzhou, China). The animals were housed in
plastic cages with water and food available ad libitum. The
temperature (22±1°C) and humidity (60±5%) were controlled in a room
with a 12-h light-dark cycle. All the rats were acclimatized to
these conditions for 1 week prior to the study.
Establishment of LIRI models
All rats were anesthetized by intraperitoneal
injection of 400 mg/kg 10% chloral hydrate (Batch no. 20150121;
Tianjin Guangfu Fine Chemical Research Institution, Tianjin,
China), and the caudal vein was cannulated for fluid and drug
administration. After tracheostomy was completed, all rats were
endotracheally intubated with a vein puncture needle and then
connected to a breathing machine (Model: HX-100E; Temo Technology
Co., Ltd, Beijing, China) specialized for small animals. Rats were
placed in a right lateral position and the left lung was exposed
through the fifth intercostal space. The hilum of the left lung was
occluded with a non-invasive microvascular clip for 30 min. The
clip was removed at the end of the ischemic period, and the left
lung was allowed to regain ventilation and blood for 2 h. After the
experiment was finished, the rats were sacrificed via
exsanguination by cardiac puncture and the left lung was removed
for further analysis.
Experimental protocol and drug
administration
A total of 48 rats were randomly allocated into six
groups (n=8 per group) as follows: i) The sham group received
saline administration (1 ml/h intravenously) without LIRI; ii) the
IR group received saline administration (1 ml/h intravenously)
following LIRI; iii) the LIRI + 1 µg/kg dexmedetomidine
preconditioning group [pre-low dose (LD)] received 1 µg/kg
dexmedetomidine prior to LIRI; iv) the LIRI + 10 µg/kg
dexmedetomidine preconditioning group [pre-high dose (HD)] received
10 µg/kg dexmedetomidine prior to LIRI; v) the IR + 1 µg/kg
dexmedetomidine postconditioning group (post-LD) received 1 µg/kg
dexmedetomidine following LIRI; and vi) the IR +1 0 µg/kg
dexmedetomidine postconditioning group (post-HD) received 10 µg/kg
dexmedetomidine following LIR.
Drug administration
Dexmedetomidine (Batch no. 15030932; Hengrui
Medicine Co., Ltd., Lianyungang, China) preconditioning [0.1
(pre-LD) or 1 (pre-HD) µg/kg/min total over 10 min] was
administered intravenously after the caudal vein was opened.
Dexmedetomidine postconditioning [0.1 (post-LD) or 1 (post-HD)
µg/kg/min total over 10 min) was administered immediately after the
reperfusion period started.
Wet/dry (W/D) lung weight ratio
The W/D weight ratio for 3 rats from each group was
calculated. The upper lobe of the left lung was immediately weighed
following harvest to obtain the wet weight and again following
desiccation in a 60°C oven for 48 h to obtain the dry weight. The
lung water content was then assessed by the W/D ratio.
Examination of lung injury by light
microscopy
The left lungs from 2 rats from each group were
perfused with 4% formaldehyde (room temperature for 24 h). The
formaldehyde infused left lung samples were embedded in paraffin
wax (58°C for 2 h), sectioned (5 µm thick) and stained with
hematoxylin and eosin. The samples were subsequently examined using
light microscopy at a magnification of ×200 and the mean value of
three fields of view was used as to assess the lung injury.
Examination of lung injury by
transmission electron microscopy
The left lungs from 2 rats from each group were
perfused with 2.5% glutaraldehyde solution (precooled at 4°C) and
preserved. Following fixation at 4°C for 24 h with 2.5%
glutaraldehyde, the samples were treated with 1/15 M phosphate
buffer for 10–15 min, followed by 50, 70, 80, 70 and 100% acetone
dehydration washes for 10–15 min each (at room temperature).
Tissues were then embedded with EPON 812 resin (at 60°C for 48 h),
sectioned into 50-nm-thick ultrathin slices, stained with citric
acid and uranyl acetate (room temperature for 30 min) and images
captured by transmission electron microscopy (H-7500; Hitachi,
Tokyo, Japan). The mean value of three fields of view were used to
assess the lung injury.
Superoxide dismutase (SOD) activity
and malondialdehyde (MDA) concentration
An ultraviolet spectrophotometer was used to perform
a colorimetric determination at a wavelength of 550 nm. Lipid
peroxidation was determined by measuring the rate of production of
thiobarbituric acid reactive substances (expressed as MDA
equivalents) Lung tissues were added to precooled PBS (4°C) and
then centrifuged (1,006.2 × g at 4°C for 15 sec) into tissue
homogenates by a low-speed homogenate slurry machine (30 sec per
time, interval ice bath for 1 min). Following centrifugation by the
aforementioned methods, the tissue homogenates were first placed
under a 37°C water bath for 40 min and then placed at room
temperature for 10 min. The activity of SOD and the concentration
of MDA were determined by a 550 nm colorimetric assay according to
the manufacturer's protocol (SOD cat no: A001-1 and MDA cat no:
A003-1; Nanjing Jiancheng Bioengineering Institute, Nanjing,
China).
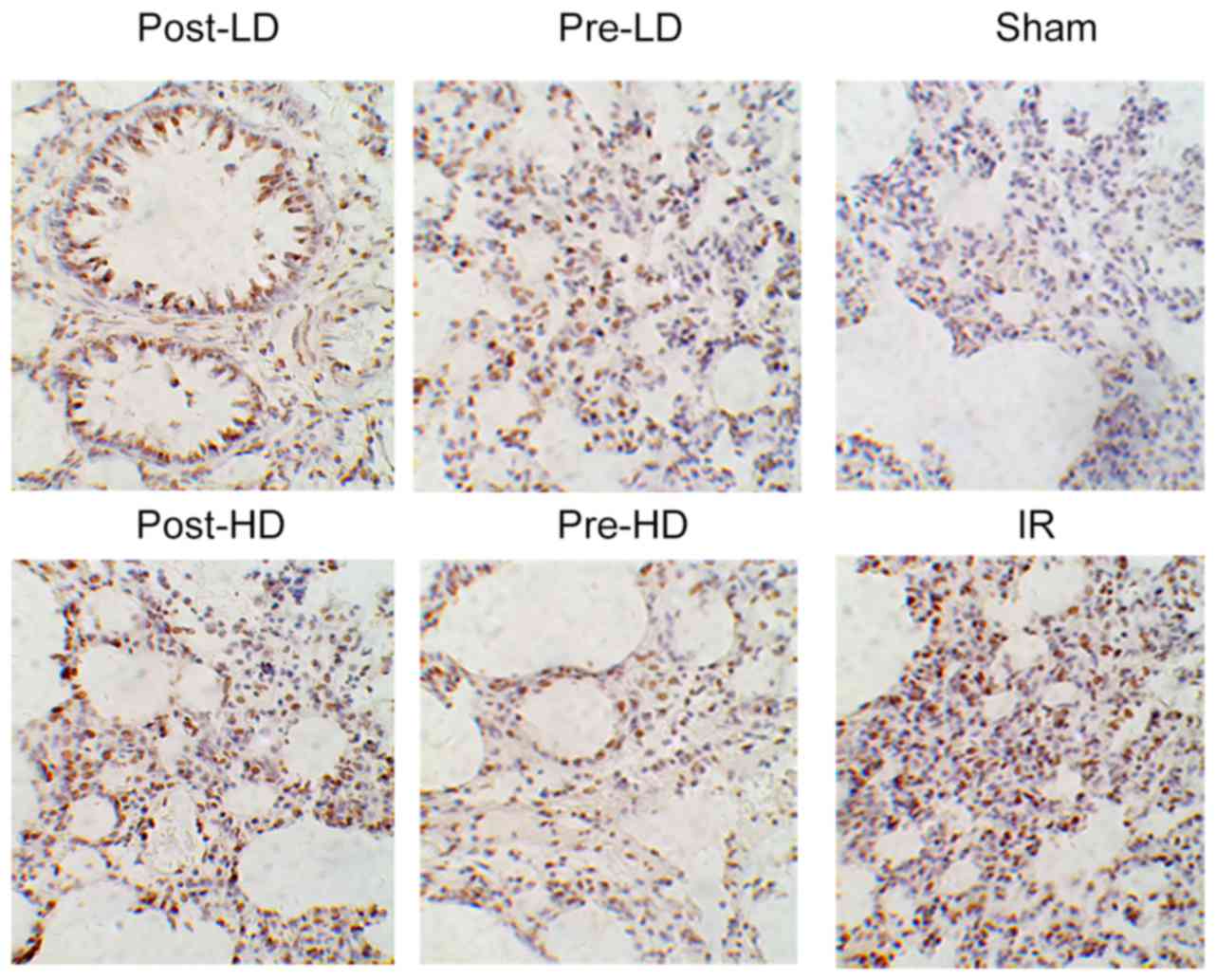
Detection of lung cell apoptosis by
the TUNEL assay
The TUNEL assay was employed according to
manufacturer's protocol of the In situ cell death detection
kit-POD (cat no. 11684817910; Roche, Basel, Switzerland). Apoptotic
cells were indicated by brown-yellow granules in the cytoplasm. The
number of apoptotic cells in five random fields of view
(magnification, ×400) was calculated. The apoptosis index (AI; %)
was expressed as follows: The number of apoptotic cells/ the total
number of cells ×100.
Western blotting
The lower lobes of the left lungs of 4 rats from
each group were used for western blotting (n=24, 4 from each
group). The lung tissues were mixed with RIPA lysis buffer (cat no.
P0013; Beyotime Institute of Biotechnology, Shanghai, China) and
centrifuged at 10,000 × g at 4°C for 10 min. According to the
manufacturer's protocol, protein quantities in the liquid
supernatant were detected using the bicinchoninic acid method
(Enhanced BCA Protein Assay kit; cat no, P0009; Beyotime Institute
of Biotechnology). Then, 12% SDS-PAGE electrophoresis was performed
using 80 ug of protein loaded per lane) and proteins were
transferred onto a nitrocellulose membrane. The membranes were
blocked with 5% skimmed milk in Tris-buffered saline with Tween 20
for 1 h at room temperature, and then incubated overnight at 4°C
with rabbit polyclonal antibodies directed against
hypoxia-inducible factor 1α (HIF-1α; cat no. sc13515; 1:1,000;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA), Bcl-2/adenovirus
E1B 19-kDa interacting protein 3 (BNIP3; cat no. sc56167; 1:1,000;
Santa Cruz Biotechnology, Inc.), BNIP3-like (BNIP3L; cat no.
ab109414; 1:1,000; Abcam, Cambridge, USA) and
microtubule-associated protein 1A/1B light chain 3B (LC3II; cat no.
sc271625; 1:1,000; Santa Cruz Biotechnology Inc.). After washing,
rabbit polyclonal antibodies (secondary antibodies: HRP-labeled
rabbit Anti-Goat IgG (H+L), cat no. ZB-2306, 1:5,000; HRP-labeled
Goat Anti-Mouse IgG (H+L), cat no. ZB-2305, 1:2,500; ZSGB-BIO,
Beijing, China) directed against BNIP3, BNIP3 L, LC3II and HIF-1α
were applied to the membranes for 1 h at room temperature, and
GAPDH rabbit polyclonal antibody (cat no. sc47724; 1:1,200; Santa
Cruz Biotechnology, Inc.) was used as a control. According to the
manufacturer's protocol, Protein bands were then visualized using
enhanced chemiluminescence detection reagents (BeyoECL Plus; cat
no. P0018; Beyotime Institute of Biotechnology), equal volume of
liquid A and B were mixed and once coated to the membrane, they
were exposed to photographic film. The images were then analyzed
and protein expression quantified using Image J2x (version 2.1.4.7)
software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All data are presented as the mean ± standard
deviation. All statistical tests were performed using SPSS software
(version 17.0; SPSS, Inc., Chicago, IL, USA). The statistical
differences between groups were assessed by one-way analysis of
variance followed by Bonferroni correction. P<0.05 was
considered to indicate a statistically significant difference.
Results
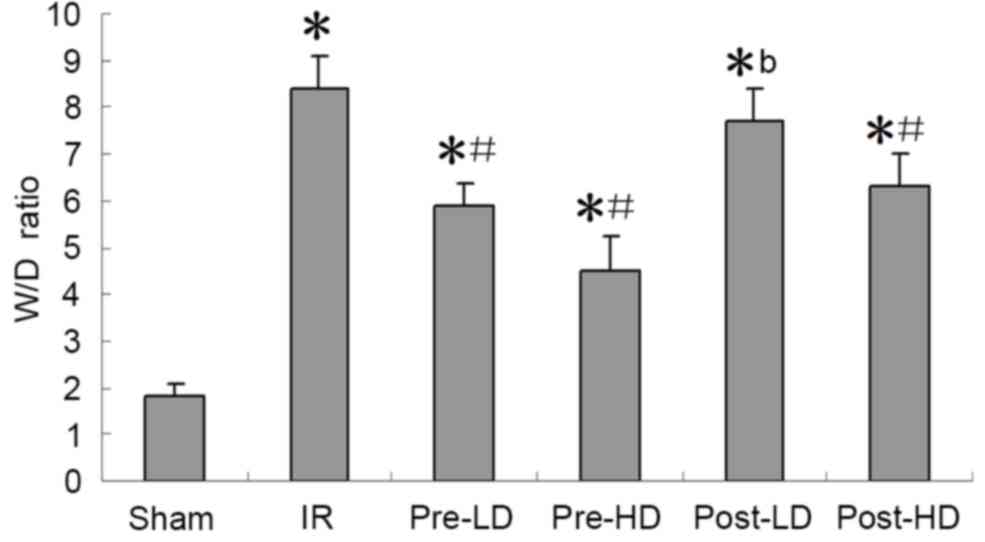
Preconditioning with HD
dexmedetomidine reduces the W/D ratio following LIRI
The W/D ratios of the IR and post-LD groups were
significantly higher compared with the sham group (P<0.05;
Fig. 1). The W/D ratio of the
post-LD group was also significantly higher compared with that of
the pre-HD group (P<0.05; Fig.
1). This indicates that preconditioning with a HD of
dexmedetomidine significantly reduces the W/D ratio following
LIRI.
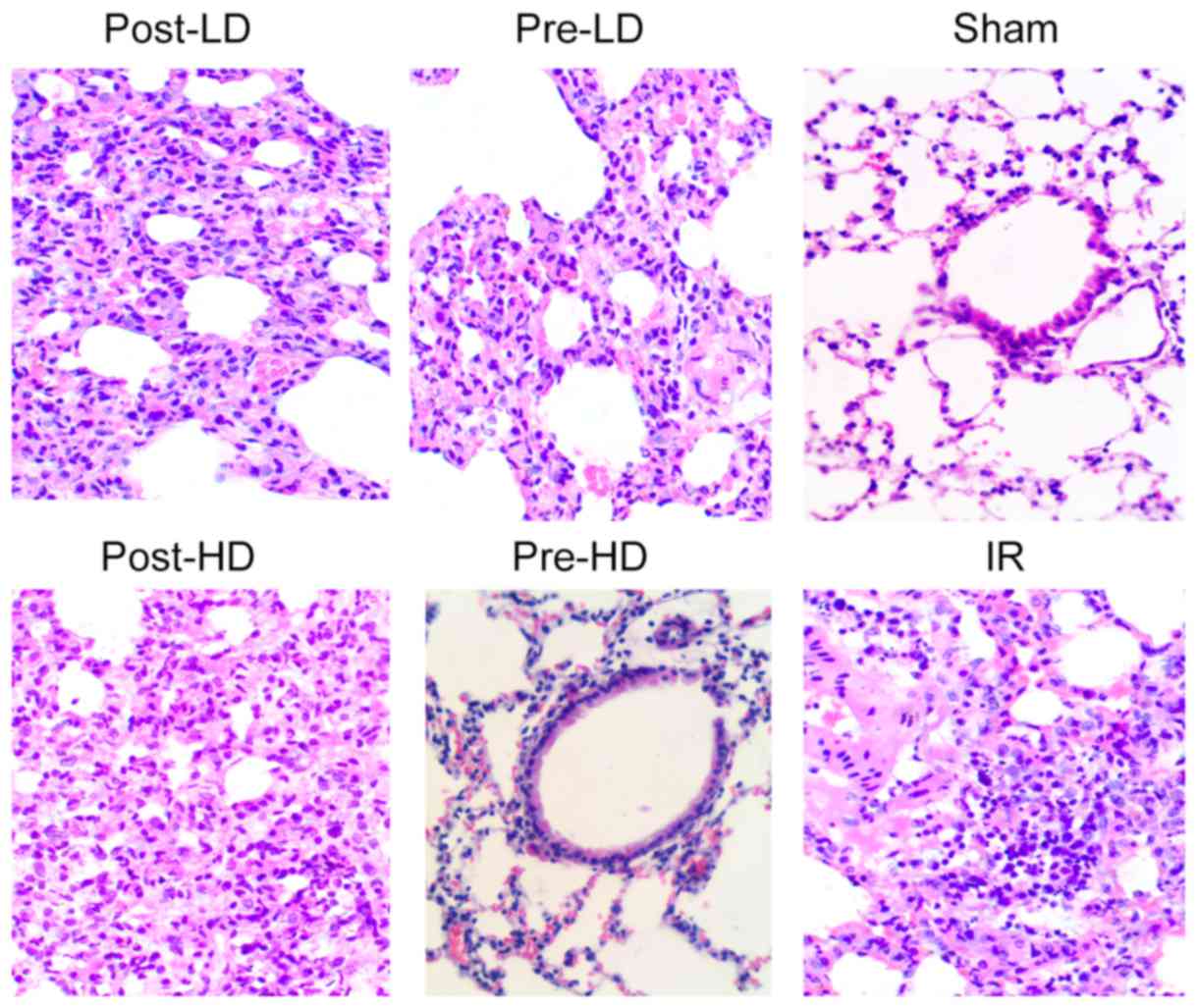
Preconditioning with HD
dexmedetomidine reduces lung injury following LIRI
Lung injury was evaluated by the standards as
follows (light microscopy): Pulmonary interstitial edema, alveolar
edema, alveolar congestion and neutrophil infiltration. As
illustrated in Fig. 2, histological
analysis revealed minimal lung injury in the sham group, while
severe lung injury was identified in the IR group and post-LD
group. Furthermore, no notable difference in lung injury was
revealed between the pre-LD and post-HD groups. By contrast, the
lung tissues harvested from the rats in the pre-HD group exhibited
notably milder injuries compared with the IR, pre-LD, post-LD and
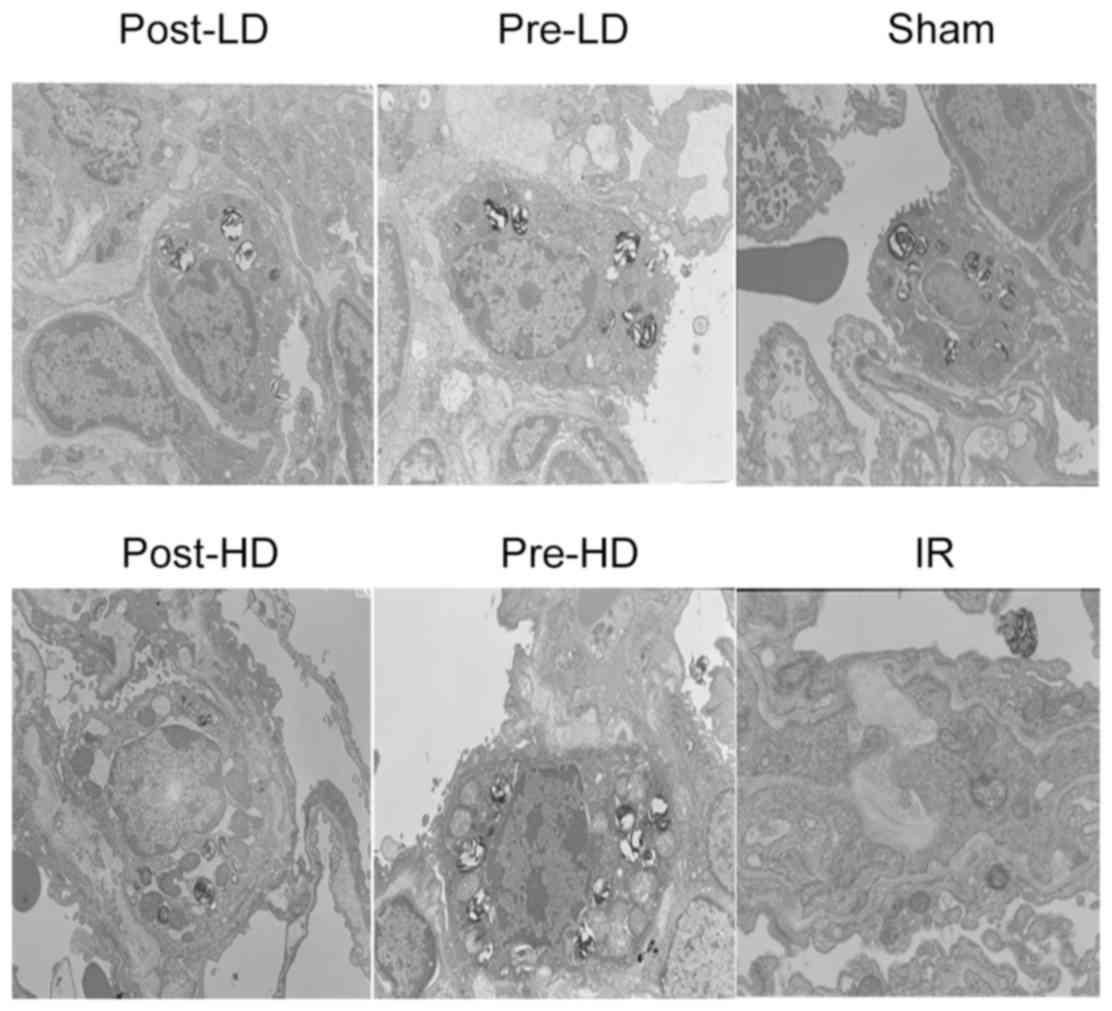
post-HD groups. As presented in Fig.
3, lung injury was assessed through electron microscopy and
similar to the results from light microscopy, no significant injury
was observed in the sham group, but a significant decrease of
alveolar type II epithelium villi and lamellar corpuscles were
observed in the IR group. Preconditioning with dexmedetomidine
attenuated these injuries.
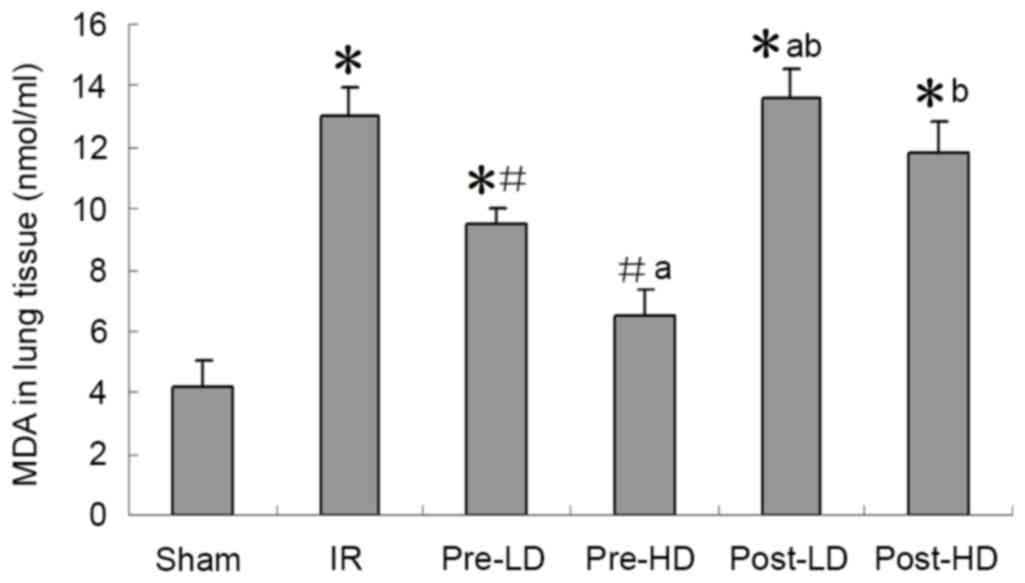
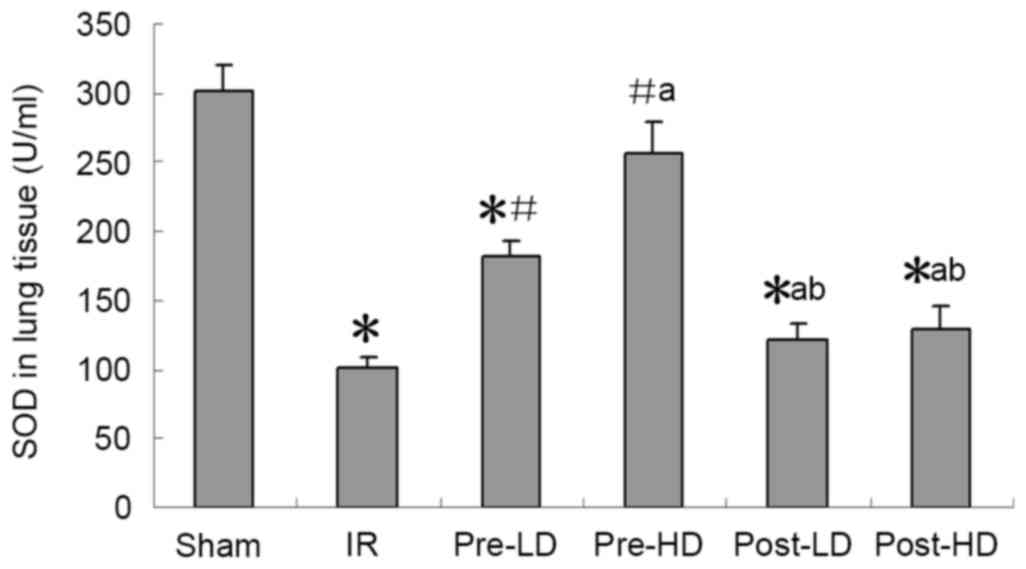
Preconditioning with HD and LD
dexmedetomidine reduces lower MDA and higher SOD following LIRI SOD
activity and MDA expression
As presented in Figs.
4 and 5, a significantly higher
MDA level and significantly lower SOD activity in the left lung
were observed in the IR group compared with the sham group (both
<0.05). Compared with the IR group, higher SOD and lower MDA
were observed in the pre-LD and pre-HD groups (all P<0.05).
However, no significant differences in MDA levels of SOD activity
were observed in the post-LD and post-HD groups compared with the
IR group.
Preconditioning with HD
dexmedetomidine reduces AI following LIRI
As demonstrated in Figs.
6 and 7, compared with the sham
group, the AI index of the other five groups were significantly
increased (P<0.05). However no significant difference in AI was
observed between the IR and post-LD groups. Furthermore, compared
with the IR group, the AI index of the pre-LD, pre-HD and post-HD
groups was significantly lower (all P<0.05). In addition, no
significant difference in AI was observed between the pre-LD and
post-HD groups. However, the AI of the pre-HD group was
significantly lower compared with the pre-LD group, while the
post-LD and post-HD groups exhibited a significant increase in AI
when compared with the pre-HD group (P<0.05; Fig. 6).
Preconditioning with HD
dexmedetomidine reduces the level of BNIP3, BNIP3L and LC3II
As illustrated in Figs.
8 and 9, expression of BNI3P,
BNI3PL and LC3II protein was significantly higher in the IR,
pre-LD, post-LD and post-HD groups compared with the sham group
(all P<0.05). Compared with the IR group, BNIP3, BNIP3 L and
LC3II protein levels were significantly lower in the pre-LD and
pre-HD groups (all P<0.05). Notably, the levels of BNIP3, BNIP3
L and LC3II protein were lower in the pre-HD group compared with
the pre-LD group, although this difference was not statistically
significant. Furthermore, BNIP3, BNIP3 L and LC3II protein levels
were significantly lower in the post-HD group compared with the IR
group (all P<0.05); however, no significant differences were
observed between the pre-LD and post-HD groups.
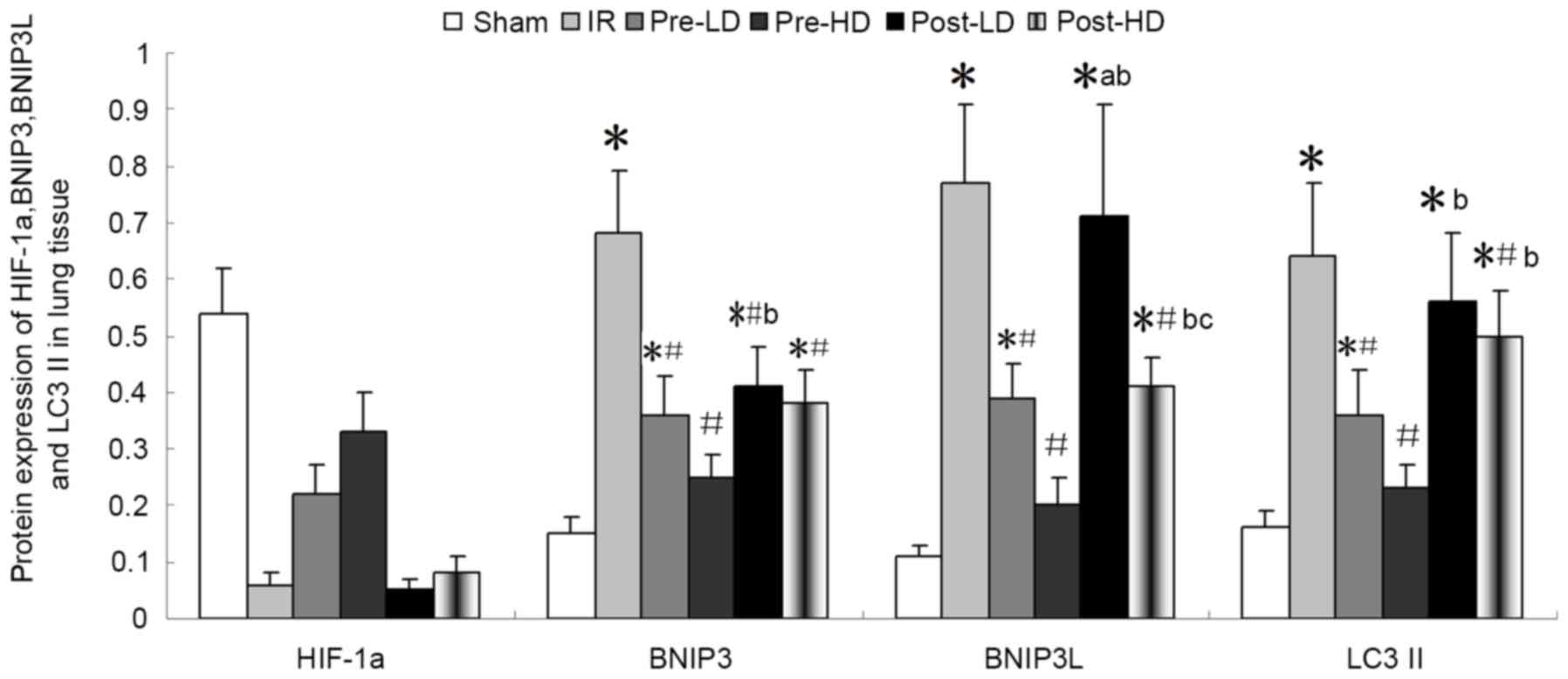 | Figure 8.Quantitative western blot analysis of
BNIP3, BNIP3 L, HIF-1α and LC3II protein expression in lung tissue
following IR injury. *P<0.05 vs. the sham group;
#P<0.05 vs. the IR group; bP<0.05 vs.
the pre-HD group; cP<0.05 vs. post-LD group. BNIP3,
Bcl-2/adenovirus E1B 19-kDa interacting protein 3; BNIP3 L, BNIP3
like; HIF-1α, hypoxia-inducible factor 1α; LC3II,
microtubule-associated protein 1A/1B light chain 3B; IR,
ischemia-reperfusion; pre-LD, low-dose dexmedetomidine
preconditioning; pre-HD, high-dose dexmedetomidine preconditioning;
post-LD, low-dose dexmedetomidine postconditioning; post-HD,
high-dose dexmedetomidine postconditioning. |
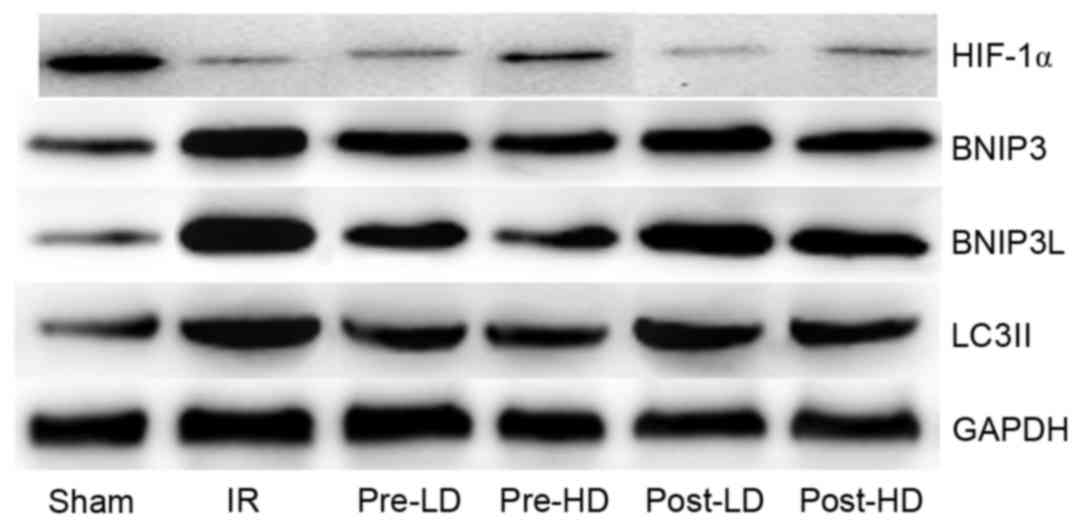 | Figure 9.Western blots of BNIP3, BNIP3 L,
HIF-1α and LC3II. BNIP3, Bcl-2/adenovirus E1B 19-kDa interacting
protein 3; BNIP3 L, BNIP3 like; HIF-1α, hypoxia-inducible factor
1α; LC3II, microtubule-associated protein 1A/1B light chain 3B; IR,
ischemia-reperfusion; pre-LD, low-dose dexmedetomidine
preconditioning; pre-HD, high-dose dexmedetomidine preconditioning;
post-LD, low-dose dexmedetomidine postconditioning; post-HD,
high-dose dexmedetomidine postconditioning. |
Preconditioning with HD
dexmedetomidine reduces the level of HIF-1α
The level of HIF-1α protein was notably lower in the
IR compared with the sham group, although this difference was not
statistically significant (Figs. 8
and 9). In addition, compared with
the IR group, HIF-1α protein levels were higher in the pre-LD and
pre-HD groups. There was not marked differences in HIF-1α levels
between the post-LD, post-HD and IR groups.
Discussion
In the present study, lung damage was evaluated by
determination of the W/D ratio and imaging techniques. High water
content in the lung is a representative symptom of acute lung
injury (17), as demonstrated by the
severe lung damage observed in the IR group. The results of the
present study demonstrated that LD and HD preconditioning with
dexmedetomidine attenuated the development of pulmonary edema, as
indicated by the significant decrease in the lung W/D ratio
compared with the IR group. However, compared with the IR group, a
significance decrease in the lung W/D ratio was observed only in
the post-HD groups and not the post-LD group. These results
indicate that preconditioning with dexmedetomidine can effectively
attenuate the development of pulmonary edema; however, only a HD of
dexmedetomidine could exert a similar effect when applied in a
postconditioning manner. HD dexmedetomidine, applied in a
post-conditioning manner had a similar effect to a LD applied in a
preconditioning manner. However, HD dexmedetomidine attenuated the
development pulmonary edema more effectively when applied in a
preconditioning manner compared with a postconditioning manner.
Lung damage observed by light and transmission
electron microscopy was consistent with the results of the W/D
ratio. The results indicated that preconditioning with
dexmedetomidine produces protection against LIRI in a
dose-dependent manner. A previous study demonstrated that the
injuries induced by intestinal IR in rats improved with the
administration of 5 µg/kg/h dexmedetomidine compared with 2.5
µg/kg/h dexmedetomidine prior to ischemia (18). Furthermore, another study
demonstrated dexmedetomidine preconditioning, but not
postconditioning or treatment with the peripheral α2-adrenergic
receptor agonist fadolmidine, ameliorated kidney IRI and
inflammatory responses (19). The
results of the present study indicate that HD dexmedetomidine
postconditioning provides slight protection to the lungs, similar
to LD dexmedetomidine applied in a preconditioning manner.
Reperfusion injury is directly associated with the
formation of reactive oxygen species (ROS), endothelial cell
injury, increased vascular permeability, and the activation and
production of neutrophils, platelets, cytokines and the complement
system (20). Furthermore, ROS serve
a key role in the development of pulmonary injury, which is
characterized by an increase in ROS and other free radicals, with
an essential role in the sequence of events leading to lung failure
(21). The importance of ROS in the
pathophysiology of IRI was demonstrated by the injection of free
radical scavengers or enzymes, including SOD, catalase and
glutathione peroxidase, which was identified to prevent the damage
that occurs during reperfusion (22,23). In
the present study, SOD activity in the lungs after IRI was
significantly higher in the pre-LD and pre-HD groups compared with
the IR group, indicating that pre-administration of dexmedetomidine
effectively attenuates oxidative stress injuries induced by IR.
Oxidative stress and lipid peroxidation serve an important role in
distant organ damage following IR. Lipid peroxidation was
determined by measuring the rate of production of thiobarbituric
acid reactive substances (expressed as MDA equivalents) (24). In the present study, MDA levels were
decreased by preconditioning with dexmedetomidine; however,
postconditioning with dexmedetomine did not have a protective
effect. The results of the present -study suggest that
dexmedetomidine alleviates oxidative stress injury induced by IR,
but only in a preconditioning manner.
Occlusion of the arterial blood supply is caused by
an embolus, resulting in ischemia and consequently a serious
imbalance between the supply and metabolic demand for oxygen,
causing tissue hypoxia. The HIF-1 transcription factor, which is
composed of oxygen-labile HIF-1α and the constitutive HIF-1β
subunit, is responsible for the response to hypoxia or ischemia.
The activation of HIF-1α serves a protective role in the IRI of
several tissues through regulating inflammation (25). HIF-1α contributes to pulmonary
vascular dysfunction in LIRI and the HIF-1α stabilizer
dimethyloxalylglycine can attenuate IR-induced inhibition of
endothelial function (26). In the
present study, postconditioning with dexmedetomidine did not
upregulate the expression of HIF-1α, while a marked upregulation of
HIF-1α was observed in the pre-LD and pre-HD groups.
Autophagy is a highly conserved process that occurs
in all eukaryotes. The functional association between autophagy and
cellular death or survival is complicated. Autophagy promotes cell
survival under conditions of stress and starvation, whereas in
other situations autophagic cell death may occur. Previous studies
(27–29) have demonstrated that autophagy can be
induced by various cellular conditions in IRI, including energy
starvation, oxidative stress, endoplasmic reticulum stress and
inflammation. Furthermore, the activation of autophagy may be a
important process that enhances tissue tolerance to ischemia
(30). The renoprotective effects of
dexmedetomidine have been identified to be mediated partly by the
maintenance of autophagy (21). It
appears that autophagy serves a protective role in LIRI and that
this protective effect is enhanced with increasing autophagy levels
(31). However, it remains
controversial whether high levels of autophagy lessen or increase
IRI, particularly in lung tissue (16). As demonstrated in the present study,
the expression of LC3II, a biomarker of autophagy, was upregulated
by IR. Furthermore, preconditioning with dexmedetomidine can reduce
autophagy levels; LC3II levels were significantly reduced by
preconditioning with dexmedetomidine, particularly with HD
dexmedetomidine.
Autophagy and apoptosis determine cellular fate.
Autophagy and apoptosis are discrete cellular processes that are
mediated by distinct groups of regulatory and executioner molecules
(32,33). The underlying mechanisms of the
association between autophagy and apoptosis are not yet fully
defined; however, recent investigations have revealed that several
apoptotic proteins modulate autophagy (34,35). The
present study demonstrated that IR-induced apoptosis could be
reduced by dexmedetomidine, particularly when administered in a
preconditioning manner.
Bcl-2/adenovirus E1B 19-kDa interacting protein 3
(BNIP3) and Bcl-2/adenovirus E1B 19-kDa interacting protein 3 like
(BNIP3L), also known as Nix, are members of the BH3-only subfamily
of Bcl-2 family proteins, and are critical for the induction of
mitochondrial autophagy, in some instances, promotes survival by
reducing radical oxygen species (ROS) and DNA damage under hypoxic
conditions (36,37). BNIP3 can also induce cell death;
however, evidence is not conclusive whether increased BNIP3
expression alone is sufficient for an apoptotic response (37). However, a previous study indicated
that (38) autophagic cell death is
induced in cancer cell lines with functioning apoptotic mechanisms,
hypothesizing that early induction of BNIP3 may be a protective
response with prolonged induction leading to cell death.
Recent studies have identified that BNIP3 and
BNIP3L, target genes of HIF-1α, are important in autophagy
(39–40). BNIP3 serves a role in the induction
of hypoxia-induced mitochondrial autophagy (39,41,42).
Autophagy can cause cell survival or death, and its role in IRI
appears to be model- and/or tissue-dependent (16,21). The
present study demonstrated that BNIP3 and BNIP3 L were
overexpressed following LIRI, and that preconditioning with
dexmedetomidine effectively attenuated IRI by downregulating BNIP3
and BNIP3L. BNIP3 and BNIP3L are known to be target genes of
HIF1-α; however, in the present study preconditioning with
dexmedetomidine upregulated the expression of HIF1-α. The trend was
not consistent with the downregulation of BNIP3 and BNIP3L that was
observed following dexmedetomidine preconditioning, thus the exact
mechanism underlying this effect remains unclear.
In conclusion, the present study demonstrated that
pulmonary IRI was associated with the upregulation of apoptosis and
autophagy. Furthermore, preconditioning with dexmedetomidine
provided protection against LIRI in a dose-dependent manner,
partially through inhibiting autophagy. Dexmedetomidine was
observed to upregulate HIF-1α and downregulate BNIP3 and BNIP3 L in
the LIRI model in the current study. The results of present study
highlight a potential clinical application for dexmedetomidine in
reducing LIRI.
Acknowledgements
The present study was supported by the National
Natural Science Fund (grant no. U1404807) and Medical Science
Research Project of Henan Province (grant no. 201602227).
References
|
1
|
Christie JD, Carby M, Bag R, Corris P,
Hertz M and Weill D; ISHLT Working Group on Primary Lung Graft
Dysfunction, : Report of the ISHLT Working Group on Primary Lung
Graft Dysfunction part II: Definition. A consensus statement of the
International Society for Heart and Lung Transplantation. J Heart
Lung Transplant. 24:1454–1459. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ng CS, Wan S, Yim AP and Arifi AA:
Pulmonary dysfunction after cardiac surgery. Chest. 121:1269–1277.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shimamoto A, Pohlman TH, Shomura S,
Tarukawa T, Takao M and Shimpo H: Toll-like receptor 4 mediates
lung ischemia-reperfusion injury. Ann Thorac Surg. 82:2017–2023.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ambrosio G and Tritto I: Reperfusion
injury: Experimental evidence and clinical implications. Am Heart
J. 138:S69–S75. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reino DC, Pisarenko V, Palange D, Doucet
D, Bonitz RP, Lu Q, Colorado I, Sheth SU, Chandler B, Kannan KB, et
al: Trauma Hemorrhagic Shock-Induced Lung Injury Involves a
Gut-Lymph-Induced TLR4 Pathway in Mice. PLoS One. 6:e148292011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jiang L, Li L, Shen J, Qi Z and Guo L:
Effect of dexmedetomidine on lung ischemia-reperfusion injury. Mol
Med Rep. 9:419–426. 2014.PubMed/NCBI
|
|
7
|
Shen J and Fu G, Jiang L, Xu J, Li L and
Fu G: Effect of dexmedetomidine pretreatment on lung injury
following intestinal ischemia-reperfusion. Exp Ther Med.
6:1359–1364. 2013.PubMed/NCBI
|
|
8
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tra T, Gong L, Kao LP, Li XL, Grandela C,
Devenish RJ, Wolvetang E and Prescott M: Autophagy in human
embryonic stem cells. PLoS One. 6:e274852011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kuma A, Hatano M, Matsui M, Yamamoto A,
Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T and Mizushima N: The
role of autophagy during the early neonatal starvation period.
Nature. 432:1032–1036. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Komatsu M, Waguri S, Ueno T, Iwata J,
Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et
al: Impairment of starvation-induced and constitutive autophagy in
Atg7-deficient mice. J Cell Biol. 169:425–434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Suzuki C, Isaka Y, Takabatake Y, Tanaka H,
Koike M, Shibata M, Uchiyama Y, Takahara S and Imai E:
Participation of autophagy in renal ischemia/reperfusion injury.
Biochem Biophys Res Commun. 368:100–106. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang M, Liu K, Luo J and Dong Z:
Autophagy is a renoprotective mechanism during in vitro hypoxia and
in vivo ischemia-reperfusion injury. Am J Pathol. 176:1181–1192.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gump JM, Staskiewicz L, Morgan MJ, Bamberg
A, Riches DW and Thorburn A: Autophagy variation within a cell
population determines cell fate through selective degradationof
Fap-1. Nat Cell Biol. 16:47–54. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang D, Li C, Zhou J, Song Y, Fang X, Ou
J, Li J and Bai C: Autophagy protects against
ischemia/reperfusion-induced lung injury through alleviating
blood-air barrier damage. J Heart Lung Transplant. 34:746–755.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang J, Wang JS, Zheng ZK, Tang J, Fan K,
Guo H and Wang JJ: Participation of autophagy in lung
ischemia-reperfusion injury in vivo. J Surg Res. 182:e79–e87. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Luce JM: Acute lung injury and the acute
respiratory distress syndrome. Crit Care Med. 26:369–376. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang XY, Liu ZM, Wen SH, Li YS, Li Y, Yao
X, Huang WQ and Liu KX: Dexmedetomidine administration before, but
not after, ischemia attenuates intestinal injury induced by
intestinal ischemia-reperfusion in rats. Anesthesiology.
116:1035–1046. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lempiäinen J, Finchenberg P, Mervaala EE,
Storvik M, Kaivola J, Lindstedt K, Levijoki J and Mervaala EM:
Dexmedetomidine preconditioning ameliorates kidney
ischemia-reperfusion injury. Pharmacol Res Perspect. 2:e000452014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
de Perrot M, Liu M, Waddell TK and
Keshavjee S: Ischemia-reperfusion-induced lung injury. Am J Respir
Crit Care Med. 167:490–511. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zimmerman BJ and Granger DN: Mechanisms of
reperfusion injury. Am J Med Sci. 307:284–292. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
De Greef KE, Ysebaert DK, Ghielli M,
Vercauteren S, Nouwen EJ, Eyskens EJ and De Broe ME: Neutrophilsand
acute ischemia-reperfusion injury. J Nephrol. 11:110–122.
1998.PubMed/NCBI
|
|
23
|
Oredsson S, Plate G and Qvarfordt P:
Experimental evaluation of oxygen free radical scavengers in the
prevention of reperfusion injury in skeletal muscle. Eur J Surg.
160:97–103. 1994.PubMed/NCBI
|
|
24
|
Deby C and Goutier R: New perspectives on
the biochemistry of superoxide anion and the efficiency of
superoxide dismutases. Biochem Pharmacol. 39:399–405. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Feinman R, Deitch EA, Watkins AC, Abungu
B, Colorado I, Kannan KB, Sheth SU, Caputo FJ, Lu Q, Ramanathan M,
et al: HIF-1 mediates pathogenic inflammatory responses to
intestinal ischemia-reperfusion injury. Am J Physiol Gastrointest
Liver Physiol. 299:G833–G843. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao X, Jin Y, Li H, Wang Z, Zhang W and
Feng C: Hypoxia-inducible factor 1 alpha contributes to pulmonary
vascular dysfunction in lung ischemia-reperfusion injury. Int J
Clin Exp Pathol. 7:3081–3088. 2014.PubMed/NCBI
|
|
27
|
Scherz-Shouval R, Shvets E, Fass E, Shorer
H, Gil L and Elazar Z: Reactive oxygen species are essential for
autophagy and specifically regulate the activity of Atg4. EMBOJ.
26:1749–1760. 2007. View Article : Google Scholar
|
|
28
|
Høyer-Hansen M, Bastholm L, Szyniarowski
P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N,
Elling F, Rizzuto R, et al: Control of macroautophagy by calcium,
calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell.
25:193–205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang J, Wang JS, Zheng ZK, Tang J, Fan K,
Guo H and Wang JJ: Participation of autophagy in lung
ischemia-reperfusion injury in vivo. J Surg Res. 182:e79–e87. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Matsui Y, Kyoi S, Takagi H, Hsu CP,
Hariharan N, Ago T, Vatner SF and Sadoshima J: Molecular mechanisms
and physiological significance of autophagy during myocardial
ischemia and reperfusion. Autophagy. 4:409–415. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang D, Li C, Zhou J, Song Y, Fang X, Ou
J, Li J and Bai C: Autophagy protects against
ischemia/reperfusion-induced lung injury through alleviating
blood-air barrier damage. J Heart Lung Transplant. 34:746–755.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Selfeating and self-killing: Crosstalk between autophagy
and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Feng Z, Zhang H, Levine AJ and Jin S: The
coordinate regulation of the p53 and mTOR pathways in cells. Proc
Natl Acad Sci USA. 102:pp. 8204–8209. 2005; View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim SY, Song X, Zhang L, Bartlett DL and
Lee YJ: Role of Bcl-xL/Beclin-1 in interplay between apoptosis and
autophagy in oxaliplatin and bortezomib-induced cell death. Biochem
Pharmacol. 88:178–188. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hamacher-Brady A, Brady NR, Logue SE,
Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA and Gustafsson AB:
Response to myocardial ischemia/reperfusion injury involves Bnip3
and autophagy. Cell Death Differ. 14:146–157. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tracy K, Dibling BC, Spike BT, Knabb JR,
Schumacker P and Macleod KF: BNIP3 is an RB/E2F target gene
required for hypoxia-induced autophagy. Mol Cell Biol.
27:6229–6242. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Azad MB, Chen Y, Henson ES, Cizeau J,
McMillan-Ward E, Israels SJ and Gibson SB: Hypoxia induces
autophagic cell death in apoptosis-competent cells through a
mechanism involving BNIP3. Autophagy. 4:195–204. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang H, Bosch-Marcc M, Shimoda LA, Tan
YS, Baek JH, Wesley JB, Gonzalez FJ and Semenza GL: Mitochondrial
autophagy is an HIF-1-dependent adaptive metabolic response to
hypoxia. J Biol Chem. 283:10892–10903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Novak I, Kirkin V, McEwan DG, Zhang J,
Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, et
al: Nix is selective autophagy receptor for mitochondrial
clearance. EMBO Rep. 11:45–51. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Band M, Joel A, Hernandez A and Avivi A:
Hypoxia-induced BNIP3 expression and mitophagy: In vivo comparision
of the rat and the hypoxia-tolerant mole, Spalax ehrenbergi. FASEB
J. 23:2327–2335. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bellot G, Garcia-Medina R, Gounon P,
Chiche J, Roux D, Pouysségur J and Mazure NM: Hypoxia-induced
autophagy is mediated through hypoxia-inducible factor induction of
BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol.
29:2570–2581. 2009. View Article : Google Scholar : PubMed/NCBI
|