Introduction
Neuropathic pain is defined as pain initiated or
caused by a primary lesion or dysfunction in the nervous system,
which often presents as spontaneous pain and produces a sense of
pain similar to that generated by noxious stimuli, including
mechanical and thermal stresses (1).
Neuropathic pain seriously impedes patient quality of life. The
electrophysiological basis of neuropathic pain is related to an
increase in the expression of Na+ and voltage-gated
Ca2+ channels on the neuronal membrane of injured nerve
sites, and the release of an excitatory neurotransmitter, which
changes the normal physiological activity of neurons and aggravates
neural responses to non-injurious and minor injurious peripheral
stimulation (2,3). A large number of neurons spontaneously
discharge and release ectopic impulses to the spinal neurons, which
increases the sensitivity of spinal neurons and transmission
between the synapses and neurotransmitters, thus increasing spinal
excitability and causing abnormalities in sensory function
(4).
γ-aminobutyric acid (GABA) in the central nervous
system serves an important role in the processing of nociceptive
information and the modulation of pain, and is the main inhibitory
neurotransmitter (5). It has
previously been demonstrated that GABA type A receptors
(GABAARs) are involved in the transmission of pain
(5,6). GABAARs are ligand-gated
chloride ion channel receptors (5,6). A
previous study by our group demonstrated that calcium-activated
chloride channels (CaCCs) serve a critical role in the generation
of GABA-induced inward currents in the dorsal root ganglions (DRGs)
of rats (7). DRGs connect the
peripheral and central nervous systems, and GABA and
GABAAR are present in the DRG neurons of humans and
animals (8). GABAA
receptors in the DRG serve an important role in alleviating the
symptoms of neuropathic pain (9,10).
Dorsal root reflexes were first systematically
investigated in the 1930s (11,12), and
were found to be triggered by primary afferent depolarization
(PAD). The stimulation of primary afferent fibers produces PAD,
which is blocked by GABAA antagonists such as picrotoxin
and bicuculline, and also by antagonists of non-N-Methyl-D-aspartic
acid (NMDA) glutamate receptors (13,14). The
most likely explanation for this is that primary afferent fibers
release excitatory amino acids, which then activate non-NMDA
glutamate receptors on GABAergic interneurons, causing them to
release GABA at axoaxonic or dendroaxonic synapses on primary
afferent terminals (13). Possible
reasons for an increase in dorsal root reflexes during inflammation
include an upregulation of the GABA system in the dorsal horn via
the action of signal transduction pathways (14). However, the superimposition of an
excitatory mechanism (dorsal root reflexes) onto an inhibitory one
(presynaptic inhibition due to PAD) may have negative effects
(11). Instead of inhibition,
hyperalgesia and allodynia may result from the central effects of
dorsal root reflexes, and neurogenic inflammation may result from
the peripheral effects (11).
Niflumic acid (NFA) is a type of non-steroidal
anti-inflammatory drug (NSAID). NSAIDs are the most widely used
pharmacological agents and exhibit a demulcent effect, principally
by reducing the synthesis of prostaglandins via inhibition of
cyclooxygenase at sites of pain and inflammation (15). NFA, a GABAAR antagonist,
is the only chloride ion channel blocker able to protect cells from
excitotoxicity (16). It has been
suggested that NSAIDs modulate GABAAR function in
heterologous expression systems (17). Chronic constriction injury (CCI)
models were first used in 1988 and are now widely used in the study
of neuropathic pain (18). The
surgical techniques used to establish CCI models have many
advantages, including a simple surgical procedure and small wound
size, and the signs and symptoms of stable spontaneous pain, heat
hyperalgesia and mechanical allodynia are similar to those observed
clinically (18). In a previous
study, the current authors demonstrated that NAF could inhibit
GABA-induced current of dorsal root ganglion neurons in both
control and CCI, but the mechanism was still unclear (19). The aim of the present study was to
investigate the effects of NFA on GABA-induced currents in the
dorsal root ganglion neurons of rats with neuropathic pain, and to
evaluate the analgesic mechanism of NFA.
Materials and methods
Materials
A total of 40 healthy male Sprague-Dawley rats aged
8–10 weeks and weighing 250–280 g, were provided by the
Experimental Animal Center of Xinjiang Medical University (Urumqi,
China). Rats were housed in separate cages in a specific
pathogen-free level barrier environment at 24±3°C, relative
humidity of 40–70% and a 12-h light-dark cycle, and were provided
with free access to food and water. The study was approved by the
ethics committee of The First Affiliated Hospital of Shihezi
University Medical College (Shihezi, China).
Establishment of the rat CCI
models
A total of 40 rats were randomly divided into three
groups: Control group (n=10), sham operation group (n=10) and CCI
group (n=20). Rats were anesthetized with an intraperitoneal
injection of 10% (w/v) sodium chloral hydrate [350 mg/kg body
weight, Sangon Biotech (Shanghai) Co., Ltd., Shanghai, China], and
the left sciatic nerve trunk was exposed under sterile conditions.
The skin was longitudinally cut and muscle was dissociated by blunt
dissection to fully expose the sciatic nerve, proximal to the
sciatic trifurcation. Approximately 5 mm of nerve was freed, and 4
tight ligatures of chrome catgut were placed around the sciatic
nerve with ~1 mm spacing. The tightness of ligatures was such that
the blood supply to the epineurium was not affected. Rats in thee
pseudo-operated groups underwent the same procedure but were not
ligated. Rats were fed normally to recover post-surgery. To assess
whether the model had been successfully established, paw withdrawal
latency was assessed, as described previously (20), and in the model group values from the
CCI ipsilateral side were significantly declined by ≥30%
(P<0.05), which indicated modeling success. A total of 10 rats
were randomly selected from each group.
Hot plate testing
To determine whether the CCI model had been
successfully established, paw withdrawal latency was assessed
(20). A Plantar Test (Hargreaves
Apparatus; cat. no. 37370) was purchased from Ugo Basile S.R.L
(Monvalle, Italy). The Plantar Test was used in accordance with the
manufacturer's protocol. Thermal hyperalgesia was measured using an
infrared intensity of 50, and expressed as paw withdrawal latency
of the left hind paw. The mean of three measurements at 4-min
intervals was regarded as the paw withdrawal latency. In order to
prevent tissue damage, the maximum latency was defined as 30 sec,
following which rats were removed from the plantar testing
apparatus. Plantar testing was performed at the following time
points: Pre-surgery and on days 1, 3, 5, 7, 10 and 14 post-surgery.
Following plantar testing at each time point, rats were prepared
for experimentation.
Electrophysiological recordings of
dorsal root ganglion neurons
Rats were anesthetized with an intraperitoneal
injection of 10% (w/v) sodium chloral hydrate (350 mg/kg body
weight) and decapitated, and the cervical, thoracic and lumbar
spine was immediately harvested. The spine was split longitudinally
and placed in an extracellular fluid substitute, the composition of
which was as follows: 150 mmol/l NaCl, 5 mmol/l KCl, 2.5 mmol/l
CaCl2, 1 mmol/l MgCl2, 10 mmol/l HEPES
(H3375; Sigma-Aldrich; Merck KGaA, Darmstadt Germany), 10 mmol/l
D-glucose [A600219-0001; Sangon Biotech (Shanghai) Co., Ltd.], and
NaOH to give a pH of 7.3–7.4 (osmotic pressure=330 mOsm). Ganglia
and the associated nerve roots were individually removed under a
stereomicroscope. The trimmed DRG was cut into pieces using eye
scissors and transferred to an eppendorf (EP) tube that contained
0.25 mg/ml trypsinase (T1426; Sigma-Aldrich; Merck KGaA) and 0.5
mg/ml collagenase (C0130; Sigma-Aldrich; Merck KGaA). The EP tube
was incubated at 37°C for 12 min, then mixed with 0.1 mg/ml trypsin
inhibitor (T6522; Sigma-Aldrich; Merck KGaA) to cease digestion and
centrifuged at 300 × g for 6 min. The supernatant was discarded and
2–3 ml extracellular fluid substitute was added for patch-clamp
experiments and left to stand for at least 30 min at room
temperature to allow cells to adhere. Whole-cell patch clamp
recordings were performed at room temperature using a whole-cell
patch clamp amplifier. Briefly, currents were recorded from single
dorsal root ganglion neurons in vitro using an Axon 700B
amplifier (Molecular Devices, LLC, Sunnyvale, CA, USA) and the
pCLAMP 10.2 hardware and software (Molecular Devices, LLC).
Microelectrodes (~1 µm diameter) were pulled using a P-97 puller
(Sutter Instrument Co., Novato, CA, USA), and the impedance of each
glass microelectrode was 3–5 MΩ. Microelectrodes were filled with
internal solution containing 140 mmol/l KCl, 1 mmol/l
CaCl2, 2 mmol/l MgCl2, 10 mmol/l HEPES and 11
mmol/l EGTA, and KOH (1 mol/l) to raise the pH to 7.3–7.4.
Separated single cells that had adhered well and exhibited round or
oval morphology, a clear shape and contour, membrane integrity, a
uniform cytoplasm and a diameter of 15–45 µm were selected under an
inverted microscope. The glass microelectrode was moved onto the
cell surface using a micromanipulator (CV-7B; Molecular Devices,
LLC, Sunnyvale, CA, USA). Vacuum suction was applied to form a
high-resistance seal between the electrode and cell membrane, and
the capacitance and series resistance were adjusted to maintain a
voltage of −60 mV, as described previously (21,22).
Pharmacological agents were prepared with sugar-free
extracellular fluid (as described above but without D-glucose). The
drug delivery system comprised a micromanipulator which allowed
rapid changing of drug delivery tubes. The diameter of every drug
delivery tube was 0.5 mm and the distance between nozzle and cell
was 100 µm. GABA (A5835; Sigma-Aldrich; Merck KGaA) (1–1,000
µmol/l) was administered from low to high concentration for 5–6 sec
to establish a ‘front control’, with an intermission of 4 min
during which cells were bathed in extracellular fluid. Bicuculline
(285269; Sigma-Aldrich; Merck KGaA) was administered at 100 µmol/l.
The same concentration of GABA was perfused as the front control.
Prior to administering the mixture of NFA (1, 10 and 100 mmol/l;
N0630-25 G; Sigma-Aldrich; Merck KGaA) and GABA (100 µmol/l), NFA
(1, 10, 100 µmol/l) was pre-perfused for ~20 sec. After the
GABA-activated current (IGABA) was steady and the cells
had been bathed for 4 min, the mixture of GABA and NFA was perfused
for 5 sec, when the IGABA recovered to the level of the
front control, the other concentrations of NFA were perfused. The
inhibitory effects of different concentrations of NFA on
IGABA were recorded and the inhibitory rate of NFA on
IGABA was calculated using the following formula:
(IGABA-Imixture)/IGABAx100
(22,23).
DRG preparation and intracellular
recording
A total of 20 male Wistar rats (age 2–3 weeks;
weight, 200–250 g) were purchased from Xinjiang Medical University.
Rats were housed in cages in a specific pathogen-free level barrier
environment at 24±3°C, relative humidity of 40–70% and a 12-h
light/dark cycle, with free access to food and water. Rats were
anesthetized with an intraperitoneal injection of 10% (w/v) sodium
chloral hydrate (350 mg/kg body weight) and decapitated, and the
cervical, thoracic and lumbar spine was immediately harvested. The
rats subsequently underwent a laminectomy at L4 or
L5. DRGs with attached dorsal roots and spinal nerves
were harvested and the fibrous sheath surrounding the DRG was
removed under the stereoscope. The isolated preparation was
transferred into a recording chamber (0.25 ml volume), and perfused
with oxygenated balanced salt solution (BSS) at room temperature.
The BSS contained 140 mmol/l NaCl, 5 mmol/l KCl, 1 mmol/l
MgCl2, 5 mmol/l glucose and 5 mmol/l Tris-HCl
(RES3098T-B701X; Sigma-Aldrich; Merck KGaA) (pH 7.4). The flow rate
was 3–5 ml/min. The preparation was pinned with small steel pins
(0.5 mm) onto a silicone gum block, which was placed in the
chamber. The sciatic nerve was placed on a pair of platinum
stimulating electrodes in the neighboring compartment, as described
previously (9).
Intracellular recordings were obtained using a glass
microelectrode (1 mm diameter) filled with 2 mol/l KCl and 1 mol/l
potassium acetate, the impedance of each glass microelectrode was
in the range of 25–60 MΩ. Membrane potentials were amplified with a
microelectrode amplifier (MEZ-8301; Nihon Kohden, Tokyo, Japan) and
membrane depolarization was filtered at 20 Hz (as previously
described) (9). Data were recorded
with a pen recorder (cat. no. XWTD-264; Shanghai Instrument Group
Co., Ltd., Shanghai, China). The values acquired for resting
membrane potentials used in the preparations were stable for 10–20
min.
Statistical analysis
Data were analyzed with SPSS 17.0 software (SPSS,
Inc., Chicago, IL, USA) and presented as the mean ± standard error
of the mean. A homogeneity test for variance was performed followed
by one-way analysis of variance, and two-group comparisons were
conducted using the least significant difference t-test. P<0.05
was considered to indicate a statistically significant difference
for all experiments.
Results
Characteristics of GABA-induced
currents in dorsal root ganglion neurons
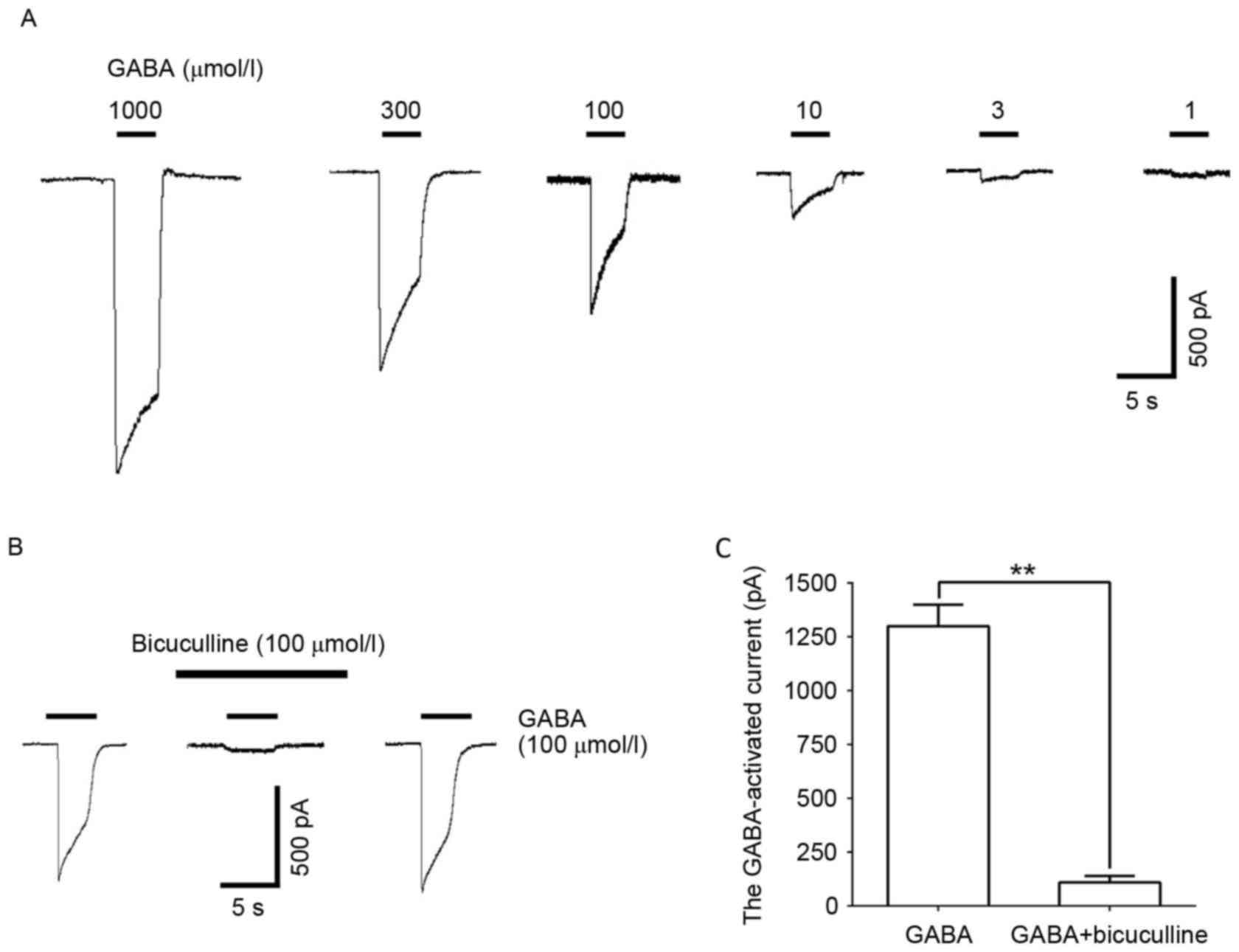
GABA (1–1,000 µmol/l) administration induced a
concentration-dependent inward current in DRG neurons (Fig. 1A). This reaction was significantly
blocked by the GABAAR selective antagonist, bicuculline
(100 µmol/l; P<0.01; Fig. 1B and
C), illustrating the GABA-induced inward current induced by the
GABAAR.
Effects of NFA on thermal hyperalgesia
in CCI rats
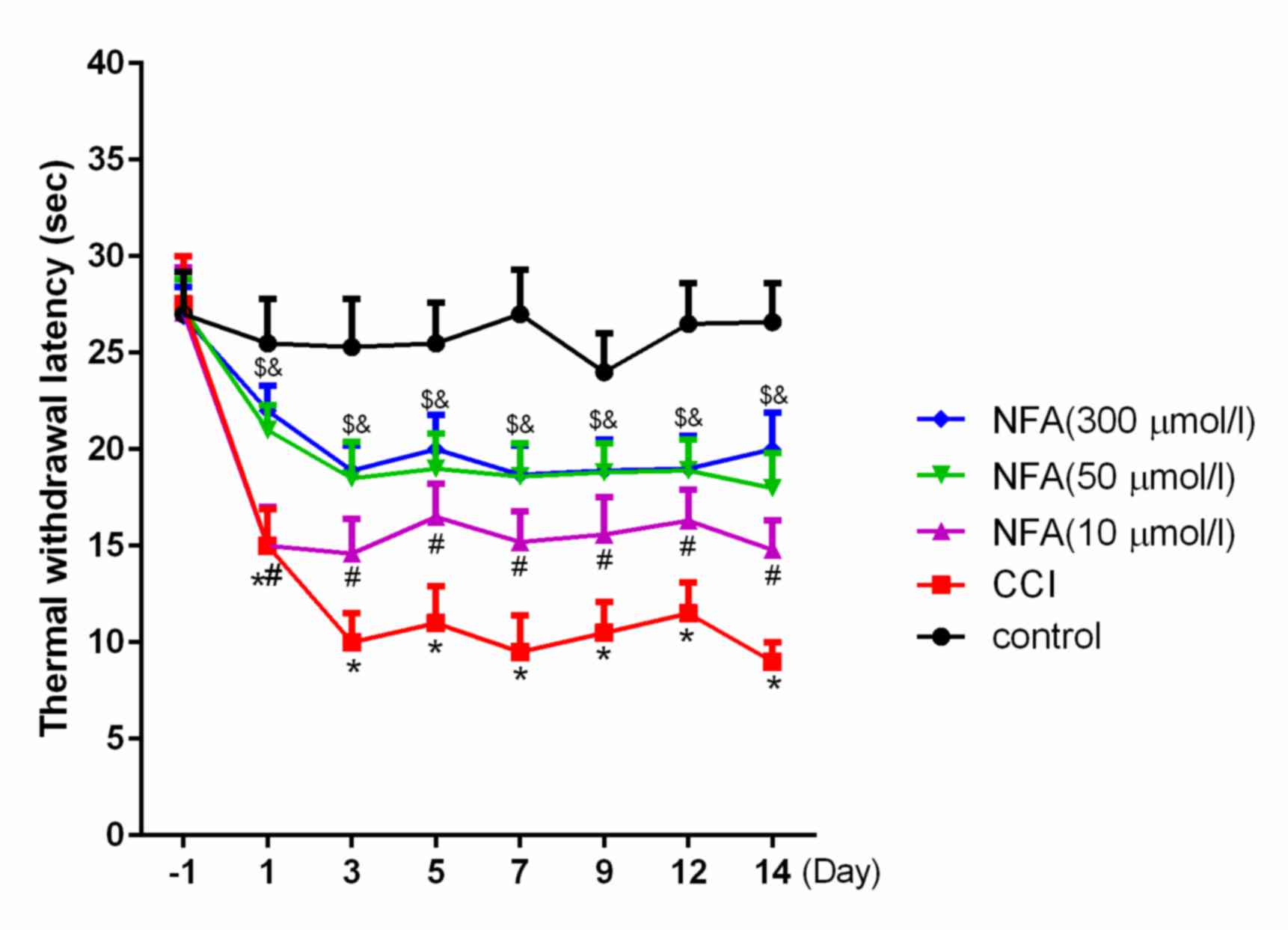
The effects of CCI on behavioral displays of
hyperalgesia were investigated using plantar testing. The thermal
withdrawal latency (TWL) of CCI rats significantly decreased from
days 1 to 14 post-surgery compared with the control (P<0.01),
and peaked on days 3–7 (Fig. 2).
This is in accordance with a previous study performed by our group
(20). The TWLs of each of the NFA
groups were significantly longer than in the CCI group (P<0.05;
Fig. 2). Furthermore, TWL in the 50
µmol/l NFA group was significantly longer than in the 10 µM NFA
group (P<0.05; Fig. 2); however,
no significant differences were observed between the 300 and 50
µmol/l NFA groups.
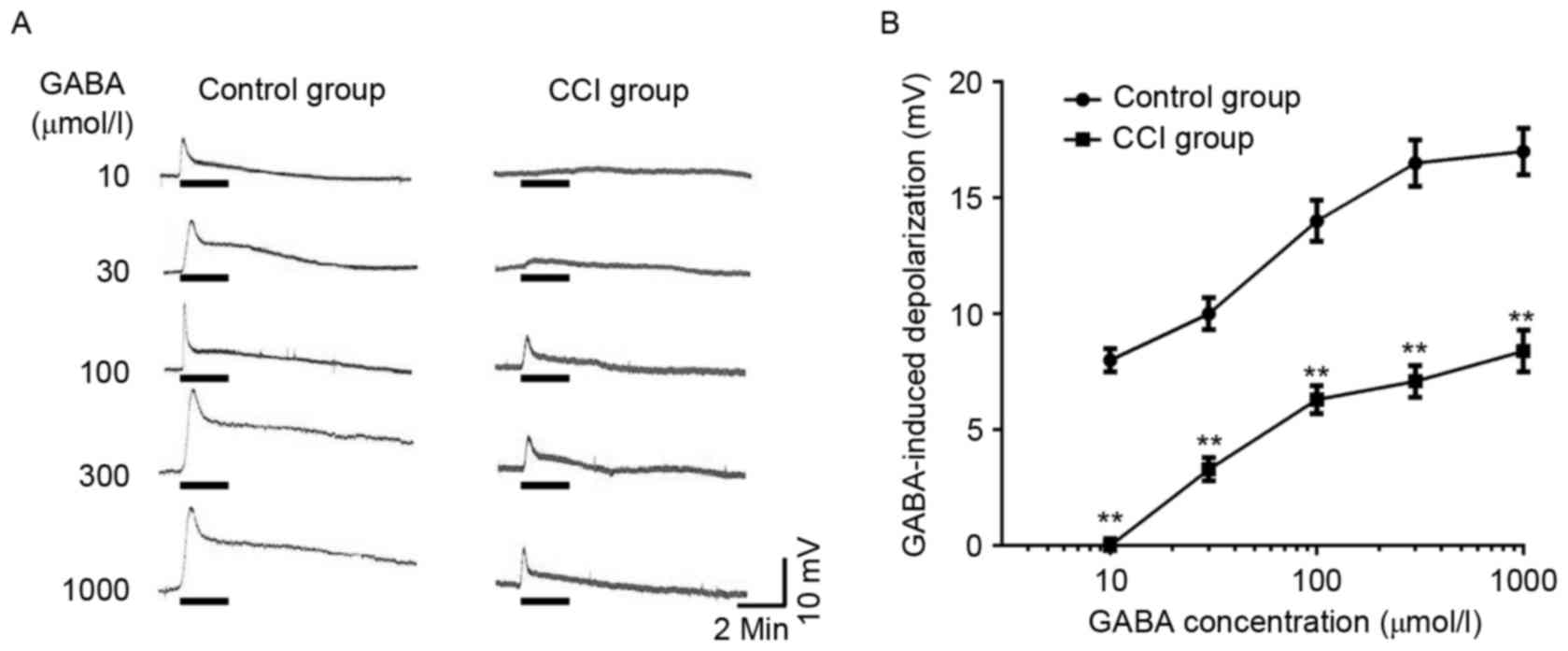
GABA-induced depolarization in the
dorsal root ganglion neurons of different groups
In DRG neurons of the normal group, the majority of
cells (90.6%) were sensitive to the application of GABA
(10−6-10−3 mol/l) and exhibited a
concentration-dependent depolarizing response. The threshold was
~10−6 mol/l GABA, and the maximal response was elicited
by 1×10−3 mol/l GABA (Fig.
3A). DRG neurons of the CCI group also exhibited a
concentration-dependent depolarizing response; however the
GABA-evoked response was reduced at each concentration of GABA when
compared with control DRG neurons (Fig.
3A and B; P<0.01). The half maximal effective concentration
(EC50) values did not differ significantly between the
normal and CCI groups. EC50 values for the normal and
CCI groups were 27.43±3.22 and 28.16±2.56 µmol/l, respectively.
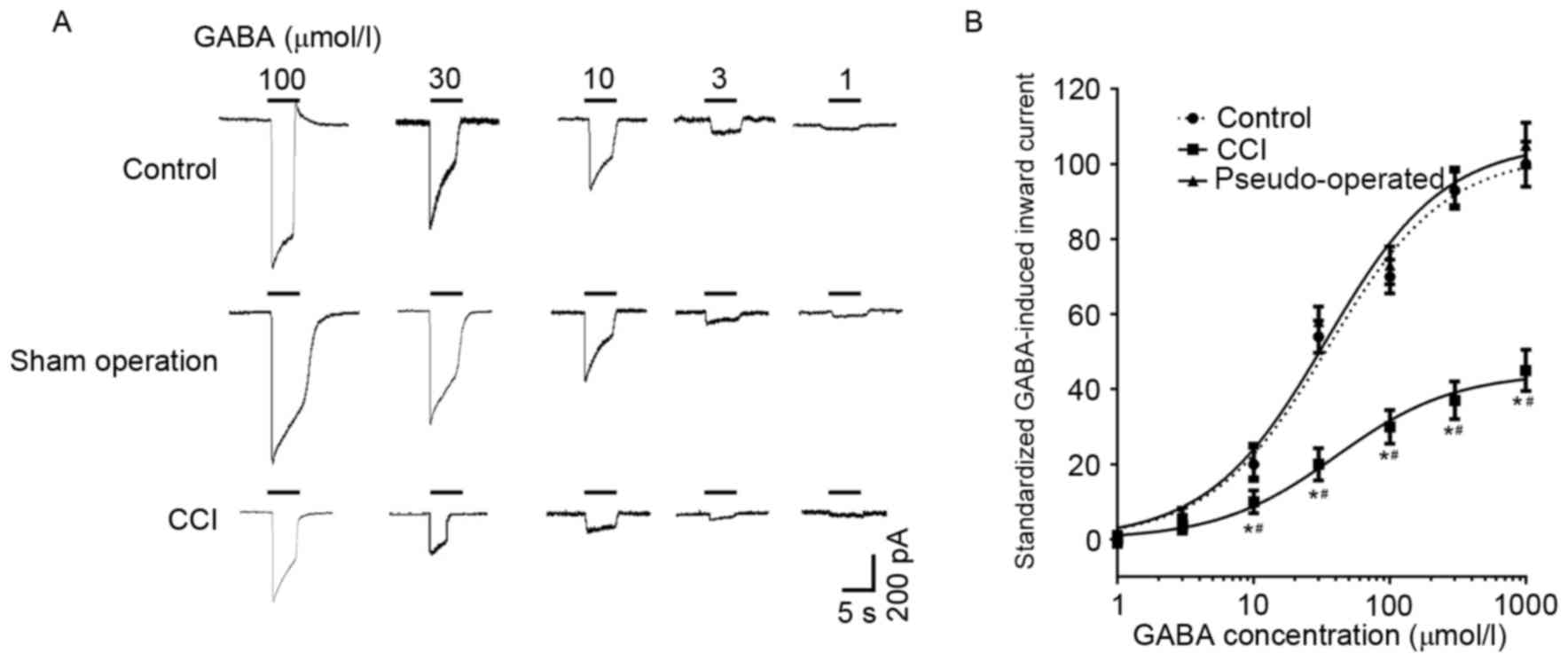
GABA-induced currents in the dorsal
root ganglion neurons of different groups
GABA induced a concentration-dependent inward
current in the L4–6 dorsal root ganglion neurons of the
control, sham operation and CCI groups (Fig. 4). The amplitudes of GABA-induced
currents in CCI rats were significantly depressed compared with the
control and pseudo-operated groups (P<0.05; Fig. 4); however, current amplitudes did not
differ significantly between the control and pseudo-operated
groups. GABA-induced (100 µmol/l) currents in the dorsal root
ganglion neurons of the control, pseudo-operated and CCI groups
were 1222.3±71.5, 1244.6±83.2 and 428.8±34.4 pA, respectively.
Effects of NFA on the inverse
potential of GABA-induced currents
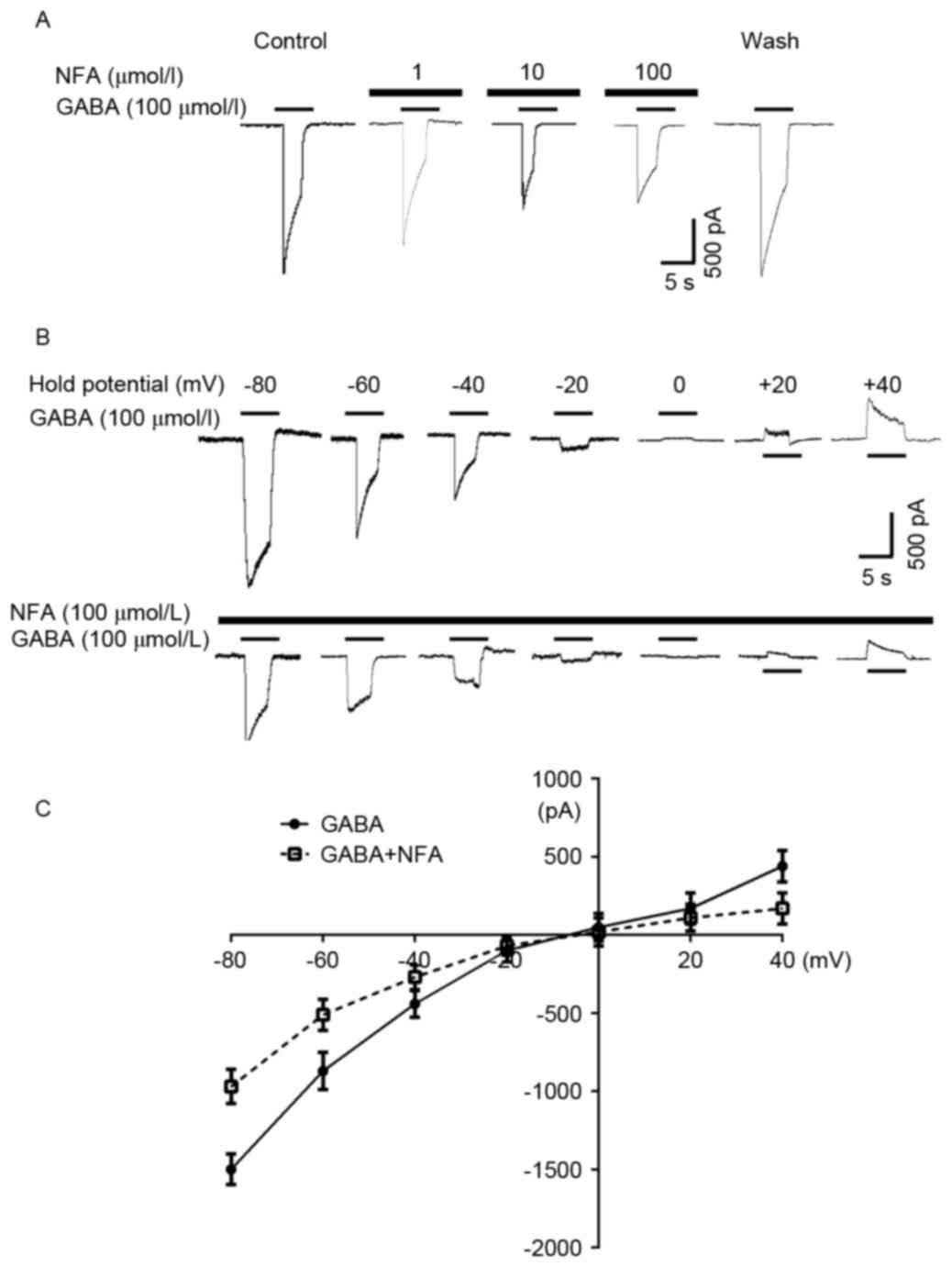
Cells were pre-incubated with NFA for 20 sec prior
to the application of GABA, resulting in a marked attenuation of
the GABA-induced inward current in the majority of the neurons
examined (96.3%). The inhibitory effect of NFA on GABA-induced
responses was concentration-dependent. The 100 µmol/l GABA-induced
inward currents were suppressed by 5.32±3.51, 33.8±5.20 and
52.2±6.32% by 1, 10, and 100 µmol/l NFA, respectively (Fig. 5A). Based on a concentration
inhibition curve determined in previous research, the inhibition
threshold was ~0.1 µmol/l NFA, maximal inhibition was achieved at
300 µmol/l NFA and the half maximal inhibitory concentration of NFA
was ~6.7 µmol/l (24). NFA did not
alter the EC50 value of GABA (~30 µmol/l) (25); however, NFA reduced the maximal GABA
current by ~60% (P<0.05, data not shown). In addition,
GABA-induced (100 µmol/l) currents were altered at different
holding potentials (−80–40 mV) in the presence and absence of 100
µmol/l NFA (Fig. 5B). The inverse
potentials of GABA-induced currents were −9.87±1.32 and −9.64±1.24
mV with and without NFA treatment, respectively (Fig. 5C).
Effects of NFA on the desensitization
of GABA-induced currents
The inhibitory effect of NFA was found to be
concentration-dependent. In previous research, the desensitization
of GABA-induced current was notable, as the amplitude of the
current decayed exponentially from a peak value and was then
maintained at a steady level, despite the constant presence and
concentration of GABA. The GABA-induced current included three
distinct phases; a peak, a desensitization phase and a steady
state. A minority of GABA-induced currents (<10%) exhibited only
the peak and desensitization phases. The desensitization of
GABA-induced currents exhibited double exponential characteristics,
and the desensitization current was a bi-exponential function
curve, and thus it was a biphasic process, including fast and slow
desensitization (25,26). As depicted in Fig. 6, the desensitization curve was a good
fit and followed the 2 term exponential equation,
f(t)=∑Aie−t/τi+C (blue lines). In this
equation, Ai was GABA-induced currents, τ was the time
constant of GABA-induced currents and C was a random constant. The
τ value of the desensitization of GABA-induced currents was
14.68±5.11 sec for fast desensitization and 175.8±42.67 sec for
slow desensitization (Fig. 6). The
suppression rate of 100 µmol/l NFA on GABA-induced current was
52.2±6.32%. Pre-application of 100 µmol/l NFA altered the τ value
of the desensitization of GABA-induced currents; the τ value
decreased to 4.64±2.21 and 43.70±14.34 sec for fast and slow
desensitization, respectively (Fig.
6). Pre-application of NFA exerted a stronger inhibitory effect
on the peak value of GABA-induced current, which may accelerate the
desensitization of GABA-induced currents.
Inhibition of GABA-induced inward
currents by NFA in different groups
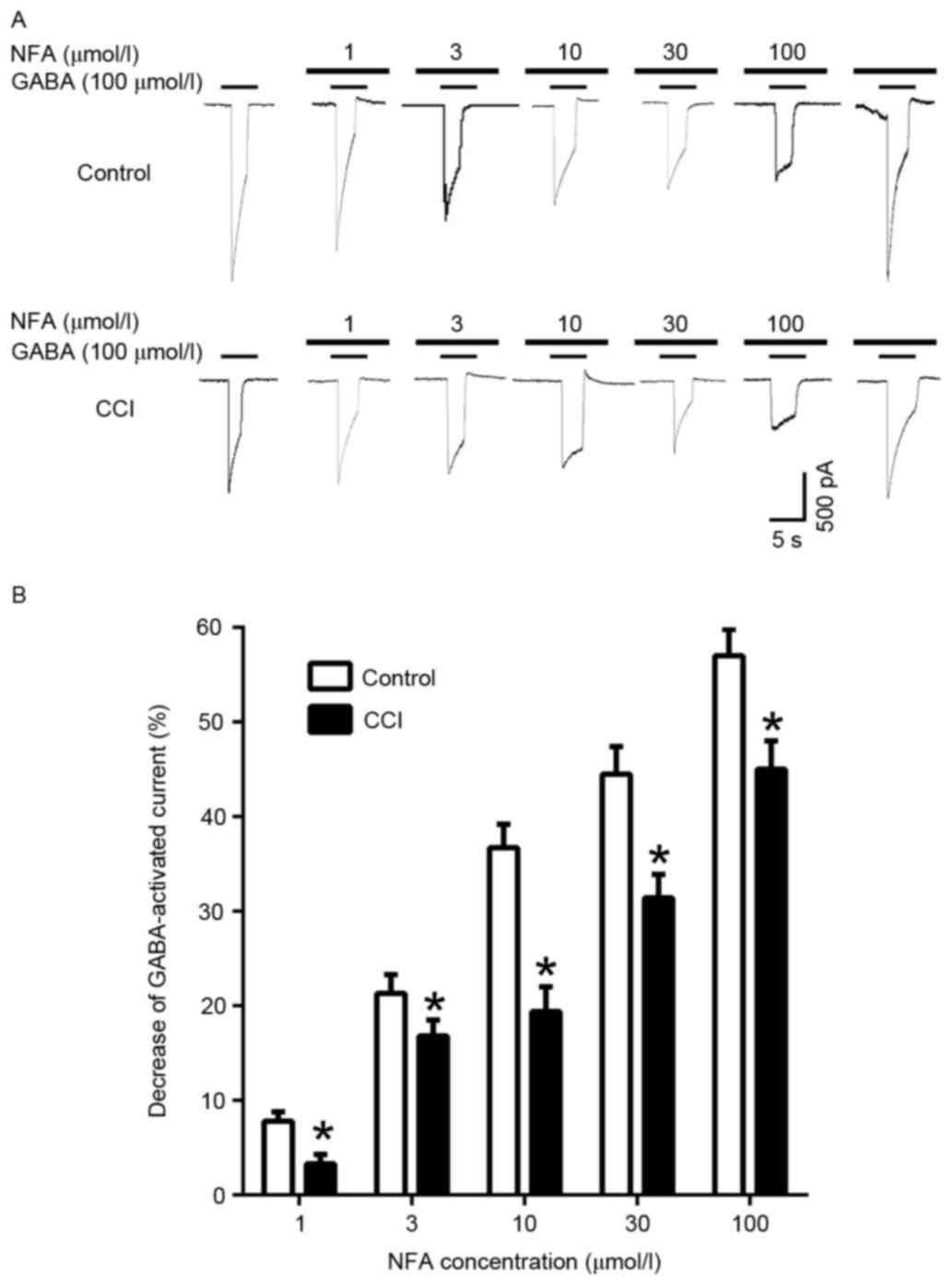
When cells were pre-incubated with NFA for 20 sec
prior to the application of GABA, NFA induced a
concentration-dependent inhibition of the GABA-induced response
(Fig. 7). In the control group, the
inward currents induced by 100 µmol/l GABA were suppressed by
7.47±2.75, 16.85±0.46, 36.65±1.13, 44.66±5.14 and 56.81±7.92% by 1,
3, 10, 30 and 100 µmol/l NFA, respectively (P<0.05). In the CCI
group the inward currents induced by 100 µmol/l GABA were
suppressed 3.05±1.92, 21.21±4.02, 19.35±5.66, 31.27±1.75 and
45.28±0.86% by 1, 3, 10, 30 and 100 µmol/l NFA, respectively
(Fig. 7B). Maximal inhibition was
achieved by 100 µmol/l NFA (Fig. 7A and
B). The GABA-induced response was inhibited by NFA in both the
control and CCI groups, however inhibition in the CCI group was
significantly greater compared with the control group at all
concentrations of NFA (P<0.05; Fig.
7B).
Discussion
In the present study, rats in the CCI group
exhibited a reduced TWL, and thus were more sensitive to heat pain,
indicating that the CCI model had been successfully established. In
a previous study, an antibody against interleukin-8 was able to
significantly increase the TWL of CCI model rats (27), suggesting that the inflammatory
reaction serves an important role in CCI models. Patients with
inflammatory pain typically exhibit a high sensitivity to heat and
may experience unbearable heat pain in the injured area (27). In the establishment of CCI models,
ligatures of chrome catgut are placed around the sciatic nerve and
sterile inflammation is induced, which damages nerve fibers and
causes neuropathic pain. As such, the CCI model includes two types
of pain; inflammatory and neuropathic (27).
The present study used a whole-cell recording
technique to measure GABA-induced inward currents induced by
GABAARs on the L4–6 dorsal root ganglion
neurons of control, pseudo-operated and CCI rats. The amplitudes of
currents in CCI rats were significantly depressed compared with
control and pseudo-operated rats. As the effects of presynaptic
GABA inhibition are primarily implemented by GABAARs
(28), these results suggest that
there is a functional change of GABAARs on the CCI
ipsilateral side. High levels of spontaneous discharge occur in
dorsal root ganglion neurons and nerve fibers following nerve
damage, due to weakening of GABAAR-induced presynaptic
inhibition. This results in the transfer of large amounts of
nociceptive information to the spinal cord and above, leading to
ultra-sensitized neurons and neuropathic pain (8). Naik et al (8) assessed the symptoms of neuropathic pain
in rats with L5 nerve injury, and observed that
neuropathic pain was alleviated by the GABAAR agonists,
muscinol and gaboxadol, in a dose-dependent manner. In turn,
neuropathic pain was aggravated by the GABAA receptor
antagonists, bicuculline and picrotoxin (8). A previous study by our group
demonstrated that chronic crush injury in DRG neurons may alter
GABAARs and initiate changes in the cytoplasmic cAMP and
cGMP signal transduction pathways, thus reducing GABAAR
functionality and presynaptic inhibition (29).
The GABAAR is a ligand-gated chloride ion
channel receptor. Using the outside-out patch-clamp mode, it has
been observed that changes in the concentrations of Cl−
on both sides of the cell membrane may alter the reversal potential
of GABA single channel currents, indicating that GABAA
channels may be selective for Cl− (11,30).
Activation of the GABAAR by GABA leading to opening of
Cl− channels, and the direction of Cl− flow
(influx or efflux) depends on the relative potential of
Cl− and the membrane resting potential (11,31). In
the peripheral nervous system, including sympathetic ganglion
cells, the intracellular concentration of Cl− is greater
than the extracellular concentration, and thus the equilibrium
potential of Cl− is smaller than the resting potential.
As such, when Cl− channels are activated by GABA, the
Cl− efflux induces membrane depolarization (11,12).
Aside from being an NSAID, NFA is recognized as a
strong CaCC blocker (32). The
present results demonstrated that NFA significantly inhibited
GABA-induced inward current in the control group. These results
suggest that CaCCs serve a critical role in the generation of
GABA-induced inward current in rat DRGs, which is in accordance
with previous experimental results (7). The present study also demonstrated that
NFA inhibits the GABA-induced response in L4–6 DRG
neurons in CCI rats, suggesting that NFA treatment may ameliorate
neuropathic pain. A previous study by our group demonstrated that
NFA exerted an analgesic effect on persistent pain in rats in a
formalin test (20). The inhibitory
effects of NFA on the GABA-induced response in CCI rats may have
been due to an increase in the number of CaCCs on the DRG neurons.
However, the inhibitory effects of NFA on the GABA-induced response
did not differ significantly between the CCI and controls groups.
In 2008, three laboratory groups cloned genes that encoded
classical CaCCs (33–35), and two of the genes were demonstrated
to encode transmembrane member (TMEM) 16A and TMEM 16B, as two CaCC
subunits. Immunofluorescence staining was used to assess TMEM 16A
expression, and the results demonstrated that TMEM 16A was
upregulated in a CCI group 14 days after surgery (unpublished
data). This may be responsible for the reduced inhibitory effect of
NFA on CCI neurons in the present study, suggesting that NFA may
alleviate neuropathic pain.
Cervero and Laird (36) proposed that, during inflammation,
dorsal root reflexes in Aβ fibers activate GABAergic
inhibitory interneurons, which in turn trigger dorsal root reflexes
in C fibers. This may lead to C fibers causing pain in response to
input from Aβ fibers, as a mechanism for allodynia
(36). As an additional outcome of
increased dorsal root reflexes during inflammation, Willis
(11) suggested that dorsal root
reflexes may propagate peripherally and release neurotransmitter
substances, including neurotransmitter peptides, into the joints,
skin and other peripheral tissues, which may induce the initial
stages of neurogenic inflammation.
Dorsal root reflexes are conducted centrifugally to
peripheral sensory endings, where they release neurotransmitters
and/or alter the excitability of sensory terminals (11). They also propagate centripetally and
release neurotransmitters that affect the excitability of
interneurons and motoneurons (11).
PAD, which is normally an inhibitory event, may be converted into
an excitatory event when dorsal root reflexes are triggered. Thus,
PAD may be a double-edged sword; generally inhibitory, but
excitatory when dorsal root reflexes are triggered (11). NFA may mediate neuropathic pain by
inhibiting dorsal root reflexes, which are triggered by
GABA-induced inward currents in the primary afferent nerve endings
leading to PAD. Thus, NSAIDs may have potential benefits in the
treatment of neuropathic pain.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 30160026) and the
Youth Science and Technology Innovation Special Foundation of
Xinjiang Production and Construction Corps (grant no.
2010JC33).
Glossary
Abbreviations
Abbreviations:
|
GABA
|
γ-aminobutyric acid
|
|
GABAAR
|
γ-aminobutyric acid type A
receptor
|
|
CaCC
|
calcium-activated chloride channel
|
|
CCI
|
chronic constriction injury
|
|
DRG
|
dorsal root ganglion
|
|
PAD
|
primary afferent depolarization
|
|
NFA
|
niflumic acid
|
|
NSAIDs
|
non-steroidal anti-inflammatory
drugs
|
|
NMDA
|
non-N-Methyl-D-aspartic acid
|
|
TWL
|
thermal withdrawal latency
|
References
|
1
|
Heydari M, Shams M, Hashempur MH, Zargaran
A, Dalfardi B and Borhani-Haghighi A: The origin of the concept of
neuropathic pain in Early Medieval Persia (9th-12th century CE).
Acta Med Hist Adriat. 13 Suppl:S9–S22. 2015.
|
|
2
|
Markman JD and Dworkin RH: Ion channel
targets and treatment efficacy in neuropathic pain. J Pain. 7 1
Suppl 1:S38–S47. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bourinet E, Francois A and Laffray S:
T-type calcium channels in neuropathic pain. Pain. 157 Suppl
1:S15–S22. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tsuda M: Microglia in the spinal cord and
neuropathic pain. J Diabetes Investig. 7:17–26. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bravo-Hernández M, Corleto JA,
Barragán-Iglesias P, González-Ramírez R, Pineda-Farias JB, Felix R,
Calcutt NA, Delgado-Lezama R, Marsala M and Granados-Soto V: The α5
subunit containing GABAA receptors contribute to chronic pain.
Pain. 157:613–626. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Maddox FN, Valeyev AY, Poth K, Holohean
AM, Wood PM, Davidoff RA, Hackman JC and Luetje CW: GABAA receptor
subunit mRNA expression in cultured embryonic and adult human
dorsal root ganglion neurons. Brain Res Dev Brain Res. 149:143–151.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao L, Li LI, Ma KT, Wang Y, Li J, Shi
WY, Zhu HE, Zhang ZS and Si JQ: NSAIDs modulate GABA-activated
currents via Ca2+-activated Cl channels in rat dorsal root ganglion
neurons. Exp Ther Med. 11:1755–1761. 2016.PubMed/NCBI
|
|
8
|
Naik AK, Pathirathna S and
Jevtovic-Todorovic V: GABAA receptor modulation in dorsal root
ganglia in vivo affects chronic pain after nerve injury.
Neuroscience. 154:1539–1553. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma KT, Si JQ, Zhang ZQ, Zhao L, Fan P, Jin
JL, Li XZ and Zhu L: Modulatory effect of CCK-8S on GABA-induced
depolarization from rat dorsal root ganglion. Brain Res.
1121:66–75. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao X, Li XL, Liu X, Wang C, Zhou DS, Ma
Q, Zhou WH and Hu ZY: Antinociceptive effects of fisetin against
diabetic neuropathic pain in mice: Engagement of antioxidant
mechanisms and spinal GABAA receptors. Pharmacol Res. 102:286–297.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Willis WD Jr: Dorsal root potentials and
dorsal root reflexes: A double-edged sword. Exp Brain Res.
124:395–421. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brooks CM and Koizumi K: Origin of the
dorsal root reflex. J Neurophysiol. 19:60–74. 1956.PubMed/NCBI
|
|
13
|
Evans RH and Long SK: Primary afferent
depolarization in the rat spinal cord is mediated by pathways
utilising NMDA and non-NMDA receptors. Neurosci Lett. 100:231–236.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hackman JC and Davidoff RA: Dorsal root
potentials in the isolated frog spinal cord: Amino acid
neurotransmitters and magnesium ions. Neuroscience. 41:61–69. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bruera E: Mechanism of action of
nonsteroidal anti-inflammatory drugs. Cancer Invest. 16:538–539.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Babot Z, Cristòfol R and Suñol C:
Excitotoxic death induced by released glutamate in depolarized
primary cultures of mouse cerebellar granule cells is dependent on
GABAA receptors and niflumic acid-sensitive chloride channels. Eur
J Neurosci. 21:103–112. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wallenstein MC: Attenuation of
epileptogenesis by nonsteroidal anti-inflammatory drugs in the rat.
Neuropharmacology. 30:657–663. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bennett GJ and Xie YK: A peripheral
mononeuropathy in rat that produces disorders of pain sensation
like those seen in man. Pain. 33:87–107. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen MG, Ma KT, Si JQ and Li L: Effects of
niflumic acid on GABA-activated currents in isolated dorsal root
ganglion neurons in rats with chronic constriction injury. J
Shihezi Univ (Natural Science). 32:193–197. 2014.(In Chinese).
|
|
20
|
Zhu H, Ma KT, Li L, Zhang ZS, Li J and Si
JQ: Changes of GABA-activated currents in isolated dorsal root
ganglion neurons in rats with neuropathic pain. Zhongguo Ying Yong
Sheng Li Xue Za Zhi. 27:376–379. 2011.(In Chinese). PubMed/NCBI
|
|
21
|
Cheng HJ, Ma KT, Li L, Zhao L, Wang Y and
Si JQ: Differential expression of alpha-adrenoceptor subtypes in
rat dorsal root ganglion after chronic constriction injury. J
Huazhong Univ Sci Technolog Med Sci. 34:322–329. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Y, Ma K, Li LI, Liu Y, Si J and Wan
YU: Effect of non-genomic actions of thyroid hormones on the
anaesthetic effect of propofol. Exp Ther Med. 10:959–965.
2015.PubMed/NCBI
|
|
23
|
Li L, Zhao L, Wang Y, Ma KT, Shi WY, Wang
YZ and Si JQ: PKCε mediates substance P inhibition of GABAA
receptors-mediated current in rat dorsal root ganglion. J Huazhong
Univ Sci Technolog Med Sci. 35:1–9. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li L, Wang Y, Ma KT, Cheng HJ, Zhao L and
Si JQ: The effect of niflumic acid and blocker of calcium channel
on the desensitization of gamma aminobutyric acid-activated
current. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 29:128–132.
2013.(In Chinese). PubMed/NCBI
|
|
25
|
Li L, Li J, Ma KT, Cheng HJ, Zhao L, Wang
Y and Si JQ: The effect of niflumic acid on gamma aminobutyric acid
activated current in DRG neurons. Zhongguo Ying Yong Sheng Li Xue
Za Zhi. 29:68–71. 2013.(In Chinese). PubMed/NCBI
|
|
26
|
Si JQ, Zhang ZQ, Li CX, Wang LF, Yang YL
and Li ZW: Modulatory effect of substance P on GABA-activated
currents from rat dorsal root ganglion. Acta Pharmacol Sin.
25:623–629. 2004.PubMed/NCBI
|
|
27
|
Bridges D, Thompson SW and Rice AS:
Mechanisms of neuropathic pain. Br J Anaesth. 87:12–26. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lian Y, Wang Y, Ma K, Zhao L, Zhang Z,
Shang Y, Si J and Li L: Expression of gamma-aminobutyric acid type
A receptor α2 subunit in the dorsal root ganglion of rats with
sciatic nerve injury. Neural Regen Res. 7:2492–2499.
2012.PubMed/NCBI
|
|
29
|
Wang Y, Li SY, Ma KT, Si JQ, Zhao L, Zhang
ZS, Zhu H and Li L: Changes in presynaptic inhibition and the
second message system of neuropathic pain model in rats. Chin J Mod
Med. 22:9–14. 2012.(In Chinese).
|
|
30
|
Gaunitz C, Schüttler A, Gillen C and
Allgaier C: Formalin-induced changes of NMDA receptor subunit
expression in the spinal cord of the rat. Amino Acids. 23:177–182.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Carlton SM, Hargett GL and Coggeshall RE:
Localization and activation of glutamate receptors in unmyelinated
axons of rat glabrous skin. Neurosci Lett. 197:25–28. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Collin T, Chat M, Lucas MG, Moreno H,
Racay P, Schwaller B, Marty A and Llano I: Developmental changes in
parvalbumin regulate presynaptic Ca2+ signaling. J Neurosci.
25:96–107. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Caputo A, Caci E, Ferrera L, Pedemonte N,
Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O and
Galietta LJ: TMEM16A, a membrane protein associated with
calcium-dependent chloride channel activity. Science. 322:590–594.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schroeder BC, Cheng T, Jan YN and Jan LY:
Expression cloning of TMEM16A as a calcium-activated chloride
channel subunit. Cell. 134:1019–1029. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang YD, Cho H, Koo JY, Tak MH, Cho Y,
Shim WS, Park SP, Lee J, Lee B, Kim BM, et al: TMEM16A confers
receptor-activated calcium-dependent chloride conductance. Nature.
455:1210–1215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cervero F and Laird JM: Mechanisms of
touch-evoked pain (allodynia): A new model. Pain. 68:13–23. 1996.
View Article : Google Scholar : PubMed/NCBI
|