Introduction
Gastric cancer (GC), as a common gastrointestinal
tumor, is one of the leading causes of cancer related mortality
worldwide, particularly in East Asian countries, including China
and Japan (1–3). Although surgical resection combined
with adjuvant therapy has achieved advances, the 5-year survival
rate remains less than 30%, causing unsatisfactory clinical outcome
of GC patients (4,5). The main reason is that most patients
are diagnosed as unresectable advanced or metastatic stage
(6), which make it difficult to
improve the early diagnosis and effective treatment for GC.
Currently, studies have shown that the initiation and development
of GC is closely associated with a sequential accumulation of
various molecular and genetic alterations (7,8).
Therefore, it is urgently needed to investigate the underlying
molecular mechanisms of initiation and metastasis to develop
appropriate approaches for improving its diagnosis and
treatment.
Cadherins are members of a large family of
transmemrane glycoproteins that play an important role in the
maintenance of normal tissue architecture by mediating specific
cell-cell adhesion, cell recognition and signaling (9,10).
Accumulating evidences have shown that perturbations in cadherins
are implicated in tumor development, especially in invasion and
metastasis (11) as putative
products of tumor suppressor genes (12–15). For
T-cadherin (T-cad, also known as CDH13 or H-cadherin) is a unique
atypical glycosylphosphatidylinositol (GPI)-anchored member of
classical cadherin superfamily, which lacks the highly conserved
transmembrane and cytoplasmic domains (16–18).
Recently, interest in the role of T-cad in human malignancies is
increasing. T-cad has been found to be downregulated in lung
cancer, ovarian cancer, bladder cancer, cervical and prostate
cancer, but abundantly expressed in hepatocellular carcinoma and
osteosarcoma (19). Moreover, T-cad
could regulate progression of tumor types including breast, hepatic
and skin carcinomas by modulating tumor cell proliferation and
migration (18). In breast cancer,
overexpression of T-cad markedly reduced cell invasive potential
and growth rate (20). Wang et
al demonstrated reduction of T-cad facilitated tumorigenicity
in prostate cancer (21). Notably,
T-cad could exert pleiotropic effects to promote tumor growth
through different mechanisms on cancer cells in vitro and
vivo (22). Although previous
studies indicated T-cad is frequently deleted in human GC and
decreased T-cad is associated with GC poor prognosis (23,24),
little is known about the biological effects of T-cad in GC.
In this study, we showed that T-cad was
downregulated in GC tissues and cell lines. Then the effects of
T-cad overexpression on the cell proliferation, cell cycle,
migration and invasion were further evaluated in GC cells in
vitro. Our data indicate that T-cad is involved in uncontrolled
cell proliferation and invasion of GC cells by, at least in part,
influencing cell growth and motility.
Materials and methods
GC tissue specimens and cell
lines
The GC tissue and adjacent noncancerous tissue were
collected from patients who underwent surgical resection in our
hospital. The fresh tissue samples were immediately snapped-frozen
in liquid nitrogen and stored at −80°C for further analysis.
Human GC cell lines, MGC80-3, SGC-7901, AGS, HGC27
and normal human immortalized normal gastric epithelial cell line
GES-1 were obtained from the Cancer Research Institution of China
Medical University. All of these cell lines were cultured in
RPMI-1640 medium (Hyclone, Logan, UT, USA) containing 10% fetal
bovine serum (FBS; HyClone), 10 units/ml penicillin and 10 mg/ml
streptomycin, and incubated in a 5% CO2 incubator at
37°C.
T-cad overexpressing in GC cell
lines
The plasmids for pcDNA3.1 and pcDNA-T-cadherin
(pcDNA-Tcad; Invitrogen Life Technologies, Carlsbad, CA, USA) were
purchased and respectively transfected into GC cell lines, MGC80-3
and AGS using Lipofectamine® 2000 (Invitrogen Life
Technologies) according to manufacturer's instructions. Then stably
transfected cells were obtained and cultured for 48 h before
confirming the expression of T-cad by qRT-PCR and western
blotting.
RNA isolation and qRT-PCR
Total RNA was isolated from tissues or cells using
the RNeasy Mini kit (Qiagen, Hilden, Germany) according to the
manufacturer's instructions. The gene-specific primers were as
follows: T-cad, forward: 5′-TTCAGCAGAAAGTGTTCCATAT-3′, reverse:
5′-GTGCATGGACGAACAGAGT-3′; GAPDH, forward:
5′-GACCCCTTCATTGACCTCAACTAC-3′, reverse:
5′-TGGTGGTGCAGGATGCATTGCTGA-3′. The qRT-PCR was performed on a Fast
Real-time PCR 7500 System (Applied Biosystems Life Technologies,
Foster City, CA, USA) by using SYBR-Green PCR Master Mix following
the reaction procedure: 1 min at 95°C, 40 cycles of 95°C for 5 sec,
and 60°C for 20 sec. Gene expression was normalized to the
expression of GAPDH by the 2−ΔΔCq method (25). Each experiment was performed in
triplicate and repeated three times.
Protein extraction and western
blot
Total proteins were extracted from tissues or cells
using RIPA lysis buffer (Beyotime Institute of Biotechnology,
Shanghai, China). Enhanced BCA Protein Assay Kit (Beyotime
Institute of Biotechnology) was used to quantify the protein
concentration. Then equivalent proteins of each sample were
separated by SDS-PAGE on 10% polyacrylamide gels and transfered to
polyvinylidene fluoride membranes (Millipore Corp., Billerica, MA,
USA). The membranes were blocked in PBS containing 5% non-fat milk
for 2 h at room temperature, and then incubated with primary
antibodies, including anti-T-cad (1:1,000; Abcam, Cambridge, MA,
USA), anti-CDK4, anti-Cyclin D1, anti-E-cadherin, anti-Vimentin and
anti-GAPDH overnight at 4°C. After washing three times with PBS for
5 min, membranes were incubated with the corresponding horseradish
peroxidase-conjugated secondary antibodies for 1 h at room
temperature. After washing, the target proteins were visualized by
an enhanced chemiluminescence detection system. GAPDH was used as
an internal control.
Cell viability and colony formation
assays
MTT assays were performed every day to determine
cell viability in GC cells over the following 5 days. Briefly,
cells were seeded in 96-well plates at a density of
2×103 cells per well and incubated with 20 µl MTT (0.5
mg/ml, Sigma, USA) for 4 h at 37°C. Then, 150 µl dimethylsulfoxide
(DMSO; Sigma, St. Louis, MO, USA) was added to dissolve the purple
formazan crystals for 1 h. Finally, the absorbance of each well was
read on an ELISA reader (Bio-Rad, Berkeley, CA, USA) at a
wavelength of 595 nm.
For the colony formation assay, cells (500 cells per
well) were cultured in 6-well plates after 48 h transfection. After
7 day culture, the cells were fixed in 4% paraformaldehyde and then
stained with crystals purple (Sigma). Surviving colonies (>50
cells per colony) were then observed and manually counted through a
light microscope. Each experiment was performed in triplicate and
repeated three times.
Cell cycle assay
The cell cycle distribution was analyzed by flow
cytometry using PI staining. Briefly, cells were seeded in 6-cm
dishes after 48 h transfection at a density of 2×105
cells per dish. Then cells were washed with cold PBS for three
times and fixed in 70% ethanol at 4°C for 24 h. After washed with
PBS again, cells were then stained with PBS containing 0.05 mg/ml
of PI and RNase A (Beyotime Institute of Biotechnology) for 30 min
in the dark. Then cells were determined for DNA content using a
FACSCalibur (BD Biosciences, San Diego, CA, USA) and data were
analyzed with the ModFit DNA software. Each experiment was
performed in triplicate and repeated three times.
Wound healing assay
For wound-healing-assay, cells were seeded into
6-well plates and culture overnight until grown to approximately
90% confluence. Then cells were scratched by a 10-µl sterile
plastic tip in a definite array. After washing the well with PBS,
the cells migrating into the wounded areas were observed and
photographed under a fluorescence microscope at 0 and 48 h time
points. The cell wound healing rate was evaluated by calculating
the percentage of the wound area compared with the area of total
cells. Each experiment was performed in triplicate and repeated
three times.
Transwell migration and invasion
assays
The cell migration assay was performed using a
Transwell chambers (8-µm pore size; Corning Inc., Acton, MA, USA).
At 48 h after transfection, a total of 1×105 cells were
trypsinized and added to the upper chamber in serum-free medium,
and culture medium containing 10% FBS was added into the lower
chamber. After incubating for 24 h, the cells that migrated into
the lower chamber were fixed with 4% paraformaldehyde and stained
with crystal violet. Finally, cells were photographed and counted
under a microscope. For the cell invasion assay, the procedure was
similar to the cell migration assay, except the Transwell chambers
were coated with 200 µl of Matrigel. Each experiment was performed
in triplicate and repeated three times.
Statistical analysis
All quantitative data were expressed as mean ±
standard deviation (SD) of three independent experiments.
Statistical analysis was performed using GraphPad Prism 5.0
software (GraphPad Software, Inc., La Jolla, CA, USA). The
differences between two groups were evaluated using the Student's
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Relative low levels of T-cad
expression in GC tissues and cell lines
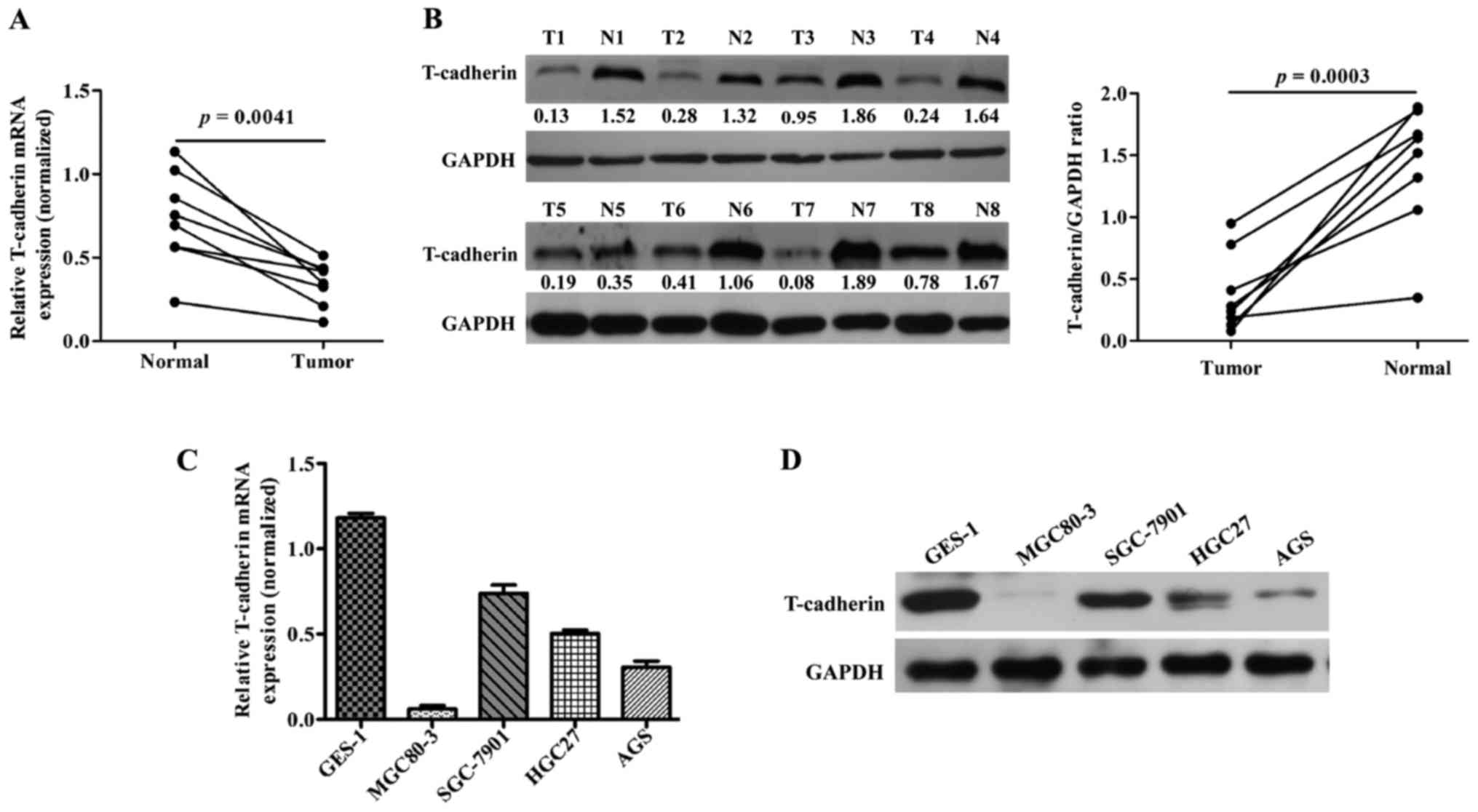
To investigate T-cad expression in GC, 8 pairs of GC
and corresponding adjacent noncancerous tissues were collected and
the basic clinicopathologic features of patients were summarized in
Table I. Then these tissues were
subjected to qRT-PCR and western blotting analyses. As shown in
Fig. 1A, the mRNA levels of T-cad
were significantly downregulated in GC tissues compared with
adjacent noncancerous tissues (P=0.0041). In line with this result,
clearly decreased levels of T-cad protein were detected in all the
tumors tissues in comparison to the paired noncancerous tissues
(Fig. 1B). In addition, we examined
the expression of T-cad in several GC cell lines. As presented in
Fig. 1C and D, the levels of T-cad
mRNA and protein in GC cell lines was much lower than that in
normal GES-1 cells, of which MGC80-3 and AGS exhibited the lowest
signals of T-cad than the other GC cell lines. These data suggested
that T-cad might serve as a tumor suppressor in GC.
 | Table I.The clinicopathologic factors of
gastric cancer (n=8). |
Table I.
The clinicopathologic factors of
gastric cancer (n=8).
|
Characteristics | Cases |
|---|
| Age (years) |
|
|
<50 | 5 |
|
≥50 | 3 |
| Gender |
|
|
Male | 2 |
|
Female | 6 |
| TNM stage |
|
| I,
II | 5 |
| III,
IV | 3 |
| Tumor site |
|
|
Upper | 2 |
|
Middle | 3 |
|
Low | 3 |
Overexpression of T-cad suppressed
cellular proliferation ability in GC cells
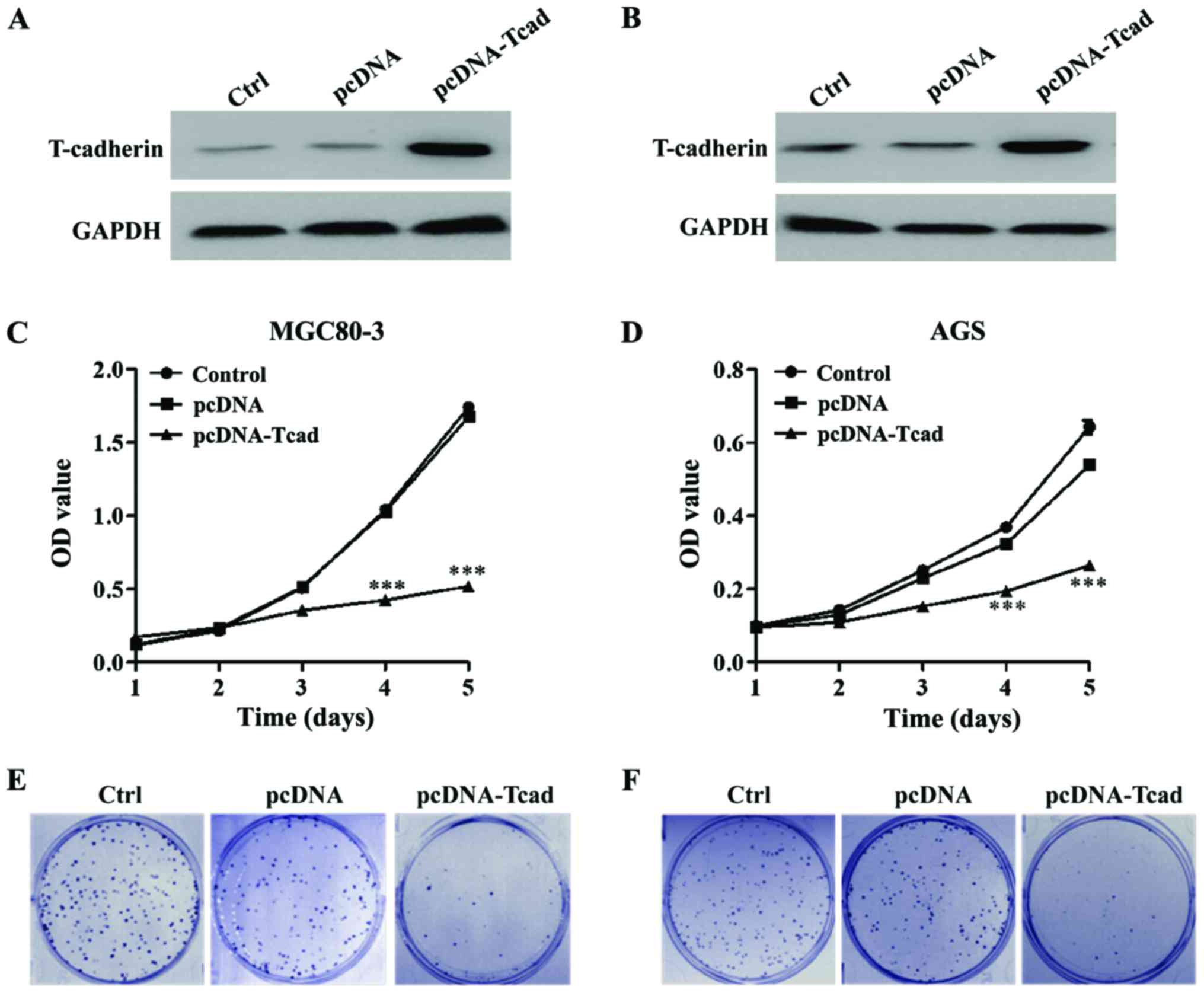
We successfully constructed stable clones
overexpressing T-cad from MGC80-3 and AGS with lower signals of
T-cad. As shown in Fig. 2A and B,
overexpression of T-cad in the two GC cell lines were confirmed by
western blotting after transfection for 48 h. Then MTT and colony
formation assays were performed to determine cell proliferation
ability. The results showed that GC cell viability was dramatically
decreased in T-cad overexpression group compared with the empty
vector group in MGC80-3 (Fig. 2C)
and AGS (Fig. 2D) cells after
transfection for 4 and 5 days (P<0.001). Consistent with the MTT
assay, colony formation assay also indicated that T-cad
overexpression led to an obvious reduction of colony number in
MGC80-3 (Fig. 2E) and AGS (Fig. 2F) cells. Collectively, these findings
supports that T-cad could inhibit cell proliferation in GC.
Overexpression of T-cad induced cell
cycle G0/G1 arrest in GC
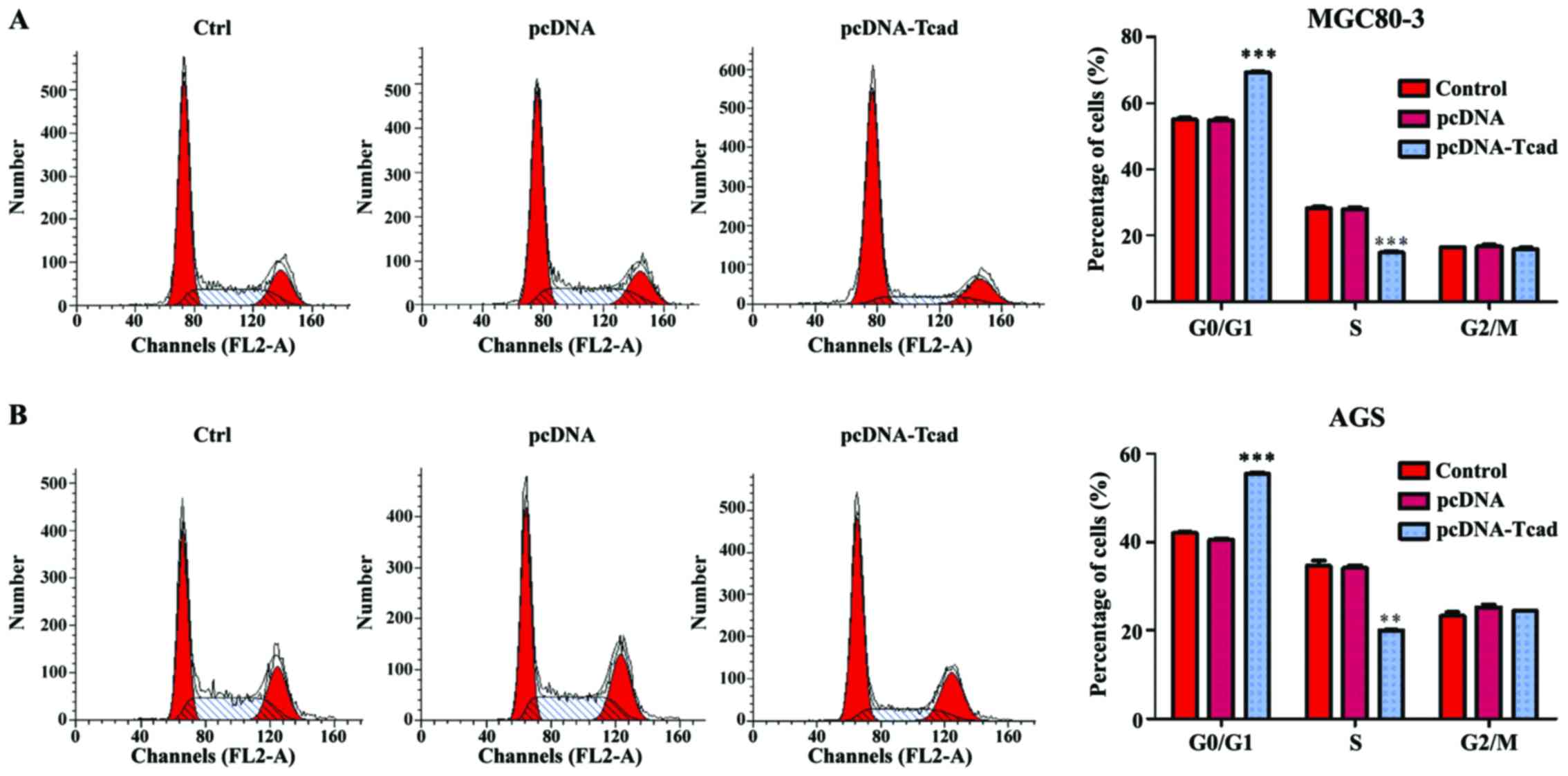
Next, we investigated the mechanisms underlying the
growth suppression effects of T-cad overexpression by analyzing
cell cycle distribution of GC cells via a flow cytometer. As shown
in Fig. 3A, the percentage of cells
in G0/G1 phase was significantly increased, but in S phase was
remarkably decreased in T-cad overexpression group, compared with
control and empty vector groups in MGC80-3 cells (P<0001).
Similar results were also found in AGS cells (Fig. 3B, P<0.001, P<0.01). The data
revealed that T-cad overexpression could arrest cell cycle at G0/G1
phase, which might be closely associated with growth suppression
effects.
Overexpression of T-cad reduced
cellular motility, migration and invasion in GC
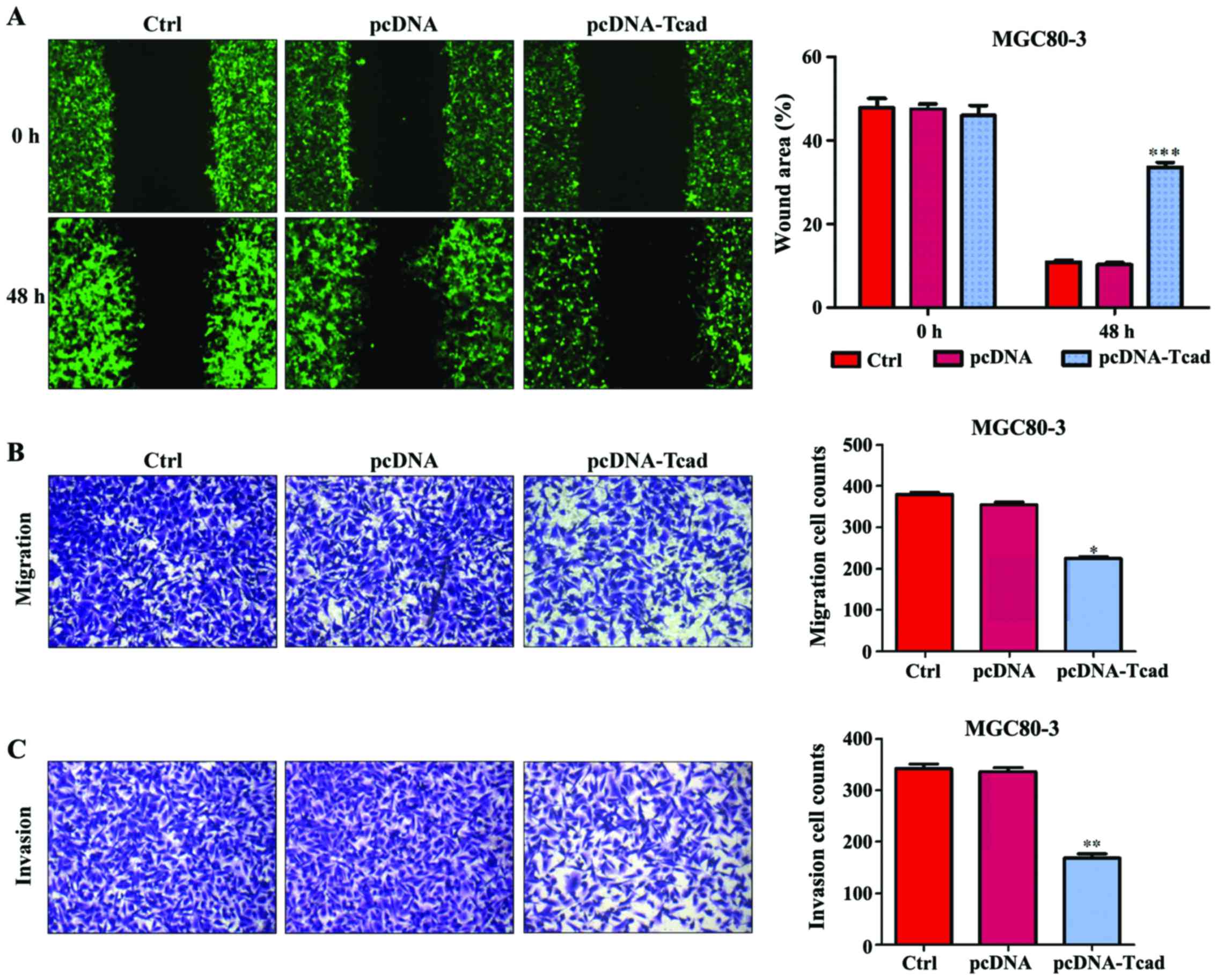
In addition to cell proliferation, we also performed
wound-healing and transwell assays to determine the effects of
T-cad on GC cell metastatic ability. As shown in Fig. 4A, an evident acceleration in the
wound closure rate was observed in control group or empty vector
groups compared with cells following pcDNA-Tcad transfection.
Quantitative analysis further demonstrated that the wound areas was
significantly larger in T-cad overexpression group compared with
control group or empty vector group in MGC80-3 cells after
incubation for 48 h (P<0001). In transwell assay, the number of
migrated cells in pcDNA-Tcad group was significantly reduced
compared with the control group or empty vector group MGC80-3 cells
(Fig. 4B, P<0.05). Subsequently,
cell invasion was determined and the results indicated that the
invasive ability of MGC80-3 cells was remarkably suppressed by
T-cad overerxpression (Fig. 4C,
P<0.01). These consistent results suggested that T-cad could
inhibit tumor cell migration and invasion in GC.
Overexpression of T-cad regulated cell
cycle and metastatic markers
To gain insights into the molecular mechanism of the
tumor-suppressive effect of T-cad, we detected the expression
alterations of some cell cycle regulators and motility markers
using Western blotting. As shown in Fig.
5, the expression levels of CDK4 and Cyclin D1, associated with
G1-S transition, were obviously downregulated in pcDNA-Tcad group.
Furthermore, inhibition of metastasis by T-cad overexpression
resulted from downregulation of Vimentin and upregulation of
E-cadherin. These results suggest that T-cad inhibits cell
proliferation, migration and invasion might by regulating the
expression of important markers involved in cell cycle, migration
and invasion.
Discussion
T-cad gene is a novel adhesion molecule found to map
to chromosome 16q24, a region often exhibiting loss of
heterozygosity in cancer including breast, prostate cancer and
others (26–28). Recently, it has been reported to be
an important independent prognostic predictor in GC. However, its
potential biological role in GC remains not fully understood. In
this study, we showed that the mRNA and protein expression levels
of T-cad were significantly lower in the GC tissues and cell lines
compared with controls, which is agreement with the previous
reports about decreased T-cad in GC (23,29).
Furthermore, we performed gain-of-function assay on GC cells to
investigate the biological effects of T-cad on cell proliferation,
migration and invasion in vitro. It was found that the
proliferative and motility activity of GC cells decreased by T-cad
overexpression. Consistent with our data, most studies have shown
that enhanced T-cad cDNA expression inhibited tumor cell growth,
whereas T-cad silencing stimulated proliferation, invasion and
metastasis in several in vitro and in vivo models
(22,30–32).
It has been suggested that T-cad may affect cell
proliferation through regulating cell cycle progression, as
demonstrated by Ivanov et al (33) and Zhong et al (34). To further investigate the underlying
mechanism of the growth inhibition effect of T-cad, we determined
whether T-cad overexpression had an impact on cell cycle
distribution. As expect, T-cad overexpression induced cell cycle
arrest at G0/G1 phase through downregulation of CDK4 and Cyclin D1
expression in GC cells. Interestingly, T-cad could negatively
regulates the cell proliferation by inducing a delay in the G2/M
phase in squamous carcinoma (35)
and astrocytomas (36). The
different cell cycle arrest mechanisms might be ascribed to
different tumor types.
In addition, T-cad encodes a cell surface
glycoprotein belonging to the cadherin family responsible for
selective cell recognition and adhesion. In human tumors, cell-cell
association is often disorganized and thought to be a cause of the
unregulated invasion and metastasis behavior of tumor cells
(37,38). Therefore, we speculate T-cad might be
associated with GC cell migration and invasion. As speculated, our
results showed that T-cad overexpression suppressed GC cell
migration and invasion by upregulating E-cadherin expression and
downregulation the expression of Vimentin and MMP-2. E-cadherin,
another of the cadherin family, is an essential adhesive tumor
suppressor as the hallmark of epithelial-mesenchymal transition
(EMT) (39). Recently, it was
reported that loss of E-cadherin promotes tumor metastatic and DNA
methylation-induced silencing is significantly correlated with
increased invasive potential of melanoma cells (40). Moreover, a significant correlation
was found between reduced levels of E-cadherin via promoter
aberrant methylation and several poor prognostic factors, namely
ulceration, head/neck localization, mitotic count, metastasis and
reduced overall/disease-free survival in cutaneous melanoma.
Likewise, these above poor prognostic factors are frequently
associated with epigenetic downregulation of E-cadherin in mucosal
or uveal melanoma (40). These
evidences further demonstrated E-cadherin plays a crucial role in
the events affeting melanoma progression and might be considered as
a prognostic factor for melanoma. In addition, the promoter of
E-cadherin frequently underwent hypermethylation in human GC
accompanied by inactivation of T-cad, suggesting their positive
correlation in GC (41,42). Vimentin, as an intermediate filament
during EMT, is required for facilitating mesenchymal cell migration
(43). Given the evidence discussed,
our findings of their correlation suggest that T-cad might play an
important role in GC metastasis by positively regulating E-cadherin
expression and negatively regulating Vimentin expression.
Collectively, our study revealed the preliminary
biological function of T-cad in GC cells and found restoration of
T-cad obviously suppressed GC cell biological behaviors by
inhibiting cell proliferation and motility. Our studies suggest
that T-cad might represent an important target for GC treatment. In
addition, some additional limitations are presented in this study.
Firstly, the mRNA determination of CDK4, Cyclin D1, Vimentin and
E-cadherin levels following ectopic expression of T-cad was nor
performed. Secondly, the luciferase assay was absent not to
demonstrate whether T-cadherin directly or indirectly regulated
these downstream molecules. Therefore, still further analysis of
T-cad regulation in GC will be the aim of or future work.
Acknowledgements
The present study was supported by funding from the
Medical Foundation of Fujian (no. 2015J01439).
References
|
1
|
Piazuelo MB and Correa P: Gastric cáncer:
Overview. Colomb Med (Cali). 44:192–201. 2013.PubMed/NCBI
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sugano K: Screening of gastric cancer in
Asia. Best Pract Res Clin Gastroenterol. 29:895–905. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xia P, Song CL, Liu JF, Wang D and Xu XY:
Prognostic value of circulating CD133(+) cells in patients with
gastric cancer. Cell Prolif. 48:311–317. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gigek CO, Chen ES, Calcagno DQ, Wisnieski
F, Burbano RR and Smith MA: Epigenetic mechanisms in gastric
cancer. Epigenomics. 4:279–294. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhu W, Ye L, Zhang J, Yu P, Wang H, Ye Z
and Tian J: PFK15, a small molecule inhibitor of PFKFB3, induces
cell cycle arrest, apoptosis and inhibits invasion in gastric
cancer. PLoS One. 11:e01637682016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yasui W, Sentani K, Sakamoto N, Anami K,
Naito Y and Oue N: Molecular pathology of gastric cancer: Research
and practice. Pathol Res Pract. 207:608–612. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baniak N, Senger JL, Ahmed S, Kanthan SC
and Kanthan R: Gastric biomarkers: A global review. World J Surg
Oncol. 14:2122016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nagafuchi A, Tsukita S and Takeichi M:
Transmembrane control of cadherin-mediated cell-cell adhesion.
Semin Cell Biol. 4:175–181. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takeichi M: Cadherin cell adhesion
receptors as a morphogenetic regulator. Science. 251:1451–1455.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wheelock MJ and Johnson KR: Cadherins as
modulators of cellular phenotype. Annu Rev Cell Dev Biol.
19:207–235. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Takeichi M: Cadherins in cancer:
Implications for invasion and metastasis. Curr Opin Cell Biol.
5:806–811. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mayer B, Johnson JP, Leitl F, Jauch KW,
Heiss MM, Schildberg FW, Birchmeier W and Funke I: E-cadherin
expression in primary and metastatic gastric cancer:
Down-regulation correlates with cellular dedifferentiation and
glandular disintegration. Cancer Res. 53:1690–1695. 1993.PubMed/NCBI
|
|
14
|
Bringuier PP, Umbas R, Schaafsma HE,
Karthaus HF, Debruyne FM and Schalken JA: Decreased E-cadherin
immunoreactivity correlates with poor survival in patients with
bladder tumors. Cancer Res. 53:3241–3245. 1993.PubMed/NCBI
|
|
15
|
Oka H, Shiozaki H, Kobayashi K, Inoue M,
Tahara H, Kobayashi T, Takatsuka Y, Matsuyoshi N, Hirano S,
Takeichi M, et al: Expression of E-cadherin cell adhesion molecules
in human breast cancer tissues and its relationship to metastasis.
Cancer Res. 53:1696–1701. 1993.PubMed/NCBI
|
|
16
|
Angst BD, Marcozzi C and Magee AI: The
cadherin superfamily: Diversity in form and function. J Cell Sci.
114:629–641. 2001.PubMed/NCBI
|
|
17
|
Takeuchi T and Ohtsuki Y: Recent progress
in T-cadherin (CDH13, H-cadherin) research. Histol Histopathol.
16:1287–1293. 2001.PubMed/NCBI
|
|
18
|
Philippova M, Joshi MB, Kyriakakis E,
Pfaff D, Erne P and Resink TJ: A guide and guard: The many faces of
T-cadherin. Cell Signal. 21:1035–1044. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Andreeva AV and Kutuzov MA: Cadherin 13 in
cancer. Genes Chromosomes Cancer. 49:775–790. 2010.PubMed/NCBI
|
|
20
|
Lee SW: H-cadherin, a novel cadherin with
growth inhibitory functions and diminished expression in human
breast cancer. Nat Med. 2:776–782. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang XD, Wang BE, Soriano R, Zha J, Zhang
Z, Modrusan Z, Cunha GR and Gao WQ: Expression profiling of the
mouse prostate after castration and hormone replacement:
Implication of H-cadherin in prostate tumorigenesis.
Differentiation. 75:219–234. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pfaff D, Philippova M, Kyriakakis E,
Maslova K, Rupp K, Buechner SA, Iezzi G, Spagnoli GC, Erne P and
Resink TJ: Paradoxical effects of T-cadherin on squamous cell
carcinoma: Up- and down-regulation increase xenograft growth by
distinct mechanisms. J Pathol. 225:512–524. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang Y, Dai Y and Huo J: Decreased
expression of T-cadherin is associated with gastric cancer
prognosis. Hepatogastroenterology. 59:1294–1298. 2012.PubMed/NCBI
|
|
24
|
Mori Y, Matsunaga M, Abe T, Fukushige S,
Miura K, Sunamura M, Shiiba K, Sato M, Nukiwa T and Horii A:
Chromosome band 16q24 is frequently deleted in human gastric
cancer. Br J Cancer. 80:556–562. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lindblom A, Rotstein S, Skoog L,
Nordenskjöld M and Larsson C: Deletions on chromosome 16 in primary
familial breast carcinomas are associated with development of
distant metastases. Cancer Res. 53:3707–3711. 1993.PubMed/NCBI
|
|
27
|
Carter BS, Ewing CM, Ward WS, Treiger BF,
Aalders TW, Schalken JA, Epstein JI and Isaacs WB: Allelic loss of
chromosomes 16q and 10q in human prostate cancer. Proc Natl Acad
Sci USA. 87:8751–8755. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen T, Sahin A and Aldaz CM: Deletion map
of chromosome 16q in ductal carcinoma in situ of the breast:
Refining a putative tumor suppressor gene region. Cancer Res.
56:5605–5609. 1996.PubMed/NCBI
|
|
29
|
Wei B, Shi H, Lu X, Shi A, Cheng Y and
Dong L: Association between the expression of T-cadherin and
vascular endothelial growth factor and the prognosis of patients
with gastric cancer. Mol Med Rep. 12:2075–2081. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pfaff D, Philippova M, Buechner SA,
Maslova K, Mathys T, Erne P and Resink TJ: T-cadherin loss induces
an invasive phenotype in human keratinocytes and squamous cell
carcinoma (SCC) cells in vitro and is associated with malignant
transformation of cutaneous SCC in vivo. Br J Dermatol.
163:353–363. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bosserhoff AK, Ellmann L, Quast AS, Eberle
J, Boyle GM and Kuphal S: Loss of T-cadherin (CDH-13) regulates AKT
signaling and desensitizes cells to apoptosis in melanoma. Mol
Carcinog. 53:635–647. 2014.PubMed/NCBI
|
|
32
|
Philippova M, Pfaff D, Kyriakakis E,
Buechner SA, Iezzi G, Spagnoli GC, Schoenenberger AW, Erne P and
Resink TJ: T-cadherin loss promotes experimental metastasis of
squamous cell carcinoma. Eur J Cancer. 49:2048–2058. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ivanov D, Philippova M, Allenspach R, Erne
P and Resink T: T-cadherin upregulation correlates with cell-cycle
progression and promotes proliferation of vascular cells.
Cardiovasc Res. 64:132–143. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhong Y, Lopez-Barcons L, Haigentz M Jr,
Ling YH and Perez-Soler R: Exogenous expression of H-cadherin in
CHO cells regulates contact inhibition of cell growth by inducing
p21 expression. Int J Oncol. 24:1573–1579. 2004.PubMed/NCBI
|
|
35
|
Mukoyama Y, Zhou S, Miyachi Y and
Matsuyoshi N: T-cadherin negatively regulates the proliferation of
cutaneous squamous carcinoma cells. J Invest Dermatol. 124:833–838.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang ZY, Wu Y, Hedrick N and Gutmann DH:
T-cadherin-mediated cell growth regulation involves G2 phase arrest
and requires p21(CIP1/WAF1) expression. Mol Cell Biol. 23:566–578.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hermiston ML and Gordon JI: Inflammatory
bowel disease and adenomas in mice expressing a dominant negative
N-cadherin. Science. 270:1203–1207. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Behrens J: The role of cell adhesion
molecules in cancer invasion and metastasis. Breast Cancer Res
Treat. 24:175–184. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pecina-Slaus N: Tumor suppressor gene
E-cadherin and its role in normal and malignant cells. Cancer Cell
Int. 3:172003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Venza M, Visalli M, Catalano T, Biondo C,
Beninati C, Teti D and Venza I: DNA methylation-induced E-cadherin
silencing is correlated with the clinicopathological features of
melanoma. Oncol Rep. 35:2451–2460. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tamura G, Yin J, Wang S, Fleisher AS, Zou
T, Abraham JM, Kong D, Smolinski KN, Wilson KT, James SP, et al:
E-Cadherin gene promoter hypermethylation in primary human gastric
carcinomas. J Natl Cancer Inst. 92:569–573. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hibi K, Kodera Y, Ito K, Akiyama S and
Nakao A: Methylation pattern of CDH13 gene in digestive tract
cancers. Br J Cancer. 91:1139–1142. 2004.PubMed/NCBI
|
|
43
|
Schoumacher M, Goldman RD, Louvard D and
Vignjevic DM: Actin, microtubules and vimentin intermediate
filaments cooperate for elongation of invadopodia. J Cell Biol.
189:541–556. 2010. View Article : Google Scholar : PubMed/NCBI
|