Introduction
Excessive inflammatory responses contribute to and
aggravate various autoimmune/chronic diseases, including
inflammatory bowel disease (IBD) (1). Crohn's disease and ulcerative colitis
are examples of major IBDs of the gastrointestinal tract; both have
a similar profile when they present in the colon, including
peripheral symptoms such as weight loss and fever, and various
colonic specific symptoms including gastric dysmotility, colonic
mucosal ulceration, shortening of the colon, and diarrhea (1–4). The
imbalance in the mucosal immune response leads to the
overproduction of inflammatory cytokines, chemokines, oxidants and
matrix metalloproteinases, which in turn results in prolonged
inflammatory responses and irreparable tissue damage (5–9).
Previous findings have shown the pathogenesis of IBD; however, its
etiology remains poorly understood (5–9).
Patients with IBD are typically treated using pharmacological
agents and strategies, such as anti-inflammatory agents,
immunomodulatory therapies, monoclonal antibody therapies and
leukapheresis, which target abnormal immune responses and
uncontrolled inflammation (4,10–14).
Sodium 4-phenylbutyrate (PBA) is an aromatic fatty
acid analog that is typically used to treat urea cycle disorders
(15). PBA has also shown potential
as a therapeutic treatment in many other diseases, including
homozygous β-thalassemia, spinal muscular atrophy, and a variety of
tumors (16,17). It has been reported that PBA may act
as a histone deacetylase inhibitor, serve as a chemical chaperone,
or act as an ammonia scavenger (17). Furthermore, PBA has been found to
suppress endoplasmic reticulum stress and exert anti-inflammatory
effects (18–21). The authors of the present study have
previously reported that PBA may function as a therapeutic reagent
for neurodegenerative disorders, and that intraperitoneal
administration of PBA suppresses the onset of experimental murine
colitis (22,23).
The aim of the present study was to investigate the
effects of orally administered PBA, on colonic inflammation in
DSS-induced colitis in mice, which is a standard mouse model of
IBD. Oral administration is a more clinically relevant route of
administration than intraperitoneal, and so the results of the
present study may have practical uses for the treatment of IBD.
Materials and methods
Experimental animals
A total of 40 5-week-old male ICR mice (28–30 g)
were purchased from Kyudo Co., Ltd. (Saga, Japan) and treated as
previously published (23). Briefly,
the mice were housed in cages (5 mice per cage) at a controlled
temperature of 20±5°C, relative humidity, 60±10% and a 12-h
light-dark cycle. The mice were fed CE-2 (Kyudo) and normal
drinking water ad libitum. The animals were divided randomly
into the following groups: DSS-non treatment control (normal
control; n=10), DSS-treated control (DSS control; n=10), or
DSS-treated with the addition of 5 (PBA 5; n=5), 7.5 (PBA 7.5;
n=5), or 10 mg (PBA10; n=10) PBA every 12 h. Survival, development
of DSS-induced colitis, and levels of inflammatory cytokines were
analyzed daily during the experiment and colon length and
histopathology were evaluated on day 12. The mice were monitored
throughout the experiment every day. All animal experiments were
conducted under university guidelines and were approved by the
Ethical Committee for Animal Care and Use of Fukuoka University
(Fukuoka, Japan).
DSS and PBA treatment
Experimental colitis was induced by the addition of
3.5% (w/v) DSS to the drinking water of the mice as previously
described (23). PBA (LKT
Laboratories, Inc., Saint Paul, MN, USA) was administered orally by
gavage at a dose of 5, 7.5 or 10 mg every 12 h.
Assessment of DSS-induced colitis
Experimental murine colitis was evaluated using a
disease activity index (DAI) as previously described (23–25).
Briefly, the DAI assigns a score to weight loss, blood in the
stool, and stool consistency. The scoring for weight loss, as the
percentage difference from weight on day 0, was as follows: 0,
<1%; 1, 1–5%; 2, 5–10%; 3, 10–20%; and 4, >20%. For stool
consistency, scoring was: 0, normal; 2, loose stool; 4, diarrhea.
Finally, scoring for stool blood was as follows: 0, negative; 2,
Hemoccult positive; 4, gross bleeding.
Measurement of cytokines using
ELISA
Cytokine levels were measured as previously
described (23). Briefly, 96-well
plates were coated overnight at 4°C with the following capture
antibodies: Anti-mouse tumor necrosis factor (TNF)-α (cat. no.
14-7325; 2.0 µg/ml), anti-mouse/rat interleukin (IL)-1β (cat. no.
14-7012; 2.0 µg/ml) and anti-mouse IL-6 (cat. no. 14-7061; 1.0
µg/ml; all eBioscience Inc.; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). ELISA/ELISPOT diluent as a blocking buffer (250
µl 5X diluent; eBioscience Inc.; Thermo Fisher Scientific, Inc.)
was added to each well for 1 h at room temperature and washed with
TBS containing 0.1% Tween-20 (TBST). The collected colonic lavage
fluid using a 1 ml syringe and mouse feeding needle; 100 µl of PBS
was slowly injected into the rectum, left for 30 sec and collected
slowly. The different between individuals was limited and ~90% of
the initial PBS was collected. The collected colonic lavage was
added at a 1:10 dilution in ELISA/ELISPOT diluent. The plates were
further incubated for 1 h at room temperature and washed with TBST.
Detection antibodies (0.8 µg/ml biotinylated anti-mouse TNF-α;
13-7326, 2.0 µg/ml biotinylated anti-mouse/rat IL-1β; 13-7112 and
1.0 µg/ml biotinylated anti-mouse IL-6; 137062; all purchased from
eBioscience Inc.; Thermo Fisher Scientific, Inc.) were added and
the plates were incubated for 1 h at room temperature. Horseradish
peroxidase-conjugated streptavidin (SNN 1004; 1:10,000; Biosource;
Thermo Fisher Scientific, Inc.) was added and the plates were
incubated again for 1 h at room temperature. Plates were washed
with TBST thoroughly, and 75 µl of 3,3′,5,5′-tetramethylbenzidine
(TMB) solution (TMB Microwell Peroxidase Substrate System; SeraCare
Life Sciences, Milford, MA, USA) was subsequently added to each
well for 10 min at room temperature, followed by an equal volume of
stop solution (1M H2SO4). The optical density
at 490 nm was read using an ELISA microplate reader (Bio-Rad Model
450 Microplate Reader; Bio-Rad Laboratories, Inc., Hercules, CA,
USA). All anti-cytokine antibodies were purchased from eBioscience
Inc.; Thermo Fisher Scientific, Inc.
Measurement of colon length and
histopathological evaluation
At the end of the experiment, all the mice were
euthanized by cervical dislocation following anesthesia with
isoflurane. A section of the colon extending from the cecocolic
junction to the anus was harvested as previously described
(23). The colon length was defined
as the length of the isolated tissue sample as above. The excised
tissue was fixed overnight in 10% neutral-buffered formalin
(Nacalai Tesque, Inc., Kyoto, Japan) at room temperature and
embedded in paraffin blocks. The paraffin-embedded tissues were cut
using a microtome and 5 µm sections were stained with hematoxylin
and eosin for microscopic examination.
Statistical analysis
Survival data were analyzed using the log-rank test.
DAI score data and ELISA data were analyzed by Student's t-test.
Colon length data were analyzed by the Tukey-Kramer post-hoc test.
Data are presented as the mean ± standard error of the mean and
statistical analysis was performed using GraphPad Prism Software,
version 6 (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
Survival rate
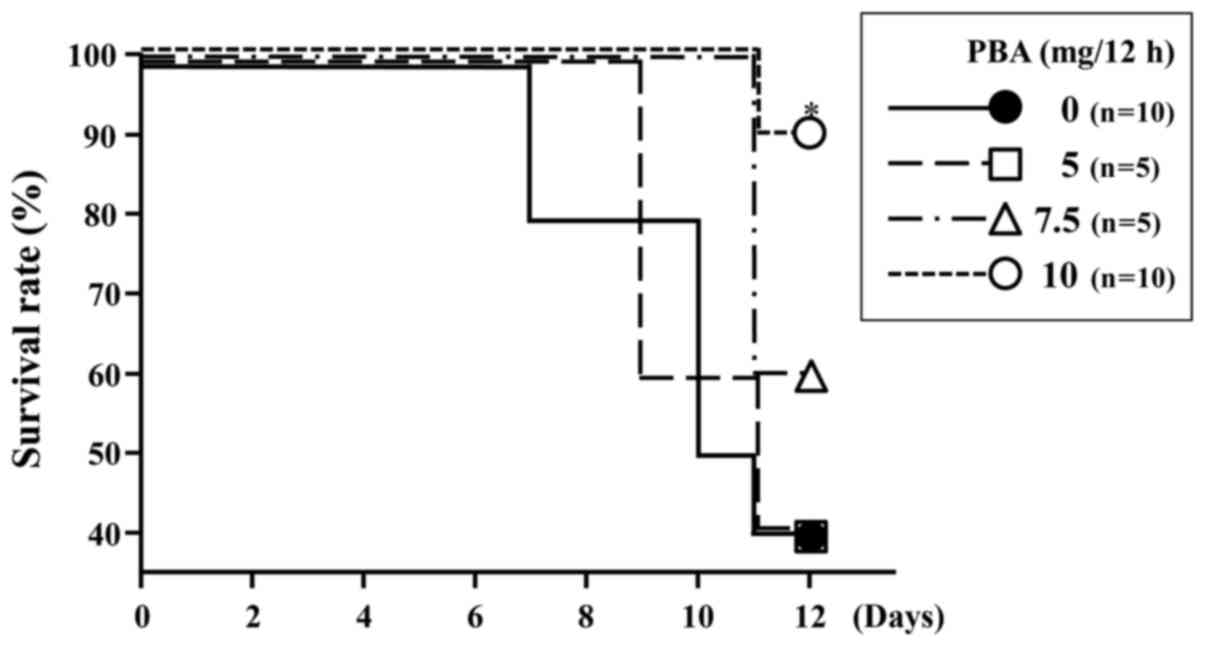
At the end of the experiment, the survival rate in
the DSS control group was 40% (4/10). The survival rates of mice
treated with DSS and PBA were 40% (2/5), 60% (3/5) and 90% (9/10)
for the PBA 5, PBA 7.5 and PBA 10 groups, respectively (Fig. 1). The survival rate for mice treated
with 10 mg PBA per 12 h was significantly higher than for rats in
the DSS control group (P=0.0156; Fig.
1). In the following experiments, only the results pertaining
to the 10 mg PBA per 12 h group are reported (henceforth denoted as
the PBA group), as this was the group yielding the highest
survival.
DAI
Over the experimental period, there were 5 cases of
severe weight loss (DAI score ≥3) in the DSS control group. By
contrast, there was only 1 case of severe weight loss in the PBA
group, which was detected on day 10 (Fig. 2A). The mice in the DSS control group
experienced a time-dependent deterioration in the condition of
their stools, and ultimately there were 7 cases of diarrhea in the
group (score=4). In the PBA group, 9/10 mice had loose stools
(score=2) and not diarrhea. One mouse in the PBA group did have
diarrhea; however, this mouse did not survive until day 12
(Fig. 2B). In the DSS control group,
occult blood (score=2) was observed in 7/10 mice on day 2 and in
the remaining 3 mice on day 4. On day 6, gross bleeding (score=4)
was observed in 5/10 mice. In the PBA group, occult blood was
detected in 5/10 mice on day 2 and 8/10 on day 4. On day 8, 2/10
mice in the PBA group exhibited gross bleeding (Fig. 2C). Overall, the DAI score of the PBA
group was significantly lower than that of the DSS control group
from day 4 until the end of the experiment (P<0.05; Fig. 2D).
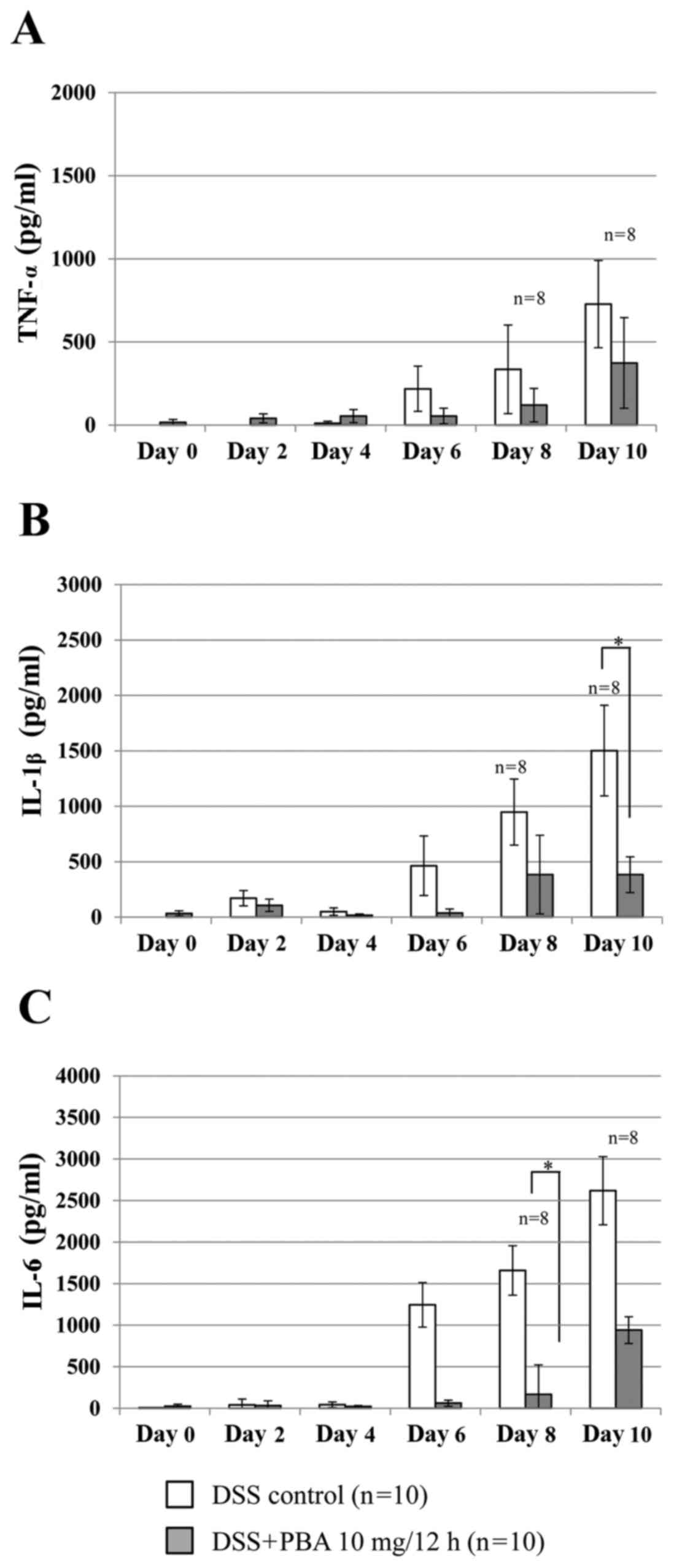
Inflammatory cytokine production
Cytokine concentration was determined using a
standard curve prepared for each plate. In the DSS control group,
TNF-α production measured in the colonic lavage fluid increased
from day 6, reaching 335.1±266.6 pg/ml by day 8 (Fig. 3A). In the PBA group, TNF-α production
was suppressed by 64.2% on day 8 and 48.6% on day 10 compared with
the DSS control group; however, this was not statistically
significant. Similarly, IL-1β production in the PBA group was
significantly suppressed on day 10 by 74.5% (382.4±160.6 pg/ml),
compared with the DSS control group, in which IL-1β production was
1502.0±409.2 pg/ml (P<0.05; Fig.
3B). Furthermore, IL-6 production in the PBA group was
significantly suppressed on day 8 by 89.9% (167.7±135.6 pg/ml),
compared with the DSS control group, in whichIL-6 production was
1658.7±600.8 pg/ml (P<0.05; Fig.
3C).
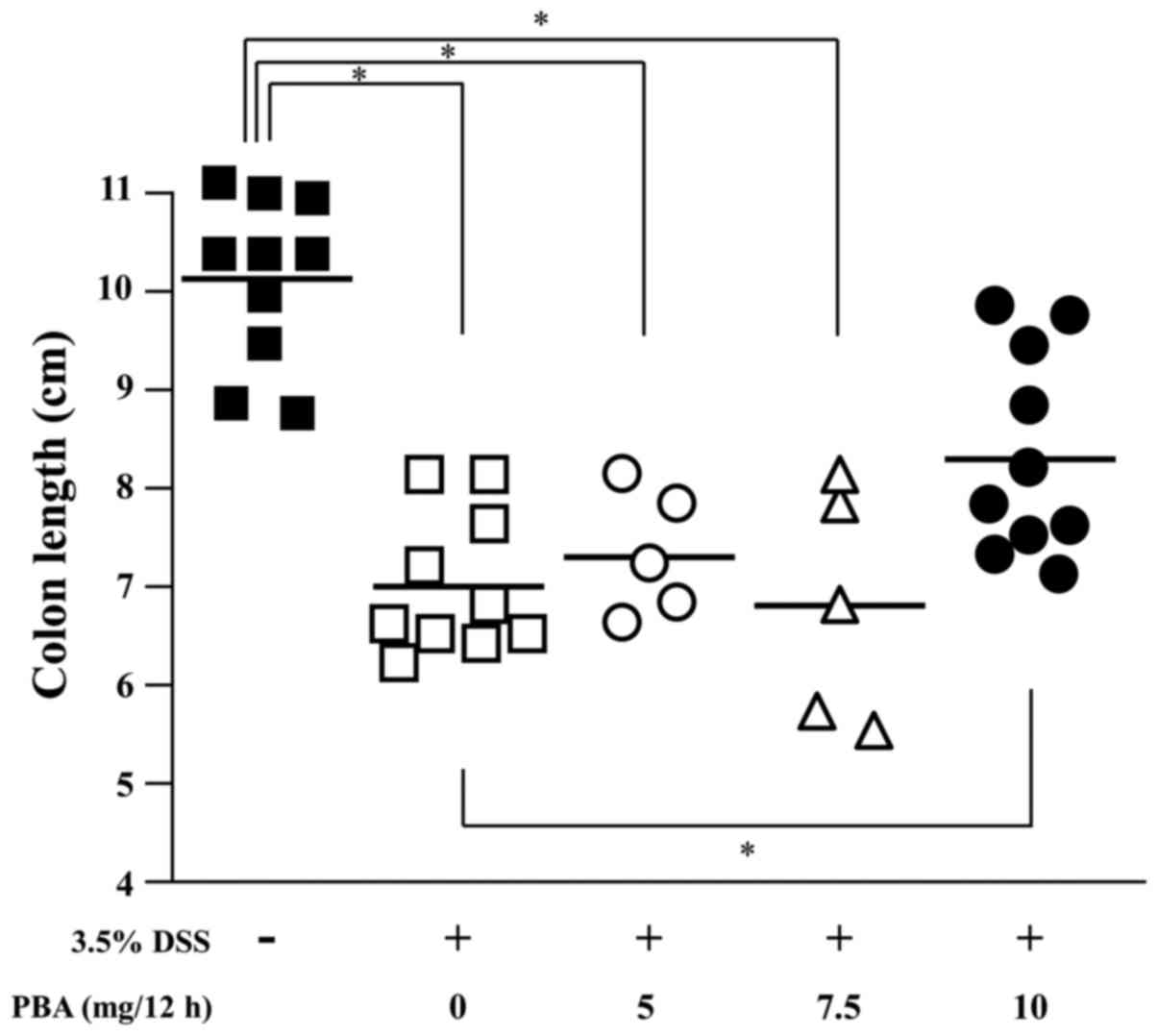
Colon length
The effect of PBA on DSS-induced shortening of the
colon was examined. The length of the colon from the cecocolic
junction to the anal verge was measured following sacrifice, and
tissues were subsequently processed for histology. The mean colon
length was 10.1±0.3 cm in healthy control mice, 7.0±0.2 cm in the
DSS control mice and 8.3±0.3 cm in the PBA-treated mice. The
difference between the mean colon length in the DSS control and PBA
groups indicates a significant suppression of DSS-induced
shortening of the colon in mice treated with 10 mg PBA/12 h
(P<0.05; Fig. 4).
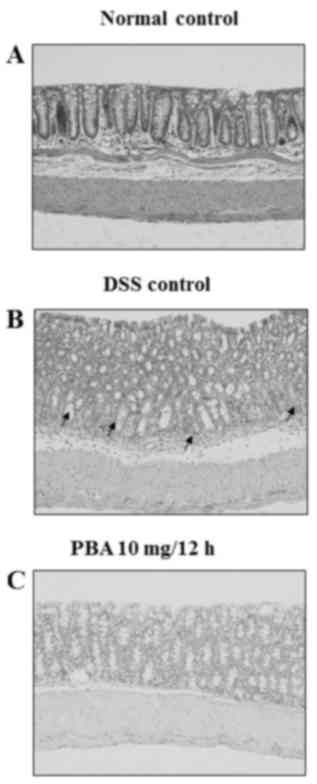
Histological findings
Hematoxylin and eosin staining revealed chronic
inflammatory infiltrates, including lymphocytes and plasma cells,
in the lamina propria around the crypts in the isolated colon
segments from the DSS control group. The surface epithelia were
partially exfoliated compared with the normal control tissue,
revealing the underlying connective tissue of the intestinal
mucosa. The crypts contained no mature goblet cells, which
indicated mucin depletion (Fig. 5A and
B). By contrast, the mucosa from mice in the PBA group had a
normal appearance and resembled that of healthy control mice
(Fig. 5A and C).
Discussion
In the present study, the effect of orally
administered PBA on experimental colitis in mice was investigated.
At the end of experiment, the survival rate of untreated mice was
low (40%) whereas, the survival rate of PBA-treated mice was as
high as 90%. Mice in the PBA group had significantly improved
scores in the DAI, with PBA treatment markedly inhibiting the
decline in weight and deteriorating stool condition. The appearance
of a positive hemoccult result or gross blood in the stools
manifested more slowly in the PBA group than in the DSS control
group. These suggest that PBA treatment delay increased the DAI
score and improved the survival rate. PBA also significantly
attenuated DSS-induced shortening of the colon and resulted in the
maintenance of mucosal integrity to the extent seen in the normal
control group. These results suggest that orally administered PBA
suppresses or limits the development of experimental colitis in a
similar manner to intraperitoneal PBA administration.
In the present study, the concentrations of three
important proinflammatory cytokines (TNF-α, IL-1β and IL-6) in
collected colonic lavage fluids increased in parallel with the
worsening of the disease state in the DSS control group. By
contrast, PBA treatment markedly reduced TNF-α, IL-1β and IL-6
production until day 8. This early inhibition appeared to be
sufficient to suppress the onset of experimental colitis, as
evidenced by the DAI, histopathological findings and improved
survival rates of PBA-treated mice. In the past decade, drugs with
anti-cytokine mechanisms of action (including Infliximab) have been
applied clinically for the treatment of patients with IBDs
(26–28). PBA has the potential to function as
an effective therapeutic treatment that is able to inhibit the
production of inflammatory cytokines.
The pathogenesis of IBD remains unclear and patients
with IBD typically employ symptomatic therapy (5–9).
Furthermore, the few medications that are available that relieve
symptoms are often associated with inescapable adverse events,
which further complicates IBD treatment (4,10–14). In
the present study, orally administered PBA was demonstrated to
delay the onset of experimental colitis. PBA treatment is not
accompanied by the particular side effects of existing IBD
therapeutics, and has the added benefit of an oral route of
administration (15–21). PBA may therefore be one of a new type
of therapeutic agents, which includes anti-TNF-α therapies
(29–33). Although the therapeutic effects of
PBA appear to involve the suppression of pro-inflammatory
cytokines, further studies are required to clarify its precise
functional mechanism. The findings of the present study may
ultimately allow for the clinical use of orally administered PBA to
relieve symptoms and delay the onset of IBD.
Acknowledgements
The present study was supported by the Central
Research Institute of Fukuoka University (grant no. 136003). The
authors would like to thank Dr Tomoyo Yasukochi, Dr Takuya
Nishinakagawa and Dr Mai Hazekawa for advice on the manuscript.
References
|
1
|
de Souza HS and Fiocchi C:
Immunopathogenesis of IBD: Current state of the art. Nat Rev
Gastroenterol Hepatol. 13:13–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Podolsky DK: Inflammatory bowel disease. N
Engl J Med. 347:417–429. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang YZ and Li YY: Inflammatory bowel
disease: Pathogenesis. World J Gastroenterol. 20:91–99. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leitner GC and Vogelsang H:
Pharmacological- and non-pharmacological therapeutic approaches in
inflammatory bowel disease in adults. World J Gastrointest
Pharmacol Ther. 7:5–20. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Coelho T, Andreoletti G, Ashton JJ,
Pengelly RJ, Gao Y, RamaKrishnan A, Batra A, Beattie RM, Williams
AP and Ennis S: Immuno-genomic profiling of patients with
inflammatory bowel disease: A systematic review of genetic and
functional in vivo studies of implicated genes. Inflamm Bowel Dis.
20:1813–1819. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dupaul-Chicoine J, Dagenais M and Saleh M:
Crosstalk between the intestinal microbiota and the innate immune
system in intestinal homeostasis and inflammatory bowel disease.
Inflamm Bowel Dis. 19:2227–2237. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ananthakrishnan AN: Environmental risk
factors for inflammatory bowel diseases: A review. Dig Dis Sci.
60:290–298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maloy KJ and Powrie F: Intestinal
homeostasis and its breakdown in inflammatory bowel disease.
Nature. 474:298–306. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Scharl M and Rogler G: Inflammatory bowel
disease pathogenesis: What is new? Curr Opin Gastroenterol.
28:301–309. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sands BE: Therapy of inflammatory bowel
disease. Gastroenterology. 118 2 Suppl 1:S68–S82. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ko JK and Auyeung KK: Inflammatory bowel
disease: Etiology, pathogenesis and current therapy. Curr Pharm
Des. 20:1082–1096. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pedersen J, Coskun M, Soendergaard C,
Salem M and Nielsen OH: Inflammatory pathways of importance for
management of inflammatory bowel disease. World J Gastroenterol.
20:64–77. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Katz JA: Management of inflammatory bowel
disease in adults. J Dig Dis. 8:65–71. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamamoto T, Umegae S and Matsumoto K:
Mucosal healing in patients with ulcerative colitis during a course
of selective leukocytapheresis therapy: A prospective cohort study.
Inflamm Bowel Dis. 16:1905–1911. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Diaz GA, Krivitzky LS, Mokhtarani M, Rhead
W, Bartley J, Feigenbaum A, Longo N, Berquist W, Berry SA,
Gallagher R, et al: Ammonia control and neurocognitive outcome
among urea cycle disorder patients treated with glycerol
phenylbutyrate. Hepatology. 57:2171–2179. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Iannitti T and Palmieri B: Clinical and
experimental applications of sodium phenylbutyrate. Drugs R D.
11:227–249. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kusaczuk M, Bartoszewicz M and
Cechowska-Pasko M: Phenylbutyric Acid: Simple structure-multiple
effects. Curr Pharm Des. 21:2147–2166. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Roy A, Ghosh A, Jana A, Liu X, Brahmachari
S, Gendelman HE and Pahan K: Sodium phenylbutyrate controls
neuroinflammatory and antioxidant activities and protects
dopaminergic neurons in mouse models of Parkinson's disease. PLoS
One. 7:e381132012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Luo ZF, Feng B, Mu J, Qi W, Zeng W, Guo
YH, Pang Q, Ye ZL, Liu L and Yuan FH: Effects of 4-phenylbutyric
acid on the process and development of diabetic nephropathy induced
in rats by streptozotocin: Regulation of endoplasmic reticulum
stress-oxidative activation. Toxicol Appl Pharmacol. 246:49–57.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park JS, Lee EJ, Lee JC, Kim WK and Kim
HS: Anti-inflammatory effects of short chain fatty acids in
IFN-gamma-stimulated RAW 264.7 murine macrophage cells: involvement
of NF-kappaB and ERK signaling pathways. Int Immunopharmacol.
7:70–77. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Morinaga M, Kon K, Saito H, Arai K, Kusama
H, Uchiyama A, Yamashina S, Ikejima K and Watanabe S: Sodium
4-phenylbutyrate prevents murine dietary steatohepatitis caused by
trans-fatty acid plus fructose. J Clin Biochem Nutr. 57:183–191.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ono K, Ikemoto M, Kawarabayashi T, Ikeda
M, Nishinakagawa T, Hosokawa M, Shoji M, Takahashi M and Nakashima
M: A chemical chaperone, sodium 4-phenylbutyric acid, attenuates
the pathogenic potency in human alpha-synuclein A30P + A53T
transgenic mice. Parkinsonism Relat Disord. 15:649–654. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ono K, Nimura S, Nishinakagawa T,
Hideshima Y, Enjyoji M, Nabeshima K and Nakashima M: Sodium
4-phenylbutyrate suppresses the development of dextran sulfate
sodium-induced colitis in mice. Exp Ther Med. 7:573–578. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wirtz S, Neufert C, Weigmann B and Neurath
MF: Chemically induced mouse models of intestinal inflammation. Nat
Protoc. 2:541–546. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cooper HS, Murthy SN, Shah RS and
Sedergran DJ: Clinicopathologic study of dextran sulfate sodium
experimental murine colitis. Lab Invest. 69:238–249.
1993.PubMed/NCBI
|
|
26
|
Dionne S, D'Agata ID, Hiscott J, Vanounou
T and Seidman EG: Colonic explant production of IL-1and its
receptor antagonist is imbalanced in inflammatory bowel disease
(IBD). Clin Exp Immunol. 112:435–442. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tountas NA, Casini-Raggi V, Yang H, Di
Giovine FS, Vecchi M, Kam L, Melani L, Pizarro TT, Rotter JI and
Cominelli F: Functional and ethnic association of allele 2 of the
interleukin-1 receptor antagonist gene in ulcerative colitis.
Gastroenterology. 117:806–813. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kwon KH, Murakami A, Hayashi R and
Ohigashi H: Interleukin-1beta targets interleukin-6 in progressing
dextran sulfate sodium-induced experimental colitis. Biochem
Biophys Res Commun. 337:647–654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rutgeerts P, Van Assche G and Vermeire S:
Optimizing anti-TNF treatment in inflammatory bowel disease.
Gastroenterology. 126:1593–1610. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yun L and Hanauer S: Selecting appropriate
anti-TNF agents in inflammatory bowel disease. Expert Rev
Gastroenterol Hepatol. 3:235–248. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bosani M, Ardizzone S and Porro GB:
Biologic targeting in the treatment of inflammatory bowel diseases.
Biologics. 3:77–97. 2009.PubMed/NCBI
|
|
32
|
Baert F, Noman M, Vermeire S, Van Assche
G, D'Haens G, Carbonez A and Rutgeerts P: Influence of
immunogenicity on the long-term efficacy of infliximab in Crohn's
disease. N Engl J Med. 348:601–608. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Farrell RJ, Alsahli M, Jeen YT, Falchuk
KR, Peppercorn MA and Michetti P: Intravenous hydrocortisone
premedication reduces antibodies to infliximab in Crohn's disease:
A randomized controlled trial. Gastroenterology. 124:917–924. 2003.
View Article : Google Scholar : PubMed/NCBI
|