Introduction
Treatment for patients with type 1 or advanced type
2 diabetes by use of exogenous insulin places them at high risk for
hypoglycemia. The physiological response to hypoglycemia involves
multiple intrinsic defense mechanisms, which consists of input
signals sent by the central nervous system and release of
epinephrine, glucagon, cortisol and growth hormone. A chief
mechanism among these is the increase in glucagon secretion. In
patients with diabetes, the counter-regulatory glucagon response is
severely compromised. Numerous in vitro and in vivo
studies have proposed that insulin, Zn2+, γ-aminobutyric
acid (GABA) and somatostatin exerts a paracrine control on glucagon
secretion under certain conditions (1,2).
Within the islets, the regulation of glucagon
secretion by glucose and paracrine factors (i.e., β-cell secretory
products) is mediated by electrical machinery comprising a variety
of ion channels that determine the depolarization or
hyperpolarization of α-cells (3).
The intra-islet insulin hypothesis holds that upon decreasing
insulin secretion by β-cells, they may send a signal to α-cells
that contributes to the release of glucagon. Another hypothesis
infers that glucagon secretion is inhibited by zinc ions due to
their effects on adenosine triphosphate (ATP)-sensitive
K+-channels of α-cells (4). In vivo studies have demonstrated
that if insulin or zinc ion infusion into the pancreatic artery is
performed, while animals are made hypoglycemic by an insulin
infusion through the jugular vein, following the end of the
infusion glucagon secretion is stimulated in animals with diabetes,
but not in animals without diabetes (4–6).
GABA serves as an inhibitor in the central nervous
system and at high concentration, it has a marked effect on the
endocrine pancreas (7–9). The former studies have verified that in
animals with repeated hypoglycemia, impaired glucagon and
epinephrine responses are associated with more GABAergic tone in
the ventromedial hypothalamus. In previous studies, activation of
the GABAA receptor (GABAAR) by alprazolam on
day 1 blunted autonomic nervous system (ANS) and neuroendocrine
responses; as a result GABAAR expression decreased on
day 2. However, these results did not fully prove whether the prior
activation had any negative impact on releasing glucagon through
directly influencing pancreatic islets or affecting neural pathways
(probably ANS) (10). However,
certain other studies demonstrated that exogenous GABA did not
cause the release of glucagon from the isolated dog pancreas. In
this species, GABA did not affect the concentration of glucagon
in vivo, regardless of whether administration was oral or by
intravenous injection (11,12). However, different findings were
obtained in human patients, as the circulating levels of glucagon
increased with oral GABA (13,14). By
contrast, in isolated mouse islets cultured with 0 mmol/l glucose,
exogenous GABA inhibited the secretion of glucagon (15). However, limited data are available on
the GABAergic tone within the islets from the hypoglycemic rats.
Therefore, it has remained to be determined whether GABA from
β-cells promotes the effects of glucose on glucagon secretion.
In order to verify this hypothesis, the present
study performed an in vivo experiment comprising three study
groups: Intrapancreatic artery infusion of GABA alone, GABA plus
insulin or insulin alone in Wistar rats with diabetes.
To investigate what channels are involved in the
initiation of glucagon secretion following termination of insulin
plus GABA infusion, the present study also performed diazoxide
(DIA), tolbutamide (TLB) and nifedipine (NIF) intrapancreatic
artery infusions plus GABA and insulin.
Materials and methods
Experimental animals
Approval of the animal experiments was obtained from
the Animal Care and Use Committee of Nanjing Medical University
(Nanjing, China). All procedures were performed with an approved
Home Office project license and in accordance with the Animals
(Scientific Procedures) Act 1986 (UK; amended 2013). All sections
of the present study are in accordance with the Animal Research:
Reporting Of In Vivo Experiments guidelines for reporting
animal research.
A total of 30 male Wistar rats (weight, 350–400 g;
age, 3–4 month old) were obtained from the animal center of Qing
long Shan (Nanjing, China). Animals were housed at 23°C with a 12-h
light/dark cycle, a relative humidity of 40–70%, and were provided
food and water ad libitum.
Diabetes was induced by intraperitoneal injection of
50 mg/kg streptozotocin (STZ; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) dissolved in sodium citrate buffer. At one and two days
after injecting STZ, blood glucose was measured. The rats were
identified as diabetic if the results of two sequential
measurements were >350 mg/dl. The animals that had not become
diabetic were injected with STZ for a second time. Animals were
kept for 2 weeks until they weighed 300–350 g, so that they could
be used for the experiments.
Glucagon response assessment
At 2 weeks after the establishment of diabetes by
STZ injection, the rats were assessed for their complete ability to
respond to hypoglycemia as in the very beginning. Diabetic rats
were fasted overnight, anesthetized and given an insulin (Eli Lilly
and Company, Indianapolis, IN, USA) infusion (0.5 units/ml at 50
µl/min) into the jugular vein so as to cause a hypoglycemic state.
The dynamic data of blood glucose, C-peptide and glucagon were
collected at the time-points of 0, 30, 60 and 90 min following
termination of the infusion. Blood glucose levels were measured
within 2 min of sample collection. Finally, the jugular catheter
was removed and the incision was closed.
Surgical procedure
Isoflurane (2%) was used to anesthetize the
non-fasted rats on the experimental day of the intrapancreatic
artery infusions. The upper abdomen was opened through a 3 cm-long
vertical incision commencing from the xiphoid process. A 25-gauge
needle was used to puncture the isolated and ligated hepatic
artery, into which a microcannula (0.008 mm inner diameter;
Biotime, Inc., Alameda, CA, USA) was inserted in a higher position
for retrograde conduction to the start of the pancreatic artery,
which was ligated over the cannula. The location of the cannula tip
was the bottom of the superior pancreatic duodenal artery, which
carried the infusion fluid through the microcannula and had no
negative impact on supplying blood from the celiac artery to the
pancreatic artery. The right jugular vein was cannulated by
heparin-filled (500 units/ml) polyethylene 50 tubing (BD
Biosciences, Franklin Lakes, NJ, USA), which was placed superior to
the vena cava. The function of the cannula was to help infuse
intravenous insulin and collect blood samples. In order not let the
opened abdomen dry out due to surgery, it was perfused with warm
saline and the surface was covered in foil.
Once the defective glucagon response in the diabetic
animals had been established, animals were considered to have
finished the glucagon response assessment. After 2 weeks of
recovery, the animals were anesthetized and rested for 30 min. In
order to lower blood glucose to 100 mg/dl, insulin (0.5 units/ml at
50 units/min) was infused into the jugular vein. After this
procedure, a basal blood sample was collected, followed by infusion
of the following solutions into the superior pancreaticoduodenal
artery: GABA (50 µmol/l), TLB (50 µmol/l), DIA (50 µmol/l), NIF (1
µmol/l) and insulin (1.5 units/ml). Glucagon levels in hypoglycemic
rats were not significantly different in rats with zinc-free
insulin infusion compared with rats without intrapancreatic artery
infusion; therefore, the control group (n=5) was infused with
zinc-free insulin infusion to explain the role of zinc. Then the
animals were split into three groups and infused with GABA or
Insulin or GABA and insulin (n=5/group). Following that another
group of animals were split into three groups and infused with
GABA, insulin and TLB or GABA or NIF (n=3/group). A total of 1 rat
without intrapancreatic artery infusion was used as blank control
rat as a reference for the control group. The standard of the
concentrations of these solutions was set according to the studies
of Franklin et al (16) and
Mehanna (17). Another blood sample
was collected after 10 min. As soon as the blood glucose was
decreased to <60 mg/dl, the infusions to the pancreatic artery
were terminated and blood samples were collected at the time-points
of 0, 15, 30, 60 and 90 min.
Assays
The aforementioned blood samples (0.3 ml from the
jugular vein at each time points) were stored in 1,000 IU/ml
Trasylol solution in heparin-coated ice-chilled tubes to prevent
glucagon degradation. As for the measurement, the Roche Accucheck
glucometer (Roche, Basel, Switzerland) was used to measure plasma
glucose. Furthermore, rat C-peptide (cat. no. K4757-100) and
glucagon (cat. no. K4756-100) (both Biovision, Inc., Milpitas, CA,
USA) enzyme immunoassay kits were used to measure plasma levels of
glucagon and C-peptide.
Statistical analysis
Data were presented as mean ± standard and
statistically analyzed using either an unpaired Student's t-test or
a one-way analysis of variance with Dunn's post hoc test. These
tests were conducted by GraphPad Prism analytical software (version
5; GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
GABA plus zinc-containing insulin
stimulate α-cells to secrete glucagon in rats with
hypoglycemia
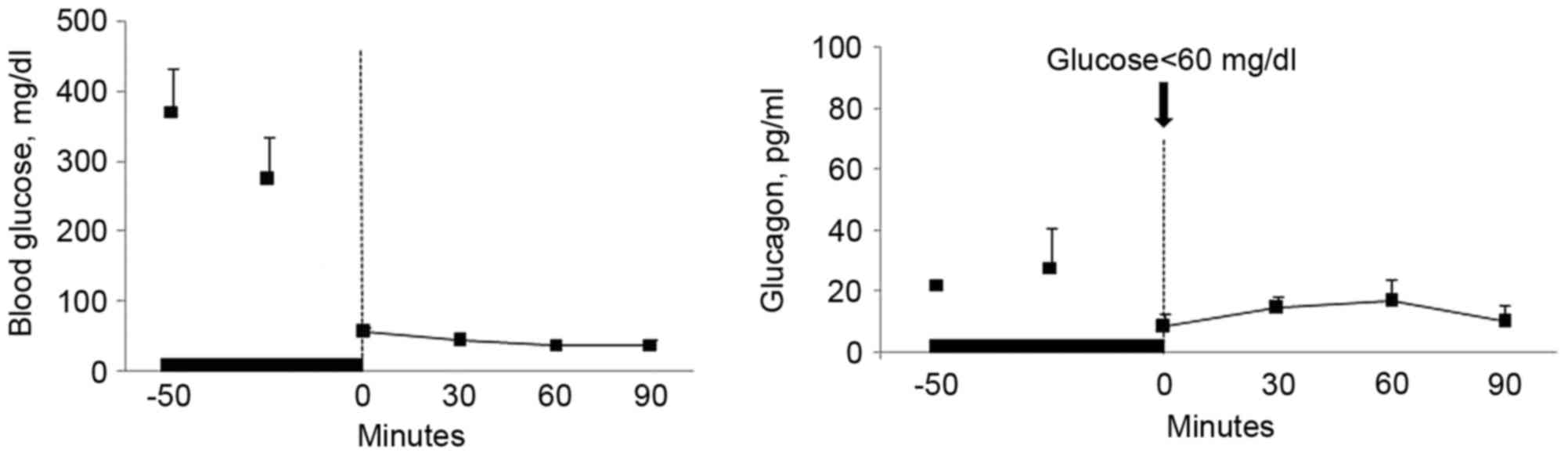
The results demonstrated that termination of
infusion of zinc-free insulin into the pancreatic artery had no
marked effect on glucagon levels in the blood of hypoglycemic rats
(Fig. 1). GABA infusion also had no
marked effect on glucagon levels in the blood of hypoglycemic rats
(Fig. 2). Although glucagon levels
in the insulin group peaked 15 min after infusion the overall level
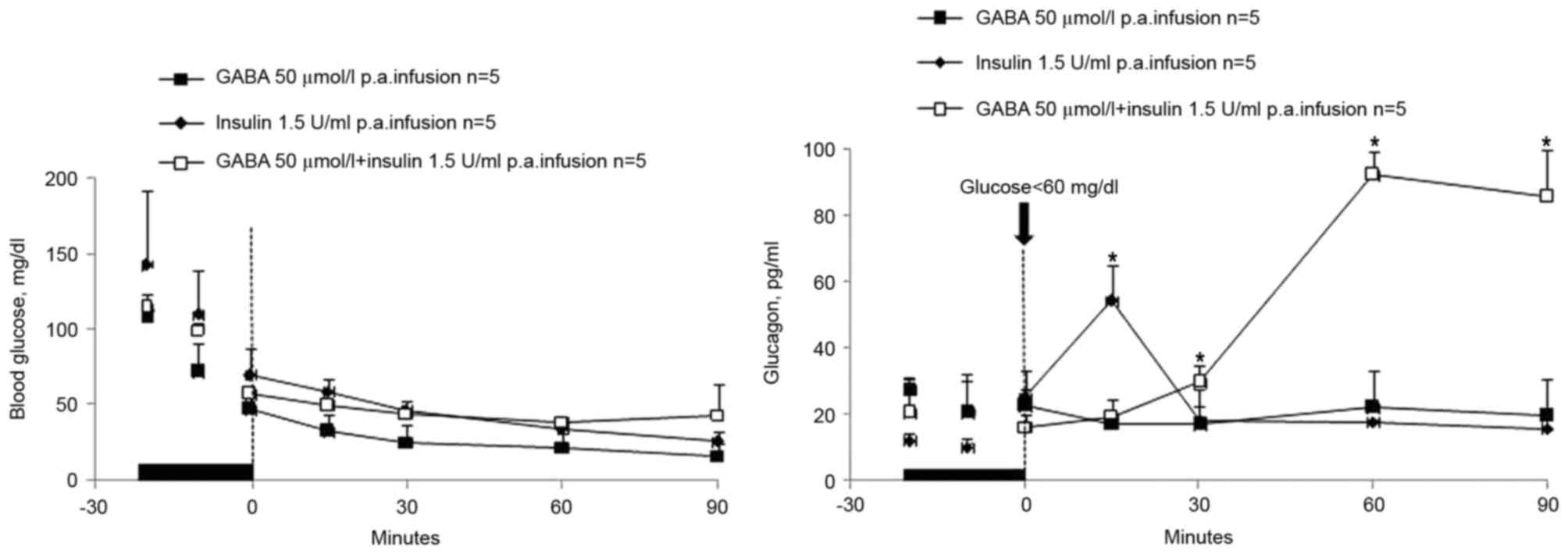
was lower that that of the GABA plus insulin group. Of note,
termination of the GABA plus insulin infusion into the pancreatic
artery promoted glucagon secretion in rats with hypoglycemia, which
was more effective than termination of the infusion of insulin
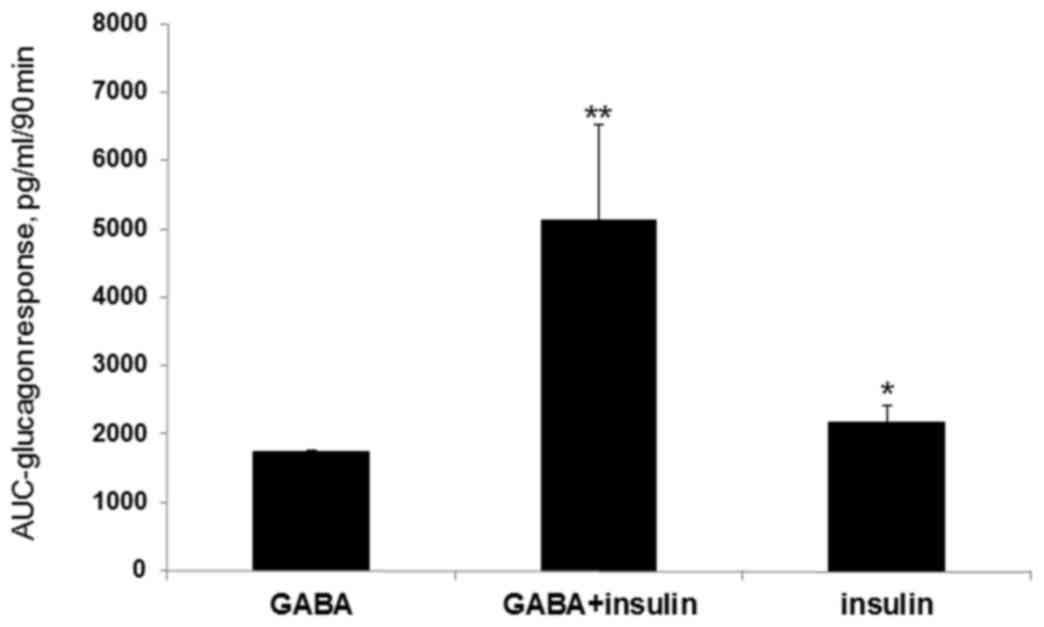
alone (AUC=2,161±260 pg/ml in a 90 min time span; P<0.02;
Figs. 2 and 3). In the GABA plus insulin group, a
significant glucagon response (AUC=5,120±1395 pg/ml in a 90 min
time span; P<0.02; Figs. 2 and
3) was observed compared with the
GABA alone group (AUC=1,749±6.87 pg/ml in a 90 min time span;
P<0.02; Figs. 2 and 3). However, C-peptide levels did not
respond to the termination of the pancreatic arterial infusions of
zinc-free insulin or GABA and were hard to measure (results not
shown).
Ion channel activator and blockers do
not affect glucagon secretion when infused with GABA and
insulin
Furthermore, DIA, TLB or NIF intrapancreatic artery
infusions plus GABA and insulin had no significant effect on
glucagon secretion (Fig. 4). These
results demonstrated that termination of delivery of insulin or
GABA plus zinc-containing insulin, but not of GABA alone, induced
β-cells to send a signal to the α-cells, which stimulated the
latter to secrete glucagon in rats with hypoglycemia. Decrements of
GABA plus zinc-containing insulin caused a greater activation
signal than those of zinc-containing insulin or GABA alone.
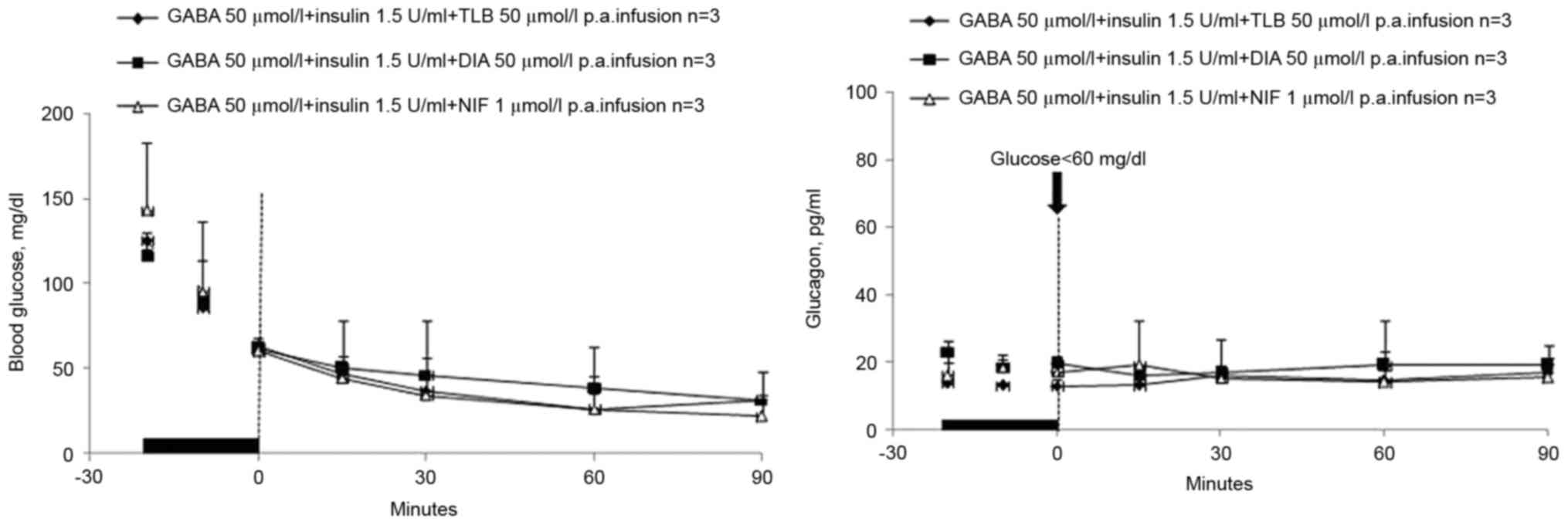 | Figure 4.DIA, TLB or NIF infusions. The
concentration levels of blood glucose and glucagon of diabetic rats
in hypoglycemia in three stages: Prior to, during and after
intrapancreatic artery infusions of GABA (50 µmol/l) + insulin (1.5
U/ml) + TLB (50 µmol/l), GABA (50 µmol/l) + insulin (1.5 U/ml) +
DIA (50 µmol/l), GABA (50 µmol/l) + insulin (1.5 U/ml) + NIF (1
µmol/l) (n=3 per group). The time-point of termination of the
infusions is set as 0 and is indicated by the vertical dashed line.
In all stages, the nadirs of glucose were <60 mg/dl, while
C-peptide was not detectable. Glucagon levels of three groups were
compared with the GABA group. GABA, γ-aminobutyric acid; DIA,
diazoxide; TLB, tolbutamide; NIF, nifedipine. |
Discussion
Various intra-islet insulin hypotheses suggested
that glucagon secretion was suppressed by insulin from β-cells. On
the contrary, in the hypoglycemic state, the marked attenuation of
insulin results in signaling to α-cells to promote glucagon
secretion (18–20).
The present study aimed to assess whether GABA had
any effect on the glucagon secretion after termination of its
infusion with or without insulin. The in vivo experiment on
Wistar rats with streptozotocin-induced diabetes demonstrated that
in the hypoglycemic state, termination of infusion with
zinc-containing insulin and zinc-containing insulin plus GABA but
not GABA alone stimulated α-cells to secrete glucagon. Termination
of infusion of GABA plus zinc-containing insulin resulted in a
greater activation signal than that of zinc-containing insulin
alone.
At the molecular level, insulin activates
intra-islet insulin signaling involving the activation of the
phosphoinositide-3 kinase/Akt signaling pathway, which induces
subsequent phosphorylation of the β-subunit of GABAAR
and the translocation of receptors from the intracellular pool to
the plasma membrane. The resulting increase in the α-cell surface
expression of GABAARs leads to α-cell membrane
hyperpolarization and the suppression of glucagon secretion
(21). As GABA is constantly
released from β-cells (22), the
increased number of GABAARs at the cell surface
increases the efficacy of the receptor-mediated inhibitory currents
(Cl2) and membrane hyperpolarization occurs. In turn, membrane
hyperpolarization shuts down voltage-dependent Ca2+
channels (23), resulting in the
inhibition of cell exocytosis and glucagon release. Göpel et
al (24) demonstrated that in
K+ATP channels, zinc activation occurred, which promoted
β-cell hyperpolarization and had a negative impact on the influx of
cytosolic Ca2+, which has an important role in secreting
glucagon. Slucca et al (25)
suggested that during hypoglycemia, β-cells actually stop releasing
zinc-insulin. The lack of zinc effects the closure of α-cell
K+ATP channels. This in turn leads to depolarization,
which leads to the opening of the voltage-dependent calcium
channels and entry of calcium, which causes glucagon exocytosis. It
many therefore be inferred that upon the interaction of β-cells
with insulin, they release GABA and Zn2+, respectively.
Under hypoglycemia, β-cells cease to cause the release of zinc,
which leads to closure of the α-cell K+ATP channels.
Furthermore, a decrement in GABA secretion decreases the
translocation of GABAAR to the plasma membrane, which
then decreases the efficacy of receptor-mediated Cl2. As a result,
the decrease of K+ATP and Cl− currents causes
depolarization, leading to the opening of calcium channels to allow
for calcium entry and glucagon exocytosis. The fact that GABA,
independent of insulin, cannot have any insulin-like effects is
well appreciated.
However, the finding of the present study that DIA,
TLB or NIF infusions plus GABA and insulin had no effect on
glucagon secretion cannot be explained. Thus, further study is
required to elucidate the effects of K+-channel
activator DIA, K+-channel blocker TLB or calcium channel
blocker NIF on the switch-off signal.
In conclusion, the present study suggested that
during hypoglycemia, α-cells receiving a signal from β-cells
secrete glucagon. Based on these findings, α-cell-produced glucagon
was detected as a result of a sudden decrease in zinc-bearing
insulin and GABA in the periportal circulation. The decrease of the
concentration of zinc-containing insulin and GABA, but not insulin
or GABA alone, may have reduced the K+ATP and
Cl− currents, which may have resulted in depolarization
and thus caused calcium entry and promoted glucagon secretion. The
results of the present study may represent a promising approach for
therapeutic applications. In the future, it will be helpful to
modulate glucagon secretion to improve diabetic complications in
patients.
References
|
1
|
MacDonald PE, De Marinis YZ, Ramracheya R,
Salehi A, Ma X, Johnson PR, Cox R, Eliasson L and Rorsman P: A
K+ATP channel-dependent pathway within alpha cells regulates
glucagon release from both rodent and human islets of Langerhans.
PLoS Biol. 5:e1432007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vieira E, Salehi A and Gylfe E: Glucose
inhibits glucagon secretion by a direct effect on mouse pancreatic
alpha cells. Diabetologia. 50:370–379. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang Q, Liang X and Wang S: Intra-isle
tglucagon secretion and action in the regulation of glucose
homeostasis. Front Physiol. 3:4852013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou H, Zhang T, Harmon JS, Bryan J and
Robertson RP: Zinc, not insulin, regulates the rat alpha-cell
response to hypoglycemia in vivo. Diabetes. 56:1107–1112. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou H, Tran PO, Yang S, Zhang T, LeRoy E,
Oseid E and Robertson RP: Regulation of alpha-cell function by the
beta-cell during hypoglycemia in Wistar rats: The ‘switch-off’
hypothesis. Diabetes. 53:1482–1487. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hope KM, Tran PO, Zhou H, Oseid E, Leroy E
and Robertson RP: Regulation of alpha-cell function by the β-cell
in isolated human and rat islets deprived of glucose: The
‘switch-off’ hypothesis. Diabetes. 53:1488–1495. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Okada Y, Taniguchi H and Schimada C: High
concentration of GABA and high glutamate decarboxylase activity in
rat pancreatic islets and human insulinoma. Science. 194:620–622.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Briel G, Gylfe E, Hellman B and Neuhoff V:
Microdetermination of free amino acids in pancreatic islets
isolated from obese-hyperglycemic mice. Acta Physiol Scand.
84:247–253. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gylfe E and Hellman B: Role of glucose as
a regulator and precursor of aminoacidsin the pancreatic
beta-cells. Endocrinology. 94:1150–1156. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hedrington MS, Farmerie S, Ertl AC, Wang
Z, Tate DB and Davis SN: Effects of antecedent GABAA activation
with alprazolam on counterregulatory responses to hypoglycemia in
healthy humans. Diabetes. 59:1074–1081. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kawai K and Unger RH: Effects of
gamma-aminobutyric acid on insulin, glucagon, and somatostatin
release from isolated perfused dog pancreas. Endocrinology.
113:111–113. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Minuk GY and Sarjeant EJ: Insulin and
glucagon secretion by the dog pancreas during intravenous and oral
administration of gamma aminobutyric acid (GABA). Horm Metab Res.
17:313–314. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cavagnini F, Pinto M, Dubini A, Invitti C,
Cappelletti G and Polli EE: Effects of gamma aminobutyric acid
(GABA) and muscimol on endocrine pancreatic function in man.
Metabolism. 31:73–77. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Passariello N, Giugliano D, Torella R,
Sgambato S, Coppola L and Frascolla N: A possible role of
gamma-aminobutyric acid in the control of the endocrine pancreas. J
Clin Endocrinol Metab. 54:1145–1149. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gilon P, Bertrand G, Loubatières-Mariani
MM, Remacle C and Henquin JC: The influence of gamma-aminobutyric
acid on hormone release by the mouse and rat endocrine pancreas.
Endocrinology. 129:2521–2529. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Franklin I, Gromada J, Gjinovci A,
Theander S and Wollheim CB: Beta cell secretory products activate
alpha cell ATP-dependent potassium channels to inhibit glucagon
release. Diabetes. 54:1808–1815. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mehanna AS: Insulin and oral antidiabetic
agents. Am J Pharmaceutical Ed. 69:1–11. 2005.
|
|
18
|
Samols E, Tyler J and Marks V:
Glucagon-insulin interrelationships. Pergamon Press; Elmsford, NY:
pp. 151–174. 1972
|
|
19
|
Maruyama H, Hisatomi A, Orci L, Orci L,
Grodsky GM and Unger RH: Insulin within islets is a physiologic
glucagon release inhibitor. J Clin Invest. 74:2296–2299. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Raju B and Cryer PE: Loss of the decrement
in intraislet insulin plausibly explains loss of the glucagon
response to hypoglycemia in insulin-deficient diabetes:
Documentation of the intraislet insulin hypothesis in humans.
Diabetes. 54:757–764. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu E, Kumar M, Zhang Y, Ju W, Obata T,
Zhang N, Liu S, Wendt A, Deng S, Ebina Y, et al: Intra-islet
insulin suppresses glucagon release via GABA-GABAA receptor system.
Cell Metab. 3:47–58. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rorsman P, Berggren PO, Bokvist K, Ericson
H, Möhler H, Ostenson CG and Smith PA: Glucose-inhibition of
glucagon secretion involves activation of GABAA-receptor chloride
channels. Nature. 341:233–236. 1989. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Göpel S, Zhang Q, Eliasson L, Ma XS,
Galvanovskis J, Kanno T, Salehi A and Rorsman P: Capacitance
measurements of exocytosisin mouse pancreatic alpha-, beta- and
delta-cells within intact islets of Langerhans. J Physiol.
556:711–726. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Göpel SO, Kanno T, Barg S, Weng XG,
Gromada J and Rorsman P: Regulation of glucagon release in mouse
α-cells by KATP channels and inactivation of TTX-sensitive Na+
channels. J Physiol. 528:509–520. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Slucca M, Harmon JS, Oseid EA, Bryan J and
Robertson RP: ATP-sensitive K+ channel mediates the zinc switch-off
signal for glucagon response during glucose deprivation. Diabetes.
59:128–134. 2010. View Article : Google Scholar : PubMed/NCBI
|